
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent binary ensemble with pattern recognition ability for identifying multiple metalloproteins with applications in serum and urine†
Demin Zhenga,
Junmei Fana,
Xinyan Huangb,
Liping Ding *a and
Yunhong Xinb
*a and
Yunhong Xinb
aKey Laboratory of Applied Surface and Colloid Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, PR China. E-mail: dinglp33@snnu.edu.cn; Tel: +86-29-8153-0789
bCollege of Physics and Information Technology, Shaanxi Normal University, Xi'an 710062, PR China
First published on 27th October 2017
Abstract
Discrimination and identification of multiple metalloproteins by a single sensor system are challenging. Based on the strategy of surfactant assembly modulation effect, a strong discriminative sensor based on a pyrene-derived fluorophore and an anionic surfactant assembly was developed. A cholic acid-modified pyrene derivative, 1, was synthesized and it can form supramolecular aggregates in aqueous solution. Its fluorescence emission could be well modulated from monomer-dominant to monomer–excimer co-emission by the anionic surfactant, sodium dodecylbenzene sulfonate (SDBS). Fluorescence measurements found that the binary ensemble based on 1/SDBS exhibited cross-reactive sensing behavior towards different metalloproteins, and principal component analysis revealed that the present ensemble sensor could discriminate 7 different metalloproteins in aqueous solution. Moreover, the discrimination of metalloproteins could be further applied in biological fluids such as serum or urine.
Introduction
Proteins are involved in all biological systems, their localization, expression level and functions are dynamically regulated in live cells.1,2 Therefore, the development of rapid and convenient detection methods for proteins is of critical importance in medical diagnostics, clinical treatment, and proteomics.3,4 Metalloproteins represent a class of proteins that contain a metal ion cofactor or clusters of metal ions.5–7 They account for approximately half of all proteins present in cells. Iron is an essential nutrient for all forms of life,8 and iron-containing proteins play vitally important roles in living systems. These proteins could be used for oxygen transport and storage9 (e.g., hemoglobins (Hb), myoglobins (Mb), etc.), electron transport10 (e.g., cytochrome c (Cyt C)), catalysis11 (e.g., peroxidases, catalases, etc.), iron transport12,13 (e.g., transferrin (TRF), lactoferrin (LF), etc.), iron storage14 (e.g., ferritins (Ferr), hemosiderin, etc.) and so on. Thus, the multiple functions and large variety of iron-containing metalloproteins evokes development of effective methods for identification and discrimination of different metalloproteins.For this purpose, fluorescent sensor arrays have been widely developed and used for differentiation of metalloproteins due to the high sensitivity, cross-reactive responses and pattern recognition ability of such sensor arrays. Fluorescent sensor arrays are usually composed of a number of cross-reactive sensor elements. Each sensor element provides a non-selective response signal to a particular analyte. Thus, the combination of the output signals from all the sensor elements could generate distinguishable recognition patterns for similar analytes and realize the discrimination among them.15 For example, Sukwattanasinitt and co-workers made use of dendritic phenylene–ethynylene fluorophores to discriminate Cyt C, Mb, bovine serum albumin (BSA), human serum albumin (HSA) and other nonmetalloproteins.16 Liu et al. put forward a strategy of using time-resolved phosphorescent sensor array based on quantum dots for recognition of a variety of proteins including three metalloproteins like horseradish peroxidase (HRP), TRF, Cyt C and some nonmetalloproteins.17 They also proposed a multidimensional sensor array based on DNA–AuNP conjugates with the triple optical plasmonic features for discrimination of TRF, Mb, HRP and so on.18 Thayumanavan et al. made use of a single surfactant assembly receptor encapsulating a variety of fluorescent transducers to generate a sensor array for discriminating Ferr, Hb, Mb and Cyt C.19 Furthermore, they exploited the use of complexes between polyelectrolytes and surfactants with varied structures and charges to generate a sensor array for discriminating metalloproteins like Cyt C, Ferr, Hb, Mb and some nonmetalloproteins.20 Rotello et al. used six functionalized poly(p-phenyleneethynylene)s (PPEs) to build a protein sensor array to discriminate metalloproteins like Cyt C, Ferr, Hb, Mb and some nonmetalloproteins.21 However, the use of sensor arrays usually needs large amount of consumption of sensor samples, and the data collection process is also tedious when different fluorophores are used. Thus, low-number element-based arrays or single discriminative sensors are highly demanded. In this respect, Yan and co-workers developed a single discriminative sensor for discrimination of proteins including Cyt C, Mb, Hb, and some nonmetalloproteins, where the multidimensional optical properties (fluorescence, phosphorescence, and light scattering) of Mn–ZnS quantum dots were collected to generate recognition patterns.22 Based on the similar strategy, Wu et al. obtained a quadruple-channel optosensing device based on Mn-doped ZnS quantum dots for protein discrimination by combing the intrinsic fluorescence of proteins as the fourth channel.23
As a different strategy, our group has made efforts using surfactant assemblies to encapsulate and modulate fluorophores to develop sensitive and discriminative sensors for proteins.24–26 In our first try, we found that cationic surfactant DTAB assemblies could well modulate the fluorescence emission of a cholesterol-modified pyrene-based fluorophore that was highly aggregated in aqueous solution. This modulation effect could be further used to sense the protein-induced assembling variation of surfactants and realized the selective detection of two negatively charged nonmetalloproteins.24 When we used a bispyrene fluorophore containing a hydrophilic spacer connecting two pyrene moieties as the probe, the modulation effect by the cationic surfactant DTAB assemblies could be used to achieve discrimination among three different types of proteins.25 However, the discrimination among the same type of proteins could not be realized by this single binary ensemble. We then developed a two-element mini sensor array to accomplish discriminating metalloproteins using the strategy of surfactant assemblies encapsulating two bispyrene fluorophores.26
As a continuous effort, in the present work, we intended to develop a single discriminative fluorescent sensor for recognizing more iron-containing metalloproteins and explore its practical applications in biological fluids. Therefore, we designed and synthesized a cationic pyrene-based fluorophore, 1, containing a cholic acid group and encapsulated it in anionic surfactant (SDBS) assemblies to function as a discriminative fluorescent sensor. The introduction of cholic acid was to provide the assembling ability of the probe and then enhance the modulation effect by the surfactant assemblies; and the presence of the cationic boronic acid group was to present charges and receptor for glycoproteins. The structure of 1 is illustrated in Scheme 1. The cross-reactive sensing behaviors of the binary ensemble based on 1/SDBS to different iron-containing proteins were examined, and the principal component analysis (PCA) revealed that the binary ensemble sensor could discriminate 7 different iron-containing proteins. Moreover, this sensing platform could also be used to detect various metalloproteins in complex biological fluids such as serum or urine.
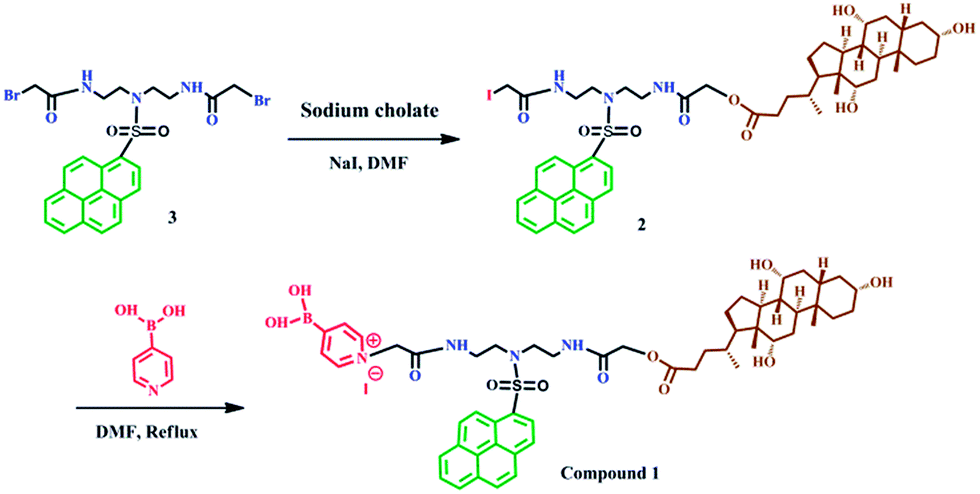 | ||
| Scheme 1 Synthesis of compound 1. | ||
Results and discussion
UV-vis absorption and fluorescence emission of 1 in neat solvents
The UV-vis absorption and steady state fluorescence emission spectra of 1 were measured in methanol and water to evaluate its basic photophysical properties. As seen in Fig. 1a, the UV-vis absorption spectrum of the fluorophore (5 µM) in methanol exhibits the typical pyrene absorption peaks at 279 and 351 nm. The molar absorption coefficient of 1 in methanol at 351 nm was determined to be (3.6 ± 0.1) × 104 L mol−1 cm−1, with R2 values of 0.996 (Fig. S1, ESI†). The emission spectra of the fluorophore (5 µM) in methanol shows strong monomer peaks at shorter wavelengths and hardly displays any excimer bands at longer wavelengths. Interestingly, similar emission profile is also observed in aqueous solution, where only monomer emission peaks appear in the spectrum. This is quite strange since cholic acid has strong aggregating ability in aqueous solution.27–29 To check if aggregation occurs in water, the TEM image of the probe (5 µM) in water was measured. As shown in Fig. 1b, the probe actually formed strong aggregates in the aqueous solution, which is about 200 nm in diameter. This is in agreement with the DLS measurements that show the diameter of the aggregate is 233 nm. The strong Tyndall effect of the probe in aqueous solution also proves the formation of aggregates (inset of Fig. 1b). Such results confirm the assembling behaviors associated with the cholic acid units. The absence of excimer emission, an extraordinary behavior, for the assembly suggested that the pyrene units may be isolated from each other by locating in different spaces of the assembly of probe 1 (Scheme 2).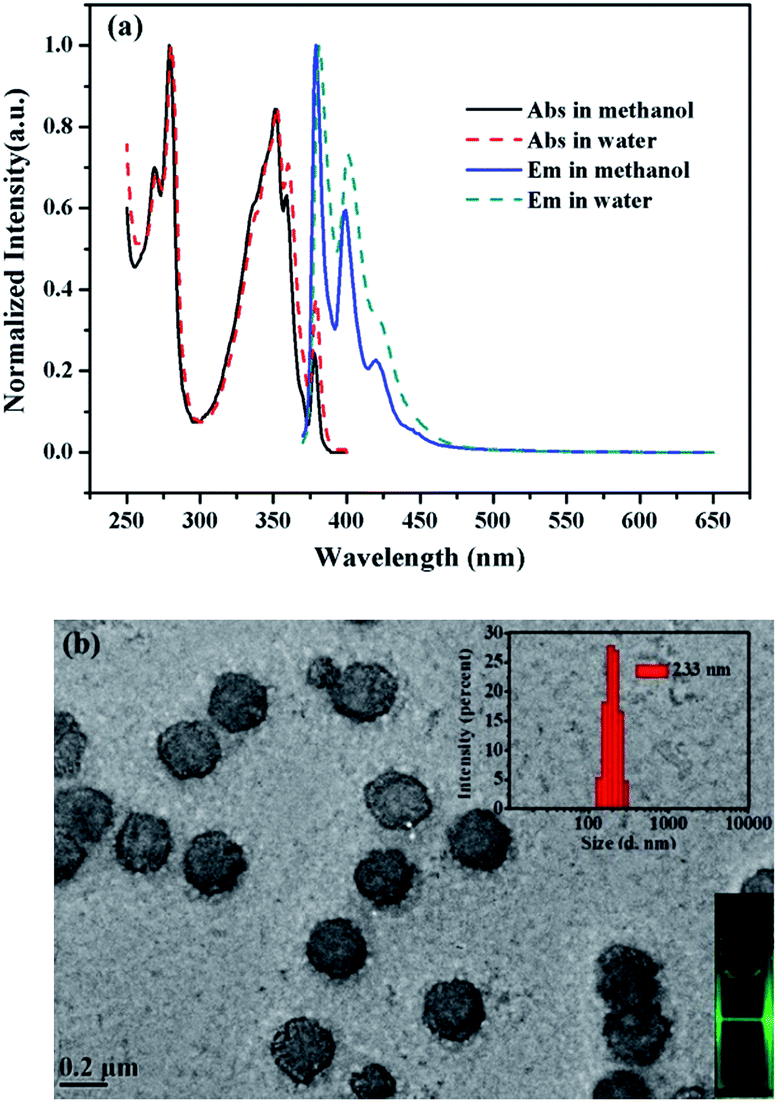 | ||
| Fig. 1 (a) UV-vis absorption and steady-state fluorescence emission spectra of 1 (5 µM) in water and methanol (λex = 354 nm). (b) TEM image, Tyndall effect (inset) and particle size distribution (inset) of assembly of 1 (5 µM) in HEPES buffer solution (10 mM, pH 7.4). | ||
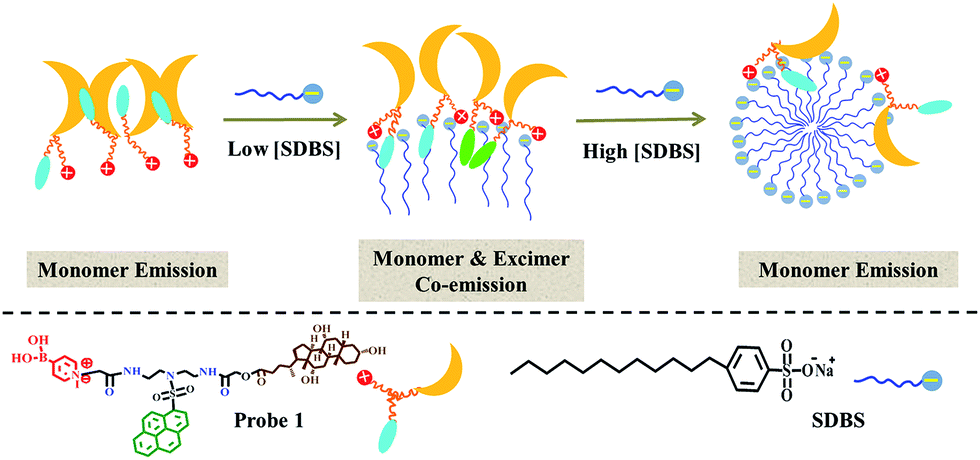 | ||
| Scheme 2 Schematic illustrating SDBS modulation effect on aggregates of 1. | ||
SDBS modulation effect on the fluorescence emission and fluorescence decay of compound 1
It is generally known that the photophysical properties of small molecular probes can be modulated by surfactant assembles due to the change of microenvironment associated with variation of aggregation states. Considering SDBS aggregates possess negative charges and probe 1 has positive charges, we systematically investigated SDBS modulation effect on the fluorescence emission of 1. All the measurements were conducted in HEPES buffer solution (10 mM, pH 7.4). As displayed in Fig. 2a, SDBS assemblies have a distinct effect on modulating the profile of the fluorescence emission of 1 and the intensity of monomer (381 nm) and excimer emission (498 nm). When SDBS concentration is as low as at 50 µM, the probe shows both strong monomer and excimer emission, which is quite different from in neat water. The increasing of SDBS concentration from 50 to 200 µM leads to further enhancement of the excimer emission along with continuously decreasing monomer emission. Further increasing SDBS concentration beyond 400 µM causes reversed fluorescence changes, where the excimer emission decreases accompanied with enhanced monomer emission. Thus, the system in 0.2 mM SDBS shows the highest excimer to monomer ratio and exhibits both distinct monomer and excimer emission, which was then selected as the fluorescent binary ensemble for sensing proteins as it may generate more signal variation to provide multiple-wavelength cross-reactive responses.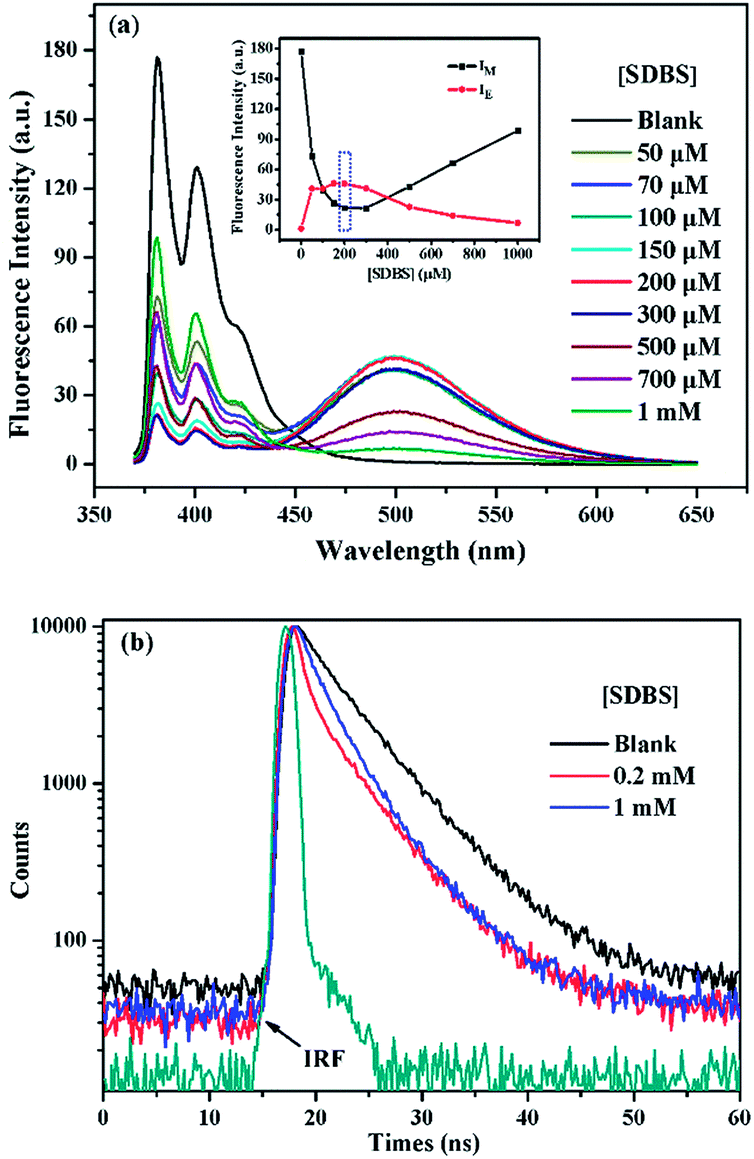 | ||
| Fig. 2 (a) Fluorescence emission of 1 (5 µM) in different concentrated SDBS solutions (10 mM HEPES, pH 7.4). Inset: fluorescence intensity of monomer at 381 nm and excimer at 498 nm of 1 (5 µM) as a function of SDBS concentration. (b) Time-resolved fluorescence decays of 1 (5 µM) in water, 0.2 mM, and 1 mM SDBS solutions (λex = 343.4 nm, λem = 381 nm). | ||
To further understand the SDBS modulation effect on the emission of 1, the time-resolved fluorescence decays of 1 (5 µM) in water, 0.2 mM and 1 mM SDBS solutions were measured. The results are illustrated in Fig. 2b. The corresponding lifetime data and the associated pre-exponential factors are depicted in Table 1. In the absence of SDBS, the pyrene-based probe exhibits bi-exponential decays with more than 89% of the pre-exponential weight obtained for the long decay time, suggesting pyrene moieties prefer to stay in less polar environment provided by the assembly of cholic acid rather than in water (Scheme 2).
| [SDBS] | τ1 (ns) | τ2 (ns) | B1 | B2 | a1 (%) | a2 (%) | χ2 | τ |
|---|---|---|---|---|---|---|---|---|
| 0 | 1.48 | 5.18 | 0.054 | 0.127 | 10.88 | 89.12 | 1.18 | 4.78 |
| 0.2 mM | 0.62 | 4.59 | 0.266 | 0.060 | 37.70 | 62.30 | 1.38 | 3.10 |
| 1 mM | 1.77 | 4.61 | 0.137 | 0.064 | 45.17 | 54.83 | 1.14 | 3.32 |
The probe exhibits quite similar decay behavior in 0.2 mM SDBS as in 1 mM SDBS, although it shows quite different fluorescence behaviors in the two environments. As depicted in Fig. 2b and in Table 1, in both cases, the probe shows clearly bi-exponential decays, suggesting the pyrene moiety exists in two different states. The strong monomer and excimer emission in 0.2 mM SDBS suggests the two lifetimes may either come from the individually existing pyrene moiety or from the π–π stacks of pyrene moieties. Whereas, the strong monomer emission in 1 mM SDBS indicates that the distance between probe molecules is enlarged by micelles, leading to separation of pyrene moieties. The two lifetimes in this solution may be due to the presence of individual pyrene moiety either inside the SDBS assemblies or outside the assemblies. The modulation effect of SDBS assemblies on the probe is schematically illustrated in Scheme 2.
Pattern recognition behaviors of 1/SDBS to metalloproteins
The sensing behaviors of 1/SDBS (5 µM/0.2 mM) to 7 metalloproteins were examined. The tested metalloproteins have diverse isoelectric points and molecular weights, which include hemoglobin (pI 6.8, Mw 64.5 kDa), myoglobin (pI 7.2, Mw 17.0 kDa), ferritin (pI 4.4, Mw 440.0 kDa), cytochrome c (pI 10.7, Mw 12.3 kDa), transferrin (pI 5.6, Mw 77 kDa), lactoferrin (pI 8.0–9.0, Mw 80 kDa) and horseradish peroxidase (pI 6.6, Mw 44.0 kDa). These proteins are all iron-containing proteins. Moreover, three of them are also glycoproteins, e.g., transferrin, lactoferrin and horseradish peroxidase.As shown in Fig. 3a–g, the ensemble sensor exhibited two main different response modes. First, the ensemble sensor presents turn-off responses to six metalloproteins including Hb, Mb, Ferr, and Cyt C, HRP and LF. As shown in panel a–f of Fig. 3, the fluorescence emission was gradually quenched along increasing the concentration of these metalloproteins. Moreover, the intensity reduction of either monomer or excimer of the ensemble sensor is varying from metalloprotein to metalloprotein. The comparison of fluorescence reduction at 381 nm by different metalloproteins was presented as an example and illustrated in Fig. S2 (ESI†). The other mode is ratiometric response and only observed for titration of TRF. As seen in Fig. 3g, along increasing TRF concentration, monomer emission was enhanced and accompanied by a decreased excimer emission via an isoemissive point at 436 nm. As illustrated in Fig. 2a, the aggregation changes of SDBS assemblies could significantly vary the florescence emission. Thus, the unique ratiometric responses to TRF could be due to the aggregation changes of the sensing ensemble. As revealed by TEM, the sensing ensemble with 0.2 mM SDBS assemblies has a much smaller diameter than the probe aggregation itself, which is ca. 5–7 nm (Fig. S3a, ESI†). The addition of TRF causes slight aggregation size changes (Fig. S3b, ESI†). Similarly, the increase of SDBS concentration (1 mM) also leads to subtle size changes (Fig. S3c, ESI†). Thus, the ratiometric responses to TRF could be possibly because the aggregation variation as caused by increasing SDBS concentration, but it could not be certain due to the lack of quantitative measurements that are hard to perform for the present system.
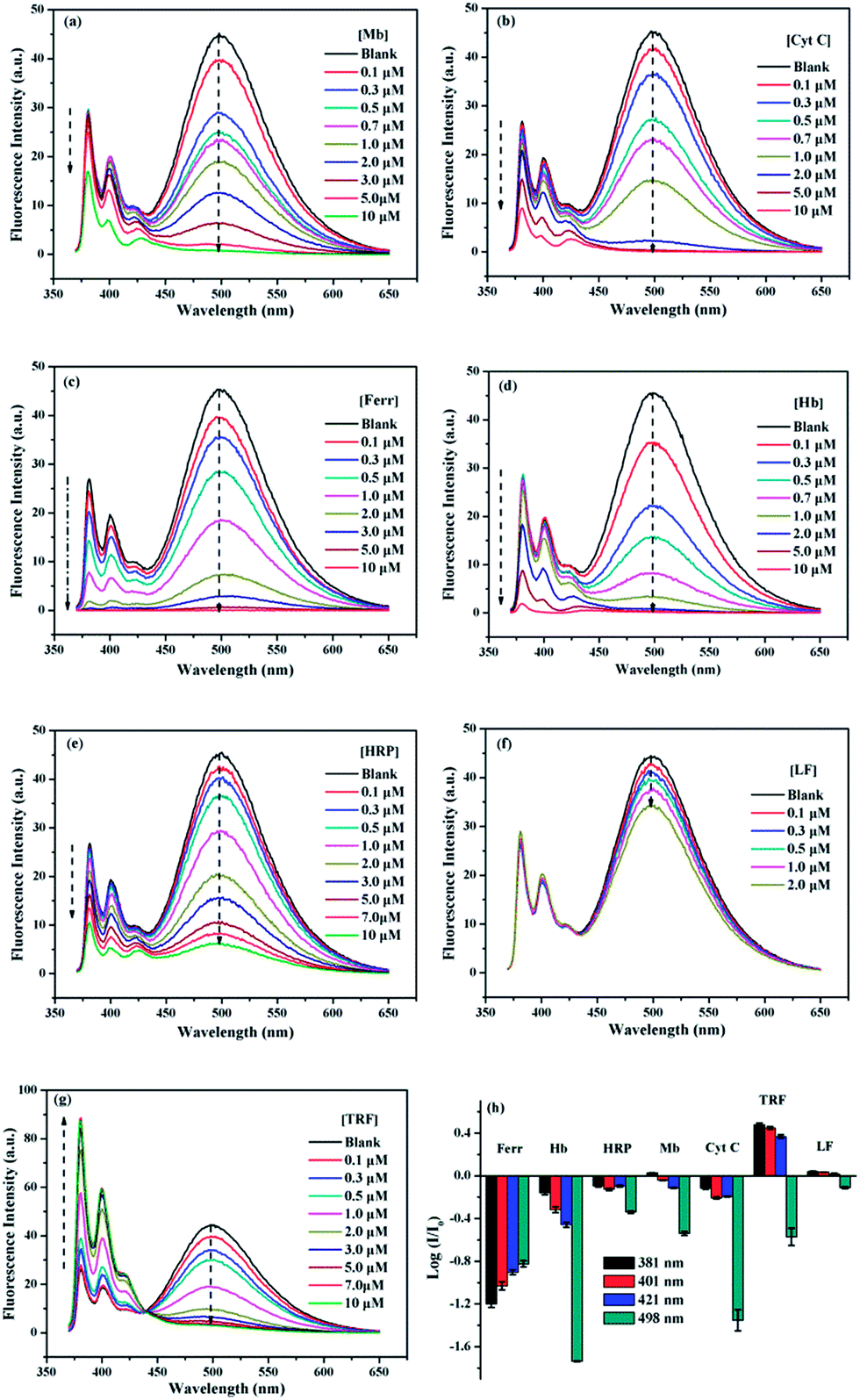 | ||
| Fig. 3 Fluorescence spectra of 1/SDBS (5 µM/0.2 mM) in aqueous buffer solution (10 mM HEPES, pH 7.4) upon titration of iron-containing metalloproteins: (a) Mb, (b) Cyt C, (c) Ferr, (d) Hb, (e) HRP, (f) LF, and (g) TRF. (h) Recognition patterns for metalloproteins (2 µM) by collecting fluorescence variation of 1/SDBS at four selected wavelengths. Each value is an average of three parallel measurements. | ||
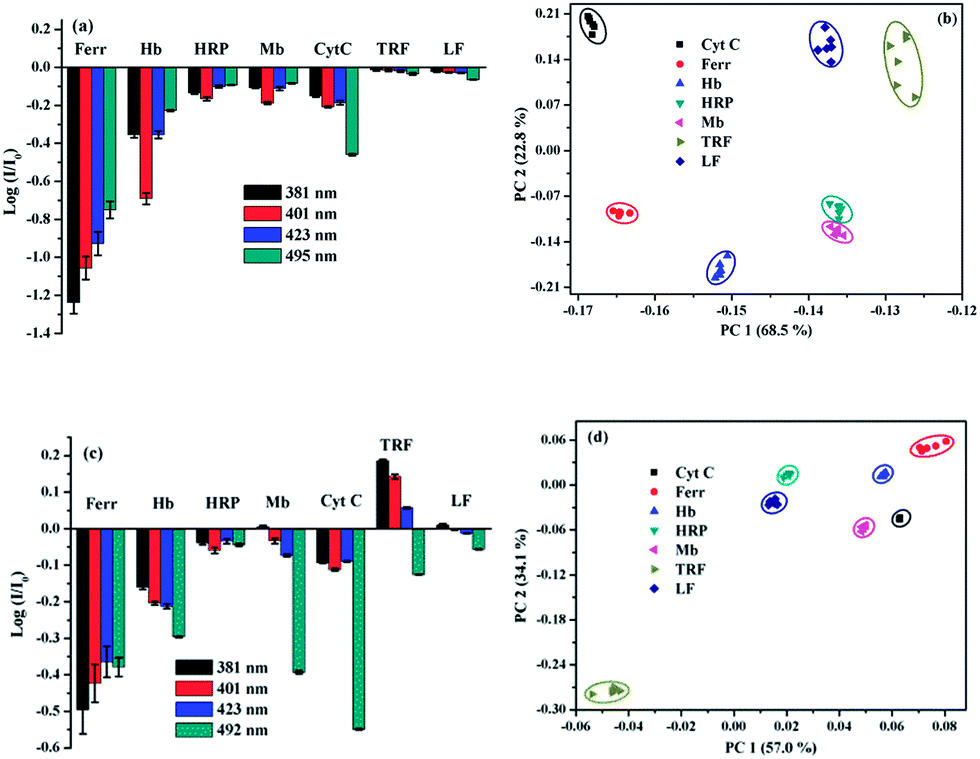
The different response modes combined with the multiple fluorescence variation of a particular wavelength render the ensemble sensor the typical multi-wavelength cross-reactive responses to metalloproteins. Therefore, we collected the logarithm data of intensity variation (log(I/I0)) at four selected wavelengths (381, 401, 421 and 498 nm), which represent two monomer emission peaks, a distorted excimer emission and the perfect excimer emission center, to provide the recognition pattern. It can be seen from Fig. 3h, a distinct recognition pattern was generated for each metalloprotein, suggesting the present ensemble sensor may be capable to discriminate the 7 proteins.
PCA has been widely utilized to analyze the pattern recognition ability of sensor arrays by reducing the dimensionality of the data set. Therefore, PCA was performed to evaluate the discriminatory power of the present sensor platform to different metalloproteins. For this purpose, the fluorescence variation of 1/SDBS at the four selected wavelengths in the presence of each metalloprotein at a particular concentration was measured 6 times repeatedly. Thus, a full set of log(I/I0) values were used to construct a matrix consisting of 1 sensor × 4 wavelengths × 7 metalloproteins × 6 replicates for PCA analysis. The resulting two-dimensional plots are shown in Fig. 4. As observed, at a low concentration of 0.5 µM (Fig. 4a), the same protein samples are well grouped together and different proteins are well separated from others except HRP and LF. At higher concentration of 5 µM, similar results were observed (Fig. 4b). Again, the same protein samples are well grouped together and different proteins are separated from others. The sensing to LF at higher concentration was not measured because of the solubility issue. For both cases, the two principle components carry more than 94% of the whole variance, suggesting the present sensor ensemble has a strong capability to discriminate multiple metalloproteins.
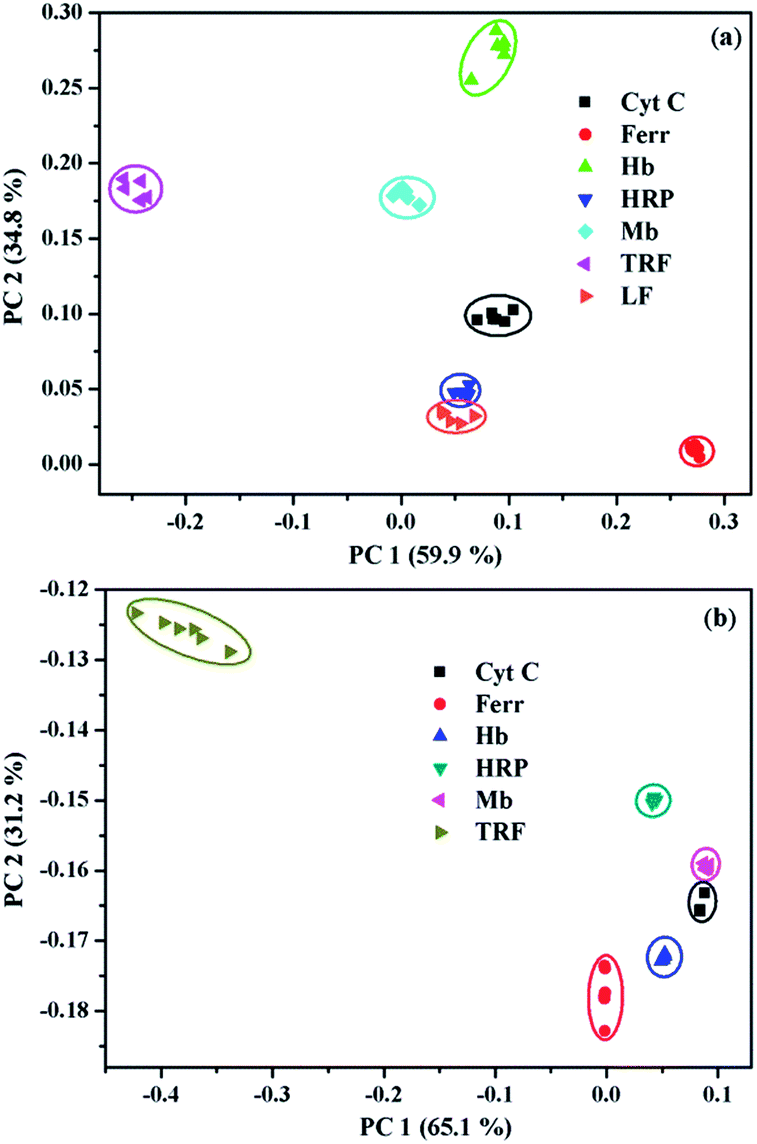 | ||
| Fig. 4 Two-dimensional PCA score plots for the discrimination of metalloproteins at (a) 0.5 µM and (b) 5 µM. | ||
Quenching mechanism of 1/SDBS to metalloproteins
The remarkable quenching responses of 1 to iron-containing metalloproteins are possibly due to the fact that the co-factors in these proteins could quench the excited state of pyrene units by energy or electron transfer.30 As to the nature of quenching by these proteins, time-resolved fluorescence decays of 1/SDBS at 498 nm in the presence of these metalloproteins were measured. The corresponding results are illustrated in Fig. 5a and S4 (ESI†). Clearly, the fluorescence decay curves show different behaviors upon titration of different metalloproteins. Fig. 5a illustrates the fluorescence decays upon titration of Mb. It can be seen all the decay curves are almost overlapped together and barely change upon increasing Mb concentration. Moreover, the τ0/τ value of 1/SDBS is independent on Mb concentration and stays close to 1 along varying Mb concentration (Fig. 5b), where τ0 and τ represent the lifetime of the sensor ensemble in the absence and presence of metalloproteins, respectively. Such result suggests that the nature of myoglobin quenching 1/SDBS is static rather than dynamic.31 This means that metalloprotein–fluorophore complex forms, and myoglobin is possibly binding with the fluorescent ensemble. Similar results were also observed for the titration of Ferr, TRF, and LF (Fig. S4a–c, ESI†). The binding with these metalloproteins could be either due to electrostatic interaction with SDBS or through binding with boronic acid groups. However, a different phenomenon was observed for the titration of HRP, Cyt C and Hb. As shown in Fig. S4d–f (ESI†), the decay becomes faster along increasing protein concentration. At the same time, the τ0/τ value increases, indicating the lifetime of 1 became shorter when HRP, Cyt C and Hb were added (Fig. 5b). These results mean that there is a certain degree of dynamic quenching to the 1/SDBS system for these three metalloproteins. The different quenching nature may cause different quenching behaviors and contributes to the cross-reactive responses. Furthermore, it is also known that these metalloproteins have different pI values and may have different electrostatic interactions with SDBS assemblies, which may further cause different variation of SDBS assemblies and lead to versatile fluorescence variation of the encapsulated probe.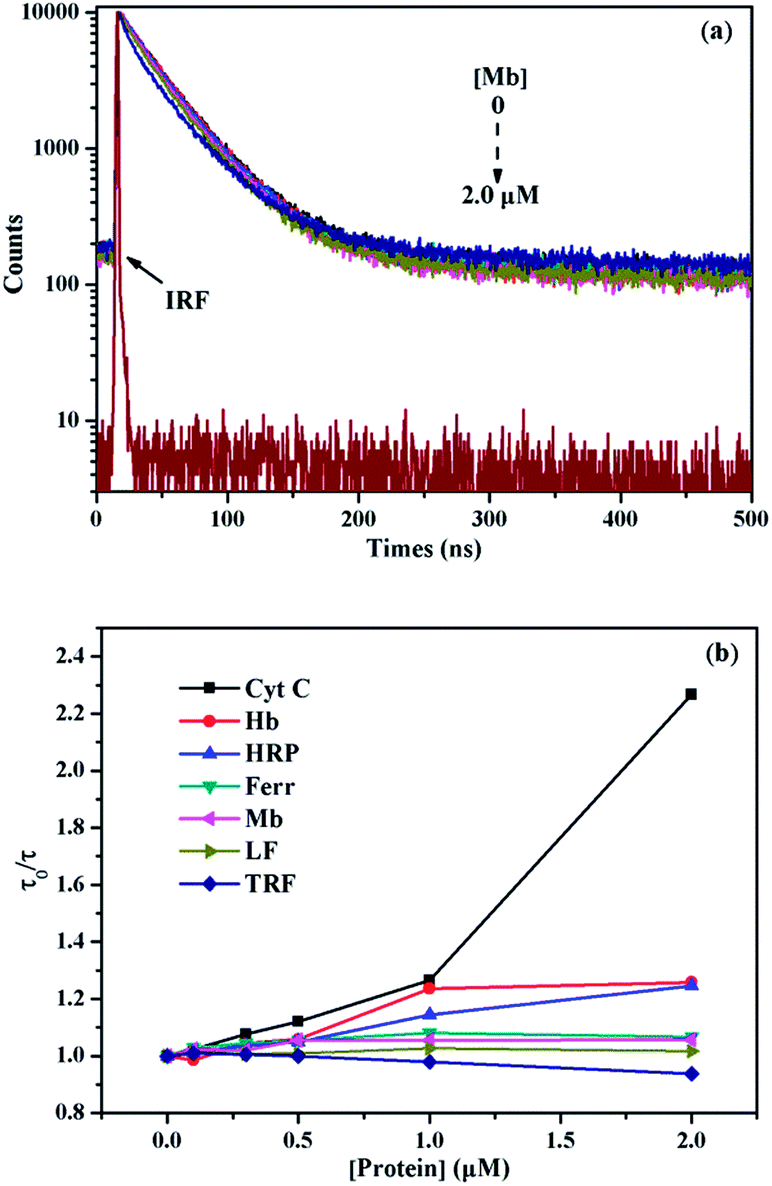 | ||
| Fig. 5 (a) Time-resolved fluorescence decays of 1/SDBS (5 µM/0.2 mM) upon titration of Mb (λex = 343.4 nm, λem = 498 nm); (b) fluorescence quenching plots of lifetime ratio (τ0/τ) of 1/SDBS (5 µM/0.2 mM) as a function of metalloprotein concentration. | ||
Then we conducted some further experiments to evaluate whether energy transfer or electron transfer is mainly accountable for the observed fluorescence quenching induced by metalloproteins. The UV-vis absorption of the tested metalloproteins was first measured and illustrated in Fig. 6a. Clearly, the absorbance spectra of HRP, Hb, Mb and Cyt C can overlap with the emission spectra of 1/SDBS (5.0 µM/0.2 mM) to some extent, suggesting Förster-type energy transfer from the fluorescent assembly to these metalloproteins is possible. However, these Fe-porphyrins are inherently poor fluorescence emitter,30 and their fluorescence emission is hardly observed to confirm the occurrence of energy transfer. Therefore, we conducted the following model study to testify the possibility of energy transfer. We used pyronine as energy acceptor as its absorbance spectrum overlaps well with the excimer emission of 1/SDBS. At the same time, it can emit fluorescence over 550–600 nm (Fig. S5, ESI†). As shown in Fig. 6b, pyronine could well quench the excimer emission of the sensor assembly with a KSV of 1.37 × 106 M−1, at the same time the emission of pyronine at 550–600 nm increased. Thus, the quenching mechanism for Hb, Mb, HRP and Cyt C could be due to the energy transfer from the fluorescent assembly to these metalloproteins. As to Ferr, TRF and LF, they do not have absorption that overlaps with the emission of the sensor system (Fig. 6a). The quenching mechanism for these three metalloproteins is more likely based on electron transfer, since there is no chromophore that could accept the excited state energy from 1/SDBS assembly (Fig. 6a).30 We then conducted another model study to check the possibility of electron transfer from the sensor ensemble to Fe3+ ions as Fe3+ can only act as electron acceptor. As displayed in Fig. 6c, Fe3+ can also quench the fluorescence of the sensor assembly with a KSV value of 2.80 × 104 M−1, suggesting electron transfer can also be responsible for the observed fluorescence quenching. Therefore, both energy and electron transfer processes may be accountable for the fluorescence quenching by metalloproteins. It is just that the two processes may contribute differently for different metalloprotein quenching, which may also account for the cross-reactive responses to metalloproteins. Based on the above observations and discussion, the process of the present sensor ensemble identifying different metalloproteins is schematically illustrated in Scheme 3.
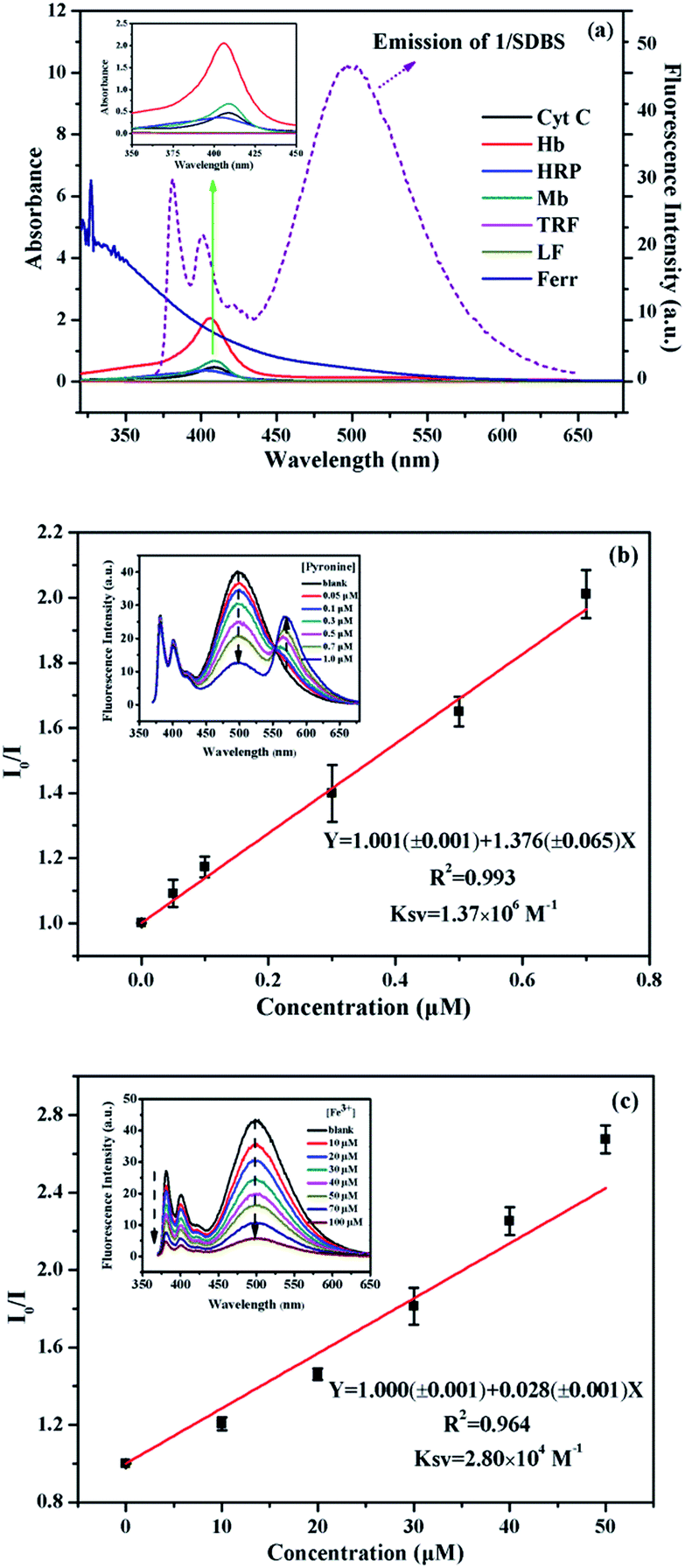 | ||
| Fig. 6 (a) UV-vis absorption of metalloproteins (5 µM) and fluorescence emission of 1/SDBS (5 µM/0.2 mM); (b) quenching plots of the fluorescence intensity ratio (I0/I) of 1/SDBS (5 µM/0.2 mM) as a function of pyronine concentration, inset: fluorescence emission of 1/SDBS upon titration of pyronine; (c) quenching plots of the fluorescence intensity ratio (I0/I) of 1/SDBS (5 µM/0.2 mM) assembly as a function of Fe3+ concentration, inset: fluorescence emission of 1/SDBS upon titration of Fe3+. | ||
 | ||
| Scheme 3 Schematic representation of 1/SDBS for discriminating different metalloproteins. | ||
Discrimination of metalloproteins in human serum and urine
As is well known, there is a direct correlation between protein levels in human serum and disease states, which makes it an attractive target for sensors and diagnostics.32,33 However, this is challenging because serum features more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 proteins, and the overall protein content is greater than 1 mM.34–36 To check the potential for practical applications, the performance of our designed 1/SDBS sensing ensemble was further evaluated for the discrimination of metalloproteins in 1.0% human serum solutions. The ensemble with 0.1 mM SDBS and 20 µM probe 1 was found to display fine monomer and excimer emission and used as the optimized sensor system in 1.0% serum solution (Fig. S6, ESI†). Thus, we measured its sensing behaviors to the 7 metalloproteins (2 µM) in 1.0% human serum. For this purpose, the logarithm data of fluorescence variation (log(I/I0)) at four selected wavelengths (381, 401, 421 and 495 nm) for the sensor ensemble to the 7 metalloproteins were collected. Fig. 7a illustrates that the array of the fluorescence variation at the four emission wavelengths can generate a distinct recognition pattern toward the measured metalloproteins. For the sake of evaluating the discriminative power of 1/SDBS (20 µM/0.1 mM) ensemble, its fluorescence variation data at four different wavelengths to the 7 metalloproteins was analyzed by PCA, where a matrix consisting of 1 sensor × 4 wavelengths × 7 metalloproteins × 6 replicates were used. As shown in Fig. 7b, the same proteins samples are all grouped together in the same cluster and separated from other protein clusters. These results demonstrate that the sensor ensemble could still well recognize and discriminate these metalloproteins even in the human serum. The sensing performances in further concentrated human serum were also measured. It is found that the sensing ensemble could also display pattern-recognition behaviors to metalloproteins in 5% human serum solution (Fig. S7a, ESI†), but show weaker discrimination ability, where only 3 metalloproteins could be well discriminated from others (Fig. S7b, ESI†). In more concentrated human serum (above 10%), the ensemble exhibited monomer-dominated emission due to the influence of proteins in the serum solution and lost the multiple-wavelength cross-reactivity to metalloproteins.
000 proteins, and the overall protein content is greater than 1 mM.34–36 To check the potential for practical applications, the performance of our designed 1/SDBS sensing ensemble was further evaluated for the discrimination of metalloproteins in 1.0% human serum solutions. The ensemble with 0.1 mM SDBS and 20 µM probe 1 was found to display fine monomer and excimer emission and used as the optimized sensor system in 1.0% serum solution (Fig. S6, ESI†). Thus, we measured its sensing behaviors to the 7 metalloproteins (2 µM) in 1.0% human serum. For this purpose, the logarithm data of fluorescence variation (log(I/I0)) at four selected wavelengths (381, 401, 421 and 495 nm) for the sensor ensemble to the 7 metalloproteins were collected. Fig. 7a illustrates that the array of the fluorescence variation at the four emission wavelengths can generate a distinct recognition pattern toward the measured metalloproteins. For the sake of evaluating the discriminative power of 1/SDBS (20 µM/0.1 mM) ensemble, its fluorescence variation data at four different wavelengths to the 7 metalloproteins was analyzed by PCA, where a matrix consisting of 1 sensor × 4 wavelengths × 7 metalloproteins × 6 replicates were used. As shown in Fig. 7b, the same proteins samples are all grouped together in the same cluster and separated from other protein clusters. These results demonstrate that the sensor ensemble could still well recognize and discriminate these metalloproteins even in the human serum. The sensing performances in further concentrated human serum were also measured. It is found that the sensing ensemble could also display pattern-recognition behaviors to metalloproteins in 5% human serum solution (Fig. S7a, ESI†), but show weaker discrimination ability, where only 3 metalloproteins could be well discriminated from others (Fig. S7b, ESI†). In more concentrated human serum (above 10%), the ensemble exhibited monomer-dominated emission due to the influence of proteins in the serum solution and lost the multiple-wavelength cross-reactivity to metalloproteins.
 | ||
| Fig. 7 (a) Recognition patterns for metalloproteins (2 µM) by collecting fluorescence variation of 1/SDBS (20 µM/0.1 mM) at four selected wavelengths in 1.0% human serum (10 mM HEPES, pH 7.4). Each value is an average of three parallel measurements. (b) Two-dimensional PCA plots for the discrimination of 7 metalloproteins (2 µM) in 1.0% human serum (10 mM HEPES, pH 7.4). (c) Recognition patterns for metalloproteins (1 µM) by collecting fluorescence variation of 1/SDBS (5 µM/0.1 mM) at four selected wavelengths in 10.0% human urine (10 mM HEPES, pH 7.4). Each value is an average of three parallel measurements. (d) Two-dimensional PCA score plots for the discrimination of 7 metalloproteins (1 µM) in 10% human urine (10 mM HEPES, pH 7.4). | ||
The testing in another biofluid, human urine, was also conducted to evaluate the potential of practical application of the present sensor ensemble. Human urine features more than 1500 proteins and the overall content is more than 1.5 µM (150 mg L−1).37–39 These proteins strongly compete with various biomolecules and make it also challenging to recognize proteins in urine fluid. To obtain the optimized sensing ensemble performed in human urines, the concentration effect of SDBS on the fluorescence emission of 1 (5 µM) was measured. The ensemble with 0.1 mM SDBS was found to display fine monomer and excimer emission (Fig. S8, ESI†). Thus, we chose 1/SDBS (5 µM/0.1 mM) ensemble as the sensing system and measured its sensing behaviors to the 7 metalloproteins at 1 µM in 10.0% human urine. The emission variation at the four wavelengths (381, 401, 423, and 492 nm) could also generate a distinct recognition pattern for each metalloprotein (Fig. 7c). The PCA analysis result shown in Fig. 7d demonstrates that the binary ensemble could also well recognize and discriminate these metalloproteins in human urine. The same protein samples are all grouped together and well separated from others. Thus, the binary ensemble based on 1/SDBS has displayed great potential in recognizing and discriminating different metalloproteins in actual physiological fluids, either human serum or human urine.
Conclusions
In summary, we developed a single discriminative sensor that can identify multiple metalloproteins in aqueous solution. The sensor ensemble is based on a specially designed cholic acid-modified pyrene derivative and an anionic surfactant SDBS assembly. Fluorescence measurements found that the fluorescence emission of the probe aggregates could be well modulated from monomer-dominated emission to monomer–excimer co-emission by SDBS assemblies. The selected binary ensemble based on the probe/SDBS assembly exhibits the typical multiple-wavelength cross-reactive responses to different metalloproteins. PCA results show that all the tested metalloproteins are well separated from one another. Time-resolved fluorescence decay measurements revealed that the nature of quenching by metalloproteins could be either static or dynamic. Control experiments proved that both energy transfer and electron transfer are responsible for the observed fluorescence quenching by metalloproteins. The binary ensemble also displays great discrimination ability of identifying different metalloproteins in biofluids like serum and urine, which makes it have great potential in practical applications.Experimental section
Materials and instruments
Pyrene (Alfa, 98%) was used after recrystallization by ethanol. Bromoacetyl bromide (J&K®, 98%), sodium cholate (J&K®, 98%) and sodium iodide (J&K®, 98%) were used as supplied. Diethylenetriamine (DETA, 98%), trifluoroacetic acid (CF3COOH, 99%), phenyl tertbutyl carbonate (98%), pyridine-4-boronic acid (90%), sodium dodecyl benzene sulfonate (SDBS, >99%), HEPES (>99.5%) and all proteins including transferrin (from human), lactoferrin (from human), cytochrome c (from horse heart), myoglobin (from equine skeletal muscle), horseradish peroxidase (from horseradish), hemoglobin (from human) and ferritin (from equine spleen, type I) were purchased from Sigma-Aldrich and used without further purification. All aqueous solutions were prepared from Milli-Q water (18.2 MΩ cm at 25 °C) and all the reagents were analytically pure.1H NMR and 13C NMR spectra of the synthesized chemicals were obtained on a Bruker Avance 600 MHz NMR spectrometer. The FTIR spectra were measured on a Fourier Transform Infrared Spectrometer (Vertex 70v, Bruker, Germany). The high resolution mass spectra (MS) were acquired on a Bruker Maxis UHR-TOF Mass Spectrometer in ESI positive mode. Steady-state fluorescence spectra were conducted on a single photon-counting fluorescence spectrometer (FS5, Edinburgh Instruments, UK) with xenon light (150 W) as excitation source, and the excitation and emission slit widths were set at 2.5 nm and 1.5 nm, respectively, and all samples were excited at 354 nm. Time-resolved fluorescence decays were conducted on a time-correlated single photon-counting fluorescence spectrometer (FLS920, Edinburgh Instruments, UK) using a laser (343.4 nm) as the excitation source. UV-vis absorption spectra were recorded on a spectrophotometer (U3900, Hitachi Instrument). The particle size distribution was performed on a Malvern Zetasizer Nano-ZS90. TEM image was obtained using FEI Tecnai G2 F20 field transmission electron microscopy at 200 kV.
Synthesis of compound 1
The synthesis procedure for obtaining compound 1 is illustrated in Scheme 1. First, compound 3 was synthesized and purified according to our previously reported method.40 Then, compound 3 was reacted with sodium cholate to obtain compound 2. The synthesis process is as follows. Sodium cholate (412 mg, 0.96 mmol) was added to a solution of 3 (584 mg, 0.96 mmol) and NaI (144 mg, 0.96 mmol) in dry DMF (80 mL), and the reaction mixture was stirred at 65 °C for 48 h. The mixture was then cooled, poured into brine solution (100 mL), and extracted with chloroform thrice, and then dried over anhydrate Na2SO4 and concentrated to afford a residue. The resulting crude product was purified by column chromatography on silica gel column with methanol/CH2Cl2 (v/v, 1/15) as elute and then recrystallized with CHCl3 and absolute ether to afford compound 2 as yellow solid (180 mg, yield 30%). 1H NMR (600 MHz, CDCl3, ppm) δ 8.90 (t, J = 8.6 Hz, 1H), 8.52 (dd, J = 21.6, 8.1 Hz, 1H), 8.21 (m, 7H), 7.10 (s, 1H), 7.05 (s, 1H), 4.51 (d, J = 20.2 Hz, 2H), 3.90 (d, J = 20.9 Hz, 1H), 3.79 (s, 1H), 3.54 (m, 11H), 2.44 (d, J = 9.2 Hz, 1H), 2.31 (m, 1H), 2.17 (s, 8H), 1.76 (m, 10H), 1.43 (ddd, J = 112.0, 52.9, 38.8 Hz, 10H), 1.00 (m, 5H), 0.83 (s, 3H), 0.63 (s, 3H). HR-MS (ESI, m/z): calcd for C48H63IN3O9S [M + H]+: 984.3330, found: 984.3301.Finally, to synthesize the final compound 1, pyridine-4-boronic acid (0.024 g, 0.199 mmol) and compound 2 (0.196 g, 0.199 mmol) were added together into a solution of 50 mL DMF for reaction. The reaction mixture was stirred at 70 °C for 72 h under nitrogen. After removing most of the solvent, the residue was precipitated in acetone (200 mL). The white precipitate was collected by filtration, washed with acetone, ether, recrystallized with DMF and acetone, then washed with CH3OH and the filtrate was evaporated to dryness to yield compound 1 (0.06 g, yield 27%). 1H NMR (600 MHz, CD3OD, ppm) δ 8.84 (d, J = 9.4 Hz, 1H), 8.60 (s, 4H), 8.46–8.29 (m, 5H), 8.25–8.05 (m, 5H), 5.35 (s, 2H), 4.04 (q, J = 15.1 Hz, 2H), 3.88 (s, 1H), 3.71 (s, 3H), 3.50 (ddd, J = 22.2, 14.1, 6.0 Hz, 4H), 3.37 (dd, J = 13.0, 6.2 Hz, 3H), 2.33–1.11 (m, 24H), 1.05–0.79 (m, 8H), 0.60 (s, 3H). 13C NMR (151 MHz, CD3OD, ppm) δ 174.70, 170.44, 166.96, 144.22, 141.51, 136.29, 132.39, 132.10, 131.91, 131.62, 131.41, 131.16, 129.57, 128.75–128.02, 126.36, 125.30, 124.97, 124.48, 73.93, 72.84, 68.99, 63.16, 62.49, 49.90, 49.47, 49.33, 49.19, 49.05, 48.91, 48.76, 48.72–47.96, 47.90, 47.39, 43.05, 40.91, 40.46, 39.24, 38.65, 36.48, 35.84, 31.73, 31.22, 29.57, 28.59, 27.86, 24.15, 23.16, 17.77, 12.98. IR (KBr plates, cm−1): 3421 (–OH), 3051 (Ar–H), 2929 (–CH2–), 1703 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1681 (Ar CC), 1307 (C–N), 1157 (OSO), 1078 (C–O–C), 1379 (B–O). HR-MS (MS-ESI, m/z) C55H72BN4O11S [M + 2CH3OH–2H2O–I]+: calcd: 1007.5006; found: 1007.4987, C54H70BN4O11S [M + CH3OH–H2O–I]+: calcd: 993.4849, found: 993.4843.
O), 1681 (Ar CC), 1307 (C–N), 1157 (OSO), 1078 (C–O–C), 1379 (B–O). HR-MS (MS-ESI, m/z) C55H72BN4O11S [M + 2CH3OH–2H2O–I]+: calcd: 1007.5006; found: 1007.4987, C54H70BN4O11S [M + CH3OH–H2O–I]+: calcd: 993.4849, found: 993.4843.
Sample preparation
The stock solution of compound 1 (2.5 × 10−4 M) was prepared in methyl alcohol. The concentration of compound 1 in the aqueous sensing system was fixed at 5 µM (VMeOH = 2%). The stock solutions of various proteins (2.5 × 10−4 M) were prepared separately in HEPES buffer solution (10 mM, pH 7.4) and stored at 0–4 °C. Biological samples were prepared in human serum (obtained from the Affiliated Hospital of Shaanxi Normal University) by diluting it with HEPES solution to the corresponding percentage in volume. In tests with human urine samples, morning urine from a health volunteer was collected and centrifuged for 10 minutes (14000 rpm) to remove the insoluble matrix. Urine was diluted with HEPES buffer solution (10 mM, pH 7.4) to 10% (v/v). For example, to 25 mL of a urine sample, 225 mL of HEPES buffer solution (10 mM, pH 7.4) were added. All these experiments were performed in compliance with the relevant laws and institutional guidelines of China and the ethics committee that approved these experiments was the Institute Review Board of the Affiliated Hospital of Shaanxi Normal University. Informed consent was also given by every participant.
Conflicts of interest
There are no conflicts of declare.Acknowledgements
The authors acknowledge the financial support from National Natural Science Foundation of China (21573140), the Program of Introducing Talents of Discipline to Universities (B14041), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R33), and the Fundamental Research Funds for the Central Universities (GK201701004).Notes and references
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- R. Kubota and I. Hamachi, Chem. Soc. Rev., 2015, 44, 4454–4471 RSC.
- X. Li, F. Wen, B. Creran, Y. Jeong, X. Zhang and V. M. Rotello, Small, 2012, 8, 3589–3592 CrossRef CAS PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- D. R. Brown, Metallomics, 2010, 2, 186–194 RSC.
- W. Maret, Metallomics, 2010, 2, 117–125 RSC.
- Y. Wei, H. Yuan, P. Xu and X. Tan, Arch. Biochem. Biophys., 2017, 616, 30–39 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- S. Shinoda and H. Tsukube, Chem. Sci., 2011, 2, 2301–2305 RSC.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS PubMed.
- J. R. Kanwar, K. Roy, Y. Patel, S.-F. Zhou, M. R. Singh, D. Singh, M. Nasir, R. Sehgal, A. Sehgal and R. S. Singh, Molecules, 2015, 20, 9703–9731 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. S. Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- P. Anzenbacher Jr, P. Lubal, P. Buček, M. A. Palacios and M. E. Kozelkova, Chem. Soc. Rev., 2010, 39, 3954–3979 RSC.
- N. Niamnont, R. Mungkarndee, I. Techakriengkrai, P. Rashatasakhon and M. Sukwattanasinitt, Biosens. Bioelectron., 2010, 26, 863–867 CrossRef CAS PubMed.
- N. Chang, J. Mao, Y. Lu, J. Yang, Y. Pu, S. Zhang and Y. Liu, Sens. Actuators, B, 2016, 233, 17–24 CrossRef CAS.
- J. Mao, Y. Lu, N. Chang, J. Yang, J. Yang, S. Zhang and Y. Liu, Analyst, 2016, 141, 4014–4017 RSC.
- B. S. Sandanaraj, R. Demont and S. Thayumanavan, J. Am. Chem. Soc., 2007, 129, 3506–3507 CrossRef CAS PubMed.
- D. C. González, E. N. Savariar and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 7708–7716 CrossRef PubMed.
- O. R. Miranda, C.-C. You, R. Phillips, I.-B. Kim, P. S. Ghosh, U. H. Bunz and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 9856–9857 CrossRef CAS PubMed.
- P. Wu, L. Miao, H. Wang, X. Shao and X. Yan, Angew. Chem., Int. Ed., 2011, 50, 8118–8121 CrossRef CAS PubMed.
- C. Li, P. Wu and X. Hou, Nanoscale, 2016, 8, 4291–4298 RSC.
- W. Hu, L. Ding, J. Cao, L. Liu, Y. Wei and Y. Fang, ACS Appl. Mater. Interfaces, 2015, 7, 4728–4736 CAS.
- J. Fan, L. Ding, Y. Bo and Y. Fang, ACS Appl. Mater. Interfaces, 2015, 7, 22487–22496 CAS.
- Y. Cao, L. Zhang, X. Huang, Y. Xin and L. Ding, ACS Appl. Mater. Interfaces, 2016, 8, 35650–35659 CAS.
- N. Carter, M. Pattabiraman and L. A. Arias, Am. J. Anal. Chem., 2012, 2, 131–136 CrossRef CAS.
- M. C. Cuquerella, J. Rohacova, M. L. Marin and M. A. Miranda, Chem. Commun., 2010, 46, 4965–4967 RSC.
- W. H. Chan, S. Y. Liu, Y. B. He and A. W. M. Lee, Tetrahedron, 2006, 62, 11687–11696 CrossRef.
- B. S. Sandanaraj, R. Demont, S. V. Aathimanikandan, E. N. Savariar and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 10686–10687 CrossRef CAS PubMed.
- M. Takayanagi, C. Iwahashi and M. Nagaoka, J. Phys. Chem. B, 2010, 114, 12340–12348 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- R. Pulido and R. H. van Huijsduijnen, FEBS J., 2008, 275, 848–866 CrossRef CAS PubMed.
- J. N. Adkins, S. M. Varnum, K. J. Auberry, R. J. Moore, N. H. Angell, R. D. Smith, D. L. Springer and J. G. Pounds, Mol. Cell. Proteomics, 2002, 1, 947–955 CAS.
- E. M. Antman, M. J. Tanasijevic, B. Thompson, M. Schactman, C. H. McCabe, C. P. Cannon, G. A. Fischer, A. Y. Fung, C. Thompson and D. Wybenga, N. Engl. J. Med., 1996, 335, 1342–1349 CrossRef CAS PubMed.
- R. Pieper, C. L. Gatlin, A. J. Makusky, P. S. Russo, C. R. Schatz, S. S. Miller, Q. Su, A. M. McGrath, M. A. Estock and P. P. Parmar, Proteomics, 2003, 3, 1345–1364 CrossRef CAS PubMed.
- D. Fliser, J. Novak, V. Thongboonkerd, A. Argiles, V. Jankowski, M. A. Girolami, J. Jankowski and H. Mischak, J. Am. Soc. Nephrol., 2007, 18, 1057–1071 CrossRef CAS PubMed.
- J. Adachi, C. Kumar, Y. Zhang, J. V. Olsen and M. Mann, Genome Biol., 2006, 7, R80 CrossRef PubMed.
- S. J. Li, M. Peng, H. Li, B. S. Liu, C. Wang, J. R. Wu, Y. X. Li and R. Zeng, Nucleic Acids Res., 2009, 37, D907–D912 CrossRef CAS PubMed.
- J. Cao, L. Ding, Y. Zhang, S. Wang and Y. Fang, J. Photochem. Photobiol., A, 2016, 314, 66–74 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and additional spectra. See DOI: 10.1039/c7ra09741g |
| This journal is © The Royal Society of Chemistry 2017 |