
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight induced construction of porous covalent organic polymeric networks for significant enhancement of CO2 gas sorption†
Soumitra Bhowmik,
Maruthi Konda and
Apurba K. Das *
*
Department of Chemistry, Indian Institute of Technology Indore, Indore 453552, India. E-mail: apurba.das@iiti.ac.in
First published on 10th October 2017
Abstract
Herein, we report morphology-controlled porous polymeric materials for enhanced CO2 capture, which was achieved using the topochemical polymerization of dipeptide functionalized diphenylbutadiynes. The topochemical reaction was executed to control the morphology of the synthesized dipeptide appended diarylbutadiyne derivatives on a solid surface. Topochemical polymerization involves the formation of polydiacetylene due to the presence of hydrogen bonding between the amide groups and intermolecular π–π stacking interactions in their self-assembled state, which was established using UV-Vis, Raman and IR spectroscopy. The change in morphology of the two dipeptide functionalized diphenylbutadiyne (DPB) was confirmed by scanning electron microscopy. Porosity was developed after UV irradiation of the diacetylene-based dipeptide appended bolaamphiphiles. Interestingly, after UV irradiation, the porous covalent organic polymers 1 and 2 show 24.22 times and 12 times enhanced N2 gas adsorption than their parent compounds 1 and 2, respectively. The surface area of the porous covalent organic polymers 1 and 2 was enhanced 21.68 times and 5.54 times than their parent compounds 1 and 2, respectively. Polymer 1 exhibits 4.23 times the CO2 capture ability than compound 1 and polymer 2 shows 4.1 times the CO2 capture ability than compound 2. This study highlights the controlled synthesis of light induced porous covalent organic polymers with high surface area used for efficient CO2 storage applications.
Introduction
In recent years, an enormous amount of greenhouse gases including CO2 are released into the atmosphere with the earth's increasing population, which has triggered a rapid increase in atmospheric temperature.1–5 Inspired by natural zeolite structures, researchers are working to develop porous materials for effective CO2 capture. Porous materials, such as covalent organic polymers (COPs),6–11 metal–organic frameworks (MOFs)12 and covalent organic frameworks (COFs)13,14 with miscellaneous properties and applications have attracted great attention in materials science research. Porous covalent polymers are used as effective CO2 sorption materials due to their ultra-high hydrothermal stability. Porous COPs are potentially beneficial for gas storage and separation,15 catalysis,16 chemical sensing, drug delivery,17 electronic devices18 and energy storage applications.19 However, considerable attention has been focused on controlling the self-assembly, pore size, surface area and functionality in nanoporous solids. Precise molecular self-assembly enables the arrangement of molecules in a well-defined molecular architecture with specific shape and size. The controlled self-assembly of peptide-based amphiphiles has attracted great attention due to their unique architectures, including fibers,20–24 tubes,25–27 spheres,28–31 vesicles32–35 and cylinders36–38 as well as their applications in drug delivery39,40 and injectable therapy to control hemorrhage.41 Peptide-based bolaamphiphiles are a unique class of amphiphiles, which can self-assemble into different architectures through non-covalent interactions upon the influence of external stimuli.42–45 Upon incorporation of a diacetylene moiety into the peptide backbones, peptide functionalized diacetylene-based amphiphiles can self-assemble into an oriented fashion and form self-assembled nanostructures. Peptide-functionalized diphenylbutadiyne-based bolaamphiphiles can also be polymerized using topochemical polymerization if they are appropriately oriented. Peptide-functionalized polydiacetylene-based amphiphiles have been used in several applications such as chromatic sensors, lipopolysaccharide detectors, biotechnology and biomedicine.46–48 Peptide-functionalized polydiacetylene-based amphiphilic fibers have also been used to sense cell adhesion via its colorimetric sensing behaviour upon UV irradiation.49 Its self-organization behaviour also changes with a change in suitable spacer length in the case of diacetylene-containing peptide bolaamphiphiles.50 Diacetylene containing peptide bolaamphiphiles have also been used as optoelectronic materials.51 The conducting poly(diphenylbutadiyne) nanostructures synthesized via photochemical polymerization show high photocatalytic activity for the photodecomposition of phenol and methyl orange under visible-light irradiation.52 Inspired by the diverse self-assembly nature of peptide bolaamphiphiles,53–59 we demonstrate the topochemical polymerization of two dipeptide appended diphenylbutadiynes on a solid surface that exhibit different morphologies.In this regard, we have synthesized two dipeptide appended diphenylbutadiynes-based bolaamphiphiles, HO-Phe-Leu-DPB-Leu-Phe-OH (compound 1) and HO-Tyr-Leu-DPB-Leu-Tyr-OH (compound 2) (DPB = diphenylbutadiyne, Phe = phenylalanine, Leu = leucine, Tyr = tyrosine), to examine their morphological patterns before and after UV irradiation (Fig. 1). The self-organization of the dipeptide appended poly(diphenylbutadiyne)s was controlled based on the dipeptide-sequence and concentration of the bolaamphiphile. The morphological alternations of the two compounds under UV irradiation have been studied using SEM. This study corresponds to a step forward in the design of porous polymer network structures from fibrillar structures.60 The porous polymeric network structures adsorbed CO2 gas almost four times higher than the fibrillar network structures of self-assembled molecules. Thus, the rational design of light induced diacetylene-based porous covalent organic polymers has a prominent impact on CO2 adsorption.
 | ||
| Fig. 1 (a) Typical topochemical polymerization mechanism. (b) Schematic representation of the preparation method used for compound 1 on a glass substrate for photochemical reaction in the solid state for 1 h. | ||
Experimental section
Materials
Methyl 4-iodo benzoate, trimethylsilylacetylene (TMSA), tetramethylethylenediamine (TMEDA), CuCl (copper(I) chloride), CuI (copper(I) iodide) and Pd(PPh3)2Cl2 (bis(triphenylphosphine) palladium(II) dichloride) were purchased from Alfa Aesar, India. L-Leucine, L-phenylalanine, L-tyrosine, HOBt (1-hydroxybenzotriazole) and DCC (N,N′-dicyclohexylcarbodiimide) were obtained commercially. For the chemical reactions and purification of the peptides, methanol, dimethylformamide (DMF), ethyl acetate and hexane were dried according to the reported literature procedures. The reactions were monitored by thin-layer chromatography (TLC). All the intermediates and final compounds were purified and characterized using NMR (400 MHz) and mass spectroscopy.General
Mass spectrometry was performed on a Bruker MicrOTOF-Q II by positive-mode electrospray ionization. All NMR spectra were recorded on a Bruker AV 400 MHz spectrometer at 298 K. The PL spectra of the films (C = 20 mmol L−1) were measured on a Horiba ScientificFluoromax-4 spectrophotometer. The FT-IR spectra of all the reported compounds were performed using a Bruker (Tensor-27) FT-IR spectrophotometer. A Raman study was performed on a Micro Raman system from Jobin Yvon Horiba LABRAM-HR visible (400–1100 nm) equipped with an Ar+ laser (488 nm, 10 mW) excitation source and CCD detector.Synthesis of compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.9) as the eluent to obtain 6 (7.5 g, 32.32 mmol, 94%) as a yellow powder. 1H-NMR (400 MHz, CDCl3): δ 7.98 (d, 2H, J = 8 Hz), 7.53 (d, 2H, J = 8 Hz), 3.91 (s, 3H, OCH3), 0.26 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 166.4, 131.8, 129.6, 129.3, 127.7, 104.0, 97.6, 52.1 ppm.:9.9) as the eluent to obtain 7 (2.624 g, 16.4 mmol, 51%) as a white powder. 1H-NMR (CDCl3, 400 MHz): δ 7.94 (d, 2H, J = 8 Hz, aromatic H), 7.49 (d, 2H, J = 8 Hz, aromatic H), 3.85 (s, 3H, OCH3), 3.16 (s, acetylene H) ppm. 13C NMR (100 MHz, CDCl3): δ 166.3, 131.9, 130.0, 129.3, 126.6, 82.7, 79.9, 52.1 ppm.
:9.9) as the eluent to obtain 6 (7.5 g, 32.32 mmol, 94%) as a yellow powder. 1H-NMR (400 MHz, CDCl3): δ 7.98 (d, 2H, J = 8 Hz), 7.53 (d, 2H, J = 8 Hz), 3.91 (s, 3H, OCH3), 0.26 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 166.4, 131.8, 129.6, 129.3, 127.7, 104.0, 97.6, 52.1 ppm.:9.9) as the eluent to obtain 7 (2.624 g, 16.4 mmol, 51%) as a white powder. 1H-NMR (CDCl3, 400 MHz): δ 7.94 (d, 2H, J = 8 Hz, aromatic H), 7.49 (d, 2H, J = 8 Hz, aromatic H), 3.85 (s, 3H, OCH3), 3.16 (s, acetylene H) ppm. 13C NMR (100 MHz, CDCl3): δ 166.3, 131.9, 130.0, 129.3, 126.6, 82.7, 79.9, 52.1 ppm.General procedure for peptide coupling
Compound 9 (1.0 equiv.) was dissolved in dry DMF (4 mL g−1) and stirred on an ice-water bath (Schemes S2 and S3, ESI†). The methyl ester protected amino acid was isolated from its corresponding methyl ester hydrochloride (4.0 equiv.) by neutralization and subsequently extracted twice with ethyl acetate (2 × 30 mL). The collected ethyl acetate extracts were dried over anhydrous Na2SO4 and concentrated to 5 mL. The solution was then added to the pre-cooled reaction mixture followed by the addition of HOBt (2.0 equiv.) and dicyclohexylcarbodiimide (DCC) (2.2 equiv.). The reaction mixture was stirred overnight. The residue was extracted by ethyl acetate (50 mL) and the DCU was removed by filtration. The organic layer was washed with 1 M HCl (3 × 50 mL), brine (2 × 50 mL), 1 M sodium carbonate (3 × 50 mL), brine (2 × 50 mL), dried over anhydrous Na2SO4 and evaporated in vacuum. Purification was carried out using silica gel column chromatography (100–200 mesh) using hexane–ethyl acetate (9:1) as the eluent to obtain the desired product.
General procedure for methyl ester hydrolysis
Methyl ester in 10 mL of MeOH was added to a round bottom flask and 2 N NaOH was added drop-wise (Schemes S2 and S3, ESI†). The reaction was monitored by thin layer chromatography (TLC). The reaction mixture was stirred overnight. 15 mL of distilled water was added to the reaction mixture and the MeOH was removed under vacuum. The aqueous part was washed with diethyl ether (2 × 30 mL), then cooled in an ice-water bath for 10 min and the pH was adjusted to 2 upon the dropwise addition of 1 N HCl. It was extracted with ethyl acetate (3 × 50 mL) and then the ethyl acetate layer was dried over anhydrous Na2SO4 and evaporated under vacuum to yield the corresponding carboxylic acid, which was used for the next step without any further purification.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, free carboxylic), 1649 (CO, amide I), 1541 (N–H, amide II). 1H-NMR (400 MHz, DMSO-d6): δ 12.68 (s, 2H, COOH), 8.51 (d, 2H, J = 8 Hz, NH), 8.14 (d, 2H, J = 8 Hz, NH), 7.89 (d, 2H, J = 8 Hz, aromatic H), 7.59 (d, 2H, J = 8 Hz, aromatic Hs), 7.22 (m, 10H of Phe aromatic ring), 4.57 (m, 2H, CαHs of Phe), 4.46 (m, 2H, CαHs of Leu), 3.05 (m, 2H, CβHs of Phe), 2.94 (m, 2H, CβHs of Phe), 1.67 (m, 4H, CβHs of Leu), 1.51 (m, 2H, CγHs of Leu), 0.90 (m, 12H, CδHs of Leu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 177.95, 177.19, 170.64, 142.67, 139.40, 136.75, 134.37, 133.33, 131.60, 129.71, 88.14, 88.04, 58.53, 56.84, 41.77, 29.55, 28.26, 26.66 ppm. ESI-MS: calcd for [C48H50N4O8 + Na]+ 833.3526, found 833.3264.O, free carboxylic), 1621 (CO, amide I), 1536 (N–H, amide II). 1H-NMR (400 MHz, DMSO-d6): δ 12.62 (m, 2H, COOH), 9.18 (s, 2H, phenolic-OH of Tyr), 8.52 (d, 2H, J = 8 Hz, NH of Tyr), 8.03 (d, 2H, J = 8 Hz, NH), 7.88 (d, 2H, J = 8 Hz, aromatic Hs), 7.58 (d, J = 8 Hz, 2H, aromatic Hs), 7.00 (d, 4H, J = 8 Hz, 4H of Tyr aromatic ring), 6.61 (d, 4H, J = 8 Hz, 4H of Phe aromatic ring), 4.55 (d, 2H, J = 8 Hz, CαHs of Tyr), 4.38 (d, 2H, J = 8 Hz, CαHs of Leu), 2.94 (m, 2H, CβHs of Tyr), 2.91 (m, 2H, CβHs of Tyr), 1.52 (m, 4H, CβHs of Leu), 1.25 (m, 2H, CγHs of Leu), 0.90 (m, 12H, CδHs of Leu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 178.06, 177.12, 170.70, 161.12, 139.43, 136.76, 135.29, 133.03, 132.61, 129.70, 120.15, 88.15, 88.06, 58.88, 56.84, 41.04, 29.57, 26.27, 26.64 ppm. ESI-MS: calcd for [C48H50N4O10 + Na]+ 865.3425, found 865.3583.
O, free carboxylic), 1649 (CO, amide I), 1541 (N–H, amide II). 1H-NMR (400 MHz, DMSO-d6): δ 12.68 (s, 2H, COOH), 8.51 (d, 2H, J = 8 Hz, NH), 8.14 (d, 2H, J = 8 Hz, NH), 7.89 (d, 2H, J = 8 Hz, aromatic H), 7.59 (d, 2H, J = 8 Hz, aromatic Hs), 7.22 (m, 10H of Phe aromatic ring), 4.57 (m, 2H, CαHs of Phe), 4.46 (m, 2H, CαHs of Leu), 3.05 (m, 2H, CβHs of Phe), 2.94 (m, 2H, CβHs of Phe), 1.67 (m, 4H, CβHs of Leu), 1.51 (m, 2H, CγHs of Leu), 0.90 (m, 12H, CδHs of Leu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 177.95, 177.19, 170.64, 142.67, 139.40, 136.75, 134.37, 133.33, 131.60, 129.71, 88.14, 88.04, 58.53, 56.84, 41.77, 29.55, 28.26, 26.66 ppm. ESI-MS: calcd for [C48H50N4O8 + Na]+ 833.3526, found 833.3264.O, free carboxylic), 1621 (CO, amide I), 1536 (N–H, amide II). 1H-NMR (400 MHz, DMSO-d6): δ 12.62 (m, 2H, COOH), 9.18 (s, 2H, phenolic-OH of Tyr), 8.52 (d, 2H, J = 8 Hz, NH of Tyr), 8.03 (d, 2H, J = 8 Hz, NH), 7.88 (d, 2H, J = 8 Hz, aromatic Hs), 7.58 (d, J = 8 Hz, 2H, aromatic Hs), 7.00 (d, 4H, J = 8 Hz, 4H of Tyr aromatic ring), 6.61 (d, 4H, J = 8 Hz, 4H of Phe aromatic ring), 4.55 (d, 2H, J = 8 Hz, CαHs of Tyr), 4.38 (d, 2H, J = 8 Hz, CαHs of Leu), 2.94 (m, 2H, CβHs of Tyr), 2.91 (m, 2H, CβHs of Tyr), 1.52 (m, 4H, CβHs of Leu), 1.25 (m, 2H, CγHs of Leu), 0.90 (m, 12H, CδHs of Leu) ppm. 13C NMR (100 MHz, DMSO-d6): δ 178.06, 177.12, 170.70, 161.12, 139.43, 136.76, 135.29, 133.03, 132.61, 129.70, 120.15, 88.15, 88.06, 58.88, 56.84, 41.04, 29.57, 26.27, 26.64 ppm. ESI-MS: calcd for [C48H50N4O10 + Na]+ 865.3425, found 865.3583.Sample preparation
Compound solutions were prepared by mixing 16.2 mg of compound 1 and 16.8 mg of compound 2, individually in 1 mL of methanol in glass vials. The compounds were completely dissolved by shaking. Then, 1 mL of each of the as-prepared solutions was drop-casted on clean and dried glass slides and was allowed to dry in air at room temperature. Then, the films were irradiated using 254 nm UV light. SEM measurements were performed on the dried films after 60 min of UV irradiation by coating with gold for both compounds 1 and 2. Field emission scanning electron microscopic study was performed on a Carl Zeiss microscope (model-Supra 55).Topochemical polymerization procedure
Topochemical polymerization was accompanied on the films upon irradiation at 254 nm using a 72 W UV lamp at 25 °C. For these experiments, the dried thin films of the compounds were irradiated for 1 h. A condenser was attached to the reactor to maintain its temperature at about 25 °C.DFT calculations
DFT calculations were performed using Gaussian 09 to evaluate the role of the dipeptide side chains in the transformation of morphology during topochemical polymerization. DFT calculations employing the B3LYP functional were carried out on monomer and short oligomers of the conjugated organic polymer. For efficient calculation, we simplified the molecular structure using a polydiacetylene chain without the peptide side chain. A long dipeptide side chain creates overlapping of the orbitals leading to steric strain. To avoid such problems, we carried out the DFT calculation of the conjugated organic polymer without considering the peptide side chain.Thermogravimetric analysis (TGA)
Thermogravimetric analysis (TGA) was performed using a METTLER TOLEDO TGA instrument. The samples were heated from 25 to 600 °C at a constant rate of 5 °C min−1 under a nitrogen atmosphere.Gas sorption measurement
Gas (N2/CO2) adsorption/desorption experiments were carried out using a Quantachrome Autosorb IQ2 Automated Gas Sorption System at 77 K (N2 sorption experiments) and 298 K (CO2 sorption experiments) over the pressure range between 0.025 bar and 1 bar (40 points system). Before the sorption measurements, monomer 1, polymer 1, monomer 2 and polymer 2 were degassed for 10 h with a heating rate of 5 °C min−1. From the gas adsorption results at low P/P0, the pore size distribution of the samples was calculated using the BJH (Barrett–Joyner–Halenda analysis) method. The BET surface area was calculated from the Brunauer–Emmett–Teller (BET) equation.Results and discussion
Topochemical polymerization on solid surface
Dipeptide appended diphenylbutadiyne-based bolaamphiphiles (compounds 1 and 2, C = 20 mmol L−1) were suspended in methanol for dissolution (Fig. 1). Compound 1 in methanol and a two-day aged methanol solution of compound 2 were then drop-casted on quartz glass to prepare their films. Compounds 1 and 2 based films were allowed to polymerize under UV light irradiation (72 W, 254 nm). A film based on compound 1 turned blue whereas the film based on compound 2 turned yellow after 60 min of UV irradiation. As a result of an effective regioselective 1,4-polymerization of the self-organized compound 1, blue colour was observed. UV-Vis spectra were collected to understand the variation in the absorption spectra of the bolaamphiphiles on solid surfaces after the topochemical polymerization reaction (Fig. 2). Prior to UV irradiation, compound 1 absorbed at 340 nm. A new peak appeared in the visible-light region at 610 nm (Fig. 2a) after polymerization. The new peak at 610 nm appeared due to the formation of polydiacetylene (the film turned to blue after 60 min of UV irradiation for compound 1).61,62 Similarly, the UV-Vis spectra of compound 2 were recorded (Fig. 2b) before and after UV irradiation. Compound 2 absorbed a peak at 334 nm prior to UV irradiation, which was attributed to the π–π* transition of diphenylbutadiyne. After 60 min of UV irradiation, a new broad peak appeared at 658 nm and the film turned to yellow. | ||
| Fig. 2 UV-Vis spectra of (a) compound 1 and (b) compound 2 before and after UV polymerization. Raman spectra of (c) compound 1 and (d) compound 2 before and after 60 min of topochemical polymerization. FTIR spectra of (e) compound 1 and (f) compound 2 before (red line) and after 60 min (black line) of topochemical polymerization. | ||
Fourier-transform Raman spectroscopy (FT-Raman) and FT-IR spectroscopy were used to monitor the polymerization reaction of compounds 1 and 2 on the solid surface (Fig. 2). Before UV irradiation, the Raman spectrum of compound 1 shows a peak at 2111 cm−1, which is assigned to the regular stretching mode of 1,3-butadiyne. After UV irradiation, the characteristic peak at 2111 cm−1 disappears and a new peak appears at 2106 cm−1 after 60 min of the polymerization reaction. The peak at 2106 cm−1 arises due to the presence of a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond within the conjugated polymer after UV irradiation. A new peak at 1565 cm−1 appears after UV irradiation due to the formation of CC bonds. It has been reported that the characteristic peaks between 1400 and 1600 cm−1 appear due to the stretching vibration of enyne (Fig. 2c).63 Compound 2 shows a characteristic peak at 2114 cm−1, which is attributed to the presence of the CC stretching vibration. However, the CC stretching vibration peak shifted to 2109 cm−1 after 1 h of UV irradiation. A new peak appears at 1607 cm−1 after UV irradiation of compound 2 due to the formation of CC bonds (Fig. 2d).63,64 These results indicate that both compounds were transformed into poly(diphenylbutadiyne) upon UV irradiation. Fig. 2e and f show the FT-IR spectra of compounds 1 and 2 prior and after the UV polymerization reaction. As shown in Fig. 2e, the characteristic peaks at 3317 and 3271 cm−1 for amide N–H stretching, 1650 cm−1 for amide I and 1537 cm−1 for amide II band were observed. Weak peaks in the region of 3100–3500 cm−1 were observed after the topochemical polymerization reaction, which are indicative of the formation of weakly hydrogen bonded NH groups. However, a sharp peak at 2110 cm−1 appeared due to the presence of CC in compound 1. The intensity of the corresponding CC peak decreases after UV irradiation. After topochemical polymerization, the amide I band at 1650 cm−1 was shifted to 1627 cm−1. The FT-IR results evidently support the structural changes during the polymerization reaction occurring after UV irradiation.65,66 The FT-IR spectra were also acquired for compound 2 before and after the topochemical polymerization reaction. Fig. 2f shows the FT-IR spectra for compound 2. The FT-IR peaks at 3284 cm−1 for amide N–H stretching, 1665 cm−1 and 1633 cm−1 for amide I and 1536 cm−1 for amide II are observed prior to the photochemical polymerization reaction of compound 2. Low intense peaks in the region of 3100–3500 cm−1 were observed after the topochemical polymerization, which are indicative of the formation of weakly hydrogen-bonded NH groups. These results suggest that intermolecular hydrogen bonding between the N–H and CO of the amide groups drives the formation of the self-assembled fiber architecture prior to UV polymerization. The self-organized structures were formed due to the polymerization of compounds 1 and 2 induced by UV irradiation.
C bond within the conjugated polymer after UV irradiation. A new peak at 1565 cm−1 appears after UV irradiation due to the formation of CC bonds. It has been reported that the characteristic peaks between 1400 and 1600 cm−1 appear due to the stretching vibration of enyne (Fig. 2c).63 Compound 2 shows a characteristic peak at 2114 cm−1, which is attributed to the presence of the CC stretching vibration. However, the CC stretching vibration peak shifted to 2109 cm−1 after 1 h of UV irradiation. A new peak appears at 1607 cm−1 after UV irradiation of compound 2 due to the formation of CC bonds (Fig. 2d).63,64 These results indicate that both compounds were transformed into poly(diphenylbutadiyne) upon UV irradiation. Fig. 2e and f show the FT-IR spectra of compounds 1 and 2 prior and after the UV polymerization reaction. As shown in Fig. 2e, the characteristic peaks at 3317 and 3271 cm−1 for amide N–H stretching, 1650 cm−1 for amide I and 1537 cm−1 for amide II band were observed. Weak peaks in the region of 3100–3500 cm−1 were observed after the topochemical polymerization reaction, which are indicative of the formation of weakly hydrogen bonded NH groups. However, a sharp peak at 2110 cm−1 appeared due to the presence of CC in compound 1. The intensity of the corresponding CC peak decreases after UV irradiation. After topochemical polymerization, the amide I band at 1650 cm−1 was shifted to 1627 cm−1. The FT-IR results evidently support the structural changes during the polymerization reaction occurring after UV irradiation.65,66 The FT-IR spectra were also acquired for compound 2 before and after the topochemical polymerization reaction. Fig. 2f shows the FT-IR spectra for compound 2. The FT-IR peaks at 3284 cm−1 for amide N–H stretching, 1665 cm−1 and 1633 cm−1 for amide I and 1536 cm−1 for amide II are observed prior to the photochemical polymerization reaction of compound 2. Low intense peaks in the region of 3100–3500 cm−1 were observed after the topochemical polymerization, which are indicative of the formation of weakly hydrogen-bonded NH groups. These results suggest that intermolecular hydrogen bonding between the N–H and CO of the amide groups drives the formation of the self-assembled fiber architecture prior to UV polymerization. The self-organized structures were formed due to the polymerization of compounds 1 and 2 induced by UV irradiation.
The change in morphology of the two compounds under UV polymerization was studied using scanning electron microscopy (SEM) (Fig. 3). Compound 1, after dissolution in methanol, was drop-casted onto quartz glass slides and revealed fibers (Fig. 3a). After 60 min of UV irradiation, the morphology alters to a porous network structure (Fig. 3b). The entire experiment was performed with a 20 mmol L−1 solution of compound 1. The experiments were performed in triplicate and similar results were obtained. Similar experiments were performed with 15 mmol L−1 and 30 mmol L−1 solutions of compound 1. Different types of morphological changes were observed with respect to the duration of UV irradiation for 15 mmol L−1 and 30 mmol L−1 solutions of compound 1 (Fig. S1 and S2, ESI†). Here, the self-organized structures of compound 1 change into polymerized porous network structures upon topochemical polymerization with different morphological features. The topochemical polymerization of compound 2 was also performed on the solid surface and in the solid state (Fig. S3, ESI†). Compound 2 (20 mmol L−1) was dissolved in methanol. Instant deposition of the methanolic solution of compound 2 did not form a self-assembled structure. However, the two-day aged solution of compound 2 forms fibrillar morphology (Fig. S3a, ESI†). The fibrillar morphology obtained for the two compounds 1 and 2 are relatively different. This differential morphology was observed probably due to the presence of the polar phenolic –OH group present in the tyrosine containing compound 2. Due to the presence of the phenolic –OH group, compound 2 exhibits a more hydrophilic nature when compared with the hydrophobic phenylalanine-based compound 1. The different lattice packing features also drive the discrepancy of the typical fibrous morphological properties in a particular solvent.67 After 60 min of UV irradiation, compound 2 shows a porous network structure (Fig. S3b, ESI†). Here, UV polymerization changes the morphology and self-organization of compound 1 and compound 2 on a solid surface.68 For both the compounds, the hydrogen bonding and π–π interactions between the molecules are involved in the formation of the self-assembled fibers. The dipeptide appended diphenylbutadiyne-based molecules are linear due to the presence of the two conjugated triple bonds. Due to an increase in degree of polymerization, the polymers become aggregated and the morphology changes after 1 h of UV irradiation (Scheme 1). After UV irradiation, the hydrogen bonds involved in the dipeptides become weaker. After 60 min of UV irradiation, the fibers are re-organized to form a porous network structure. This was evidenced in the FT-IR spectra with the peak intensity of the NH stretching of the amide bonds between 3000–3300 cm−1 decreasing after 1 h of UV irradiation. At this time, the formation of polydiacetylene was also evidenced by Raman spectroscopy as new peaks corresponding to the formation of enyne developed between 1400–1600 cm−1.69,70 Intermolecular hydrogen bonding becomes weaker and the formation of polymeric enyne is responsible for the structural changes observed for compound 1. Similarly, compound 2 shows the fibrillar morphology prior to UV irradiation due to the linear configuration in the presence of the two conjugated triple bonds. After 1 h of UV irradiation, the hydrogen bonds become weaker as evidenced by the FTIR spectra. The fibrillar structures turn into polymerized porous networks with an alternating ene-yne configuration. From the DFT study, we have observed that after the topochemical polymerization reaction, steric crowding between peptide side chains disrupts the hydrogen bonding. Due to disruption of the hydrogen bonding, the fibrillar morphology transforms to a porous network morphology. The closest distance between the two molecules due to the formation of the diyne was 3.6 Å. This result supports the crystal structure of a reported diyne system.71 The distance between the two peptide molecules should be 4.7 Å.72 Hence, the hydrogen bonds between the amide groups in the peptide molecules are disrupted. As a result, the fibrillar structures transform to porous network structures.
 | ||
| Fig. 3 SEM images at different times of the UV reaction of compound 1: (a) before the UV reaction and (b) after 60 min of UV irradiation (polymer 1). | ||
 | ||
| Scheme 1 Possible self-assembly mechanism of the dipeptide appended diphenylbutadiyne based compound 1. | ||
Thermogravimetric analysis (TGA) was performed to analyse the thermal stability of compound 1, polymer 1, compound 2 and polymer 2 (Fig. S4, ESI†). The decomposition temperature of compounds 1 and 2 was observed between 150–200 °C with an initial weight loss of 2–6%. However, the decomposition temperature of polymers 1 and 2 was observed between 500–600 °C with an initial weight loss of 2–6%. Hence, the thermal stability of the polymers was higher than their corresponding monomeric compounds.
In current research, porous covalent organic polymers with high surface areas have gained particular interest in the area of greenhouse gas storage.73 To analyse their porous features, gas adsorption studies were executed with the dried self-assembled structures of compounds 1 and 2 grown from a methanol solution and the 60 min UV irradiated polymers of 1 and 2. Polymer 1 exhibits 24.22 times more N2 gas uptake than compound 1 (Table 1). The BET (Brunauer–Emmett–Teller) surface area of compound 1 was calculated to be 4.905 m2 g−1 whereas the BET surface area becomes 106.38 m2 g−1 for polymer 1. Thus, the surface area of polymer 1 exhibits a 21.68 fold higher surface area than compound 1. The pore volume of compound 1 was calculated to be 0.0144 cm3 g−1, whereas the pore volume of polymer 1 was revealed to be 0.3845 cm3 g−1 (Fig. 4). The BET surface area of compound 2 was measured to be 10.599 m2 g−1, whereas the BET surface area becomes 58.800 m2 g−1 for polymer 2. Thus, the surface area of polymer 2 was 5.54 times higher than the surface area of compound 2. The pore volume of compound 2 was measured to be 0.0156 cm3 g−1 whereas the pore volume of polymer 2 was 0.1966 cm3 g−1. Interestingly, the porous polymer structures show enhanced sorption preferences than their corresponding monomers.74–77 The porosity developed on the surface after light induced polymerization of compounds 1 and 2 was in good agreement with the SEM images. According to the IUPAC nomenclature, the N2 gas isotherms measured for compounds 1 and 2, and polymers 1 and 2 can be classified by type I isotherms corresponding to a hierarchically porous material. As shown in Fig. 4c, the CO2 uptake of compound 1 was measured to be 6.8 cm3 g−1. However, the CO2 uptake was calculated to be 29.71 cm3 g−1 for polymer 1. Thus, polymer 1 exhibits 4.36 times more CO2 storage capacity than compound 1. The CO2 uptake of compound 2 was measured to be 6.73 cm3 g−1. However, the CO2 uptake observed for polymer 2 was measured to be 28.08 cm3 g−1 (Fig. 4d). Thus, polymer 2 shows 4.17 times more CO2 storage capacity than compound 2 (Table 1). The CO2 uptake capacities of the synthesized covalent organic polymers (1.32 mmol g−1 for polymer 1 and 1.25 mmol g−1 for polymer 2) at 1 bar are comparable to the previously reported adsorption capacities of COPs and porous organic solids. Porous organic solids such as TCMP-3 and TCMP-0 adsorb 1.29 mmol g−1 and 1.34 mmol g−1 CO2, respectively. Other COPs such as CMP-1 (1.18 mmol g−1), CMP-1-(COOH) (0.95 mmol g−1), CMP-1-(NH2) (0.95 mmol g−1), CMP-1(CH3)2 (0.94 mmol g−1), CMP-1-(OH) (1.07 mmol g−1), CMP-5 (1.1 mmol g−1), CMP-5 (0.63 mmol g−1), PMF-1 (1.33 mmol g−1) and TCMP-5 (0.681 mmol g−1) also adsorb CO2.78–82 In addition, the advantage of our as-prepared polymer-adsorbents is that they can be synthesized easily via a light-assisted polymerization reaction. Captivatingly, these polydiacetylene-adsorbents can capture CO2 gas in a significant amount. The development of the porous architectures with enhanced surface areas after topochemical polymerization was responsible for their enhanced CO2 sorption.
| Parameter | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (diameter in nm) | Isotherm type | Maximum amount of gas adsorbed (cm3 g−1) |
|---|---|---|---|---|---|
| N2 sorption | |||||
| Compound 1 | 4.905 | 0.0144 | 1.744 | I | 9.38 |
| Polymer 1 | 106.368 | 0.3845 | 2.7196 | I | 227.22 |
| Compound 2 | 10.599 | 0.0156 | 2.158 | I | 10.34 |
| Polymer 2 | 58.800 | 0.1966 | 2.7127 | I | 128.28 |
|
|||||
| CO2 sorption | |||||
| Compound 1 | — | 0.01427 | 0.7378 | 6.8 | |
| Polymer 1 | — | 0.05944 | 0.8734 | 29.71 | |
| Compound 2 | — | 0.0141 | 0.8066 | 6.73 | |
| Polymer 2 | — | 0.05891 | 1.0477 | 28.08 | |
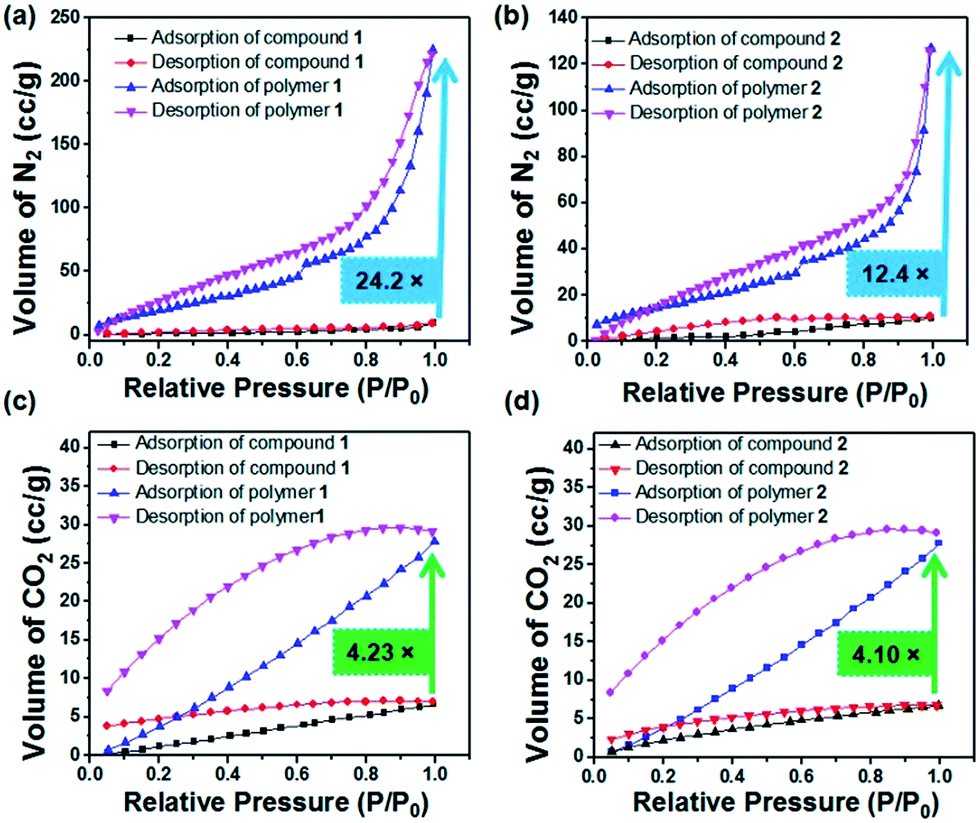 | ||
| Fig. 4 (a) N2 adsorption and desorption isotherms at 77 K (P0 = 1 atm) obtained for compound 1 and polymer 1. (b) N2 adsorption and desorption isotherms at 77 K (P0 = 1 atm) obtained for compound 2 and polymer 2. (c) CO2 adsorption and desorption isotherms at 298 K (P0 = 1 atm). (d) CO2 adsorption and desorption isotherms at 298 K (P0 = 1 atm) obtained for compound 2 and polymer 2. | ||
Conclusion
In summary, porous covalent organic polymers were developed using the topochemical polymerization of dipeptide appended diphenylbutadiyne-based bolaamphiphiles on a solid surface. Topochemical polymerization leads to change in the self-organized morphological features from fibers to porous network structures. The surface area, pore volume and porosity of the polymers were increased several times when compared to their corresponding compounds prior to polymerization. Polymers 1 and 2 exhibit 4.2 and 4.1 times more CO2 sorption behaviour than compounds 1 and 2, respectively. The porous polymer networks could interact with the N2 and CO2 gases through their amide bonds and the close proximity of the molecules through the covalent enyne bonds. The polymers are thermally stable when compared to their monomers, which was revealed by our TGA experiments. The light induced development of covalent organic polymers presented in this study unbolts new possibilities for tuning the porous properties of organic polymers for a variety of applications, including the design of catalysts and promising adsorbents for industrial applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AKD sincerely acknowledges NanoMission, Department of Science & Technology (Project No. SR/NM/NS-1458/2014), New Delhi, India for financial support. SB is thankful to MHRD, New Delhi for his doctoral fellowship and MK is thankful to IIT Indore for his postdoctoral research fellowship. The sophisticated instrumentation centre (SIC), IIT Indore is acknowledged for providing access to their instrumentation. The authors thank the UGC-DAE Consortium for Scientific Research, Indore for their help in recording the Raman data.References
- R. Babarao and J. Jiang, Energy Environ. Sci., 2008, 1, 139–143 CAS.
- Z. Xiang, R. Mercado, J. M. Huck, H. Wang, Z. Guo, W. Wang, D. Cao, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2015, 137, 13301–13307 CrossRef CAS PubMed.
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 CAS.
- L. Zhai, N. Huang, H. Xu, Q. Chen and D. Jiang, Chem. Commun., 2017, 53, 4242–4245 RSC.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691–2718 CAS.
- Y.-L. Wong, J. M. Tobin, Z. Xu and F. Vilela, J. Mater. Chem. A, 2016, 4, 18677–18686 CAS.
- K. Park, K. Lee, H. Kim, V. Ganesan, K. Cho, S. K. Jeong and S. Yoon, J. Mater. Chem. A, 2017, 5, 8576–8582 CAS.
- G. Mukherjee, J. Thote, H. B. Aiyappa, S. Kandambeth, S. Banerjee, K. Vanka and R. Banerjee, Chem. Commun., 2017, 53, 4461–4464 RSC.
- X. Shi, Y. Yao, Y. Xu, K. Liu, G. Zhu, L. Chi and G. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 7481–7488 CAS.
- Z. Xiang, X. Zhou, C. Zhou, S. Zhong, X. He, C. Qin and D. Cao, J. Mater. Chem., 2012, 22, 22663–22669 RSC.
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 CAS.
- P. Ghosh, S. K. Saha, A. Roychowdhury and P. Banerjee, Eur. J. Inorg. Chem., 2015, 2015, 2851–2857 CrossRef CAS.
- P. Bhanja, K. Bhunia, S. K. Das, D. Pradhan, R. Kimura, Y. Hijikata, S. Irle and A. Bhaumik, ChemSusChem, 2017, 10, 921–929 CrossRef CAS PubMed.
- H. Ma, H. Ren, S. Meng, Z. Yan, H. Zhao, F. Sun and G. Zhu, Chem. Commun., 2013, 49, 9773–9775 RSC.
- Z. Xiang, D. Cao, W. Wang, W. Yang, B. Han and J. Lu, J. Phys. Chem. C, 2012, 116, 5974–5980 CAS.
- J. Sun and X. Bao, Chem.–Eur. J., 2008, 14, 7478–7488 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 3207 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- J. Hotz and W. Meier, Langmuir, 1998, 14, 1031–1036 CrossRef CAS.
- S. Lei, X. Gao, D. Cheng, L. Fei, W. Lu, J. Zhou, Y. Xiao, B. Cheng, Y. Wang and H. Huang, Eur. J. Inorg. Chem., 2017, 2017, 1892–1899 CrossRef CAS.
- A. Perino, M. Schmutz, S. Meunier, P. J. Mesini and A. Wagner, Langmuir, 2011, 27, 12149–12155 CrossRef CAS PubMed.
- N. Laggoune, F. Delattre, J. Lyskawa, F. Stoffelbach, J. M. Guigner, S. Ruellan, G. Cooke and P. Woisel, Polym. Chem., 2015, 6, 7389–7394 RSC.
- V. V. Atuchin, T. A. Gavrilova, V. G. Kostrovsky, L. D. Pokrovsky and I. B. Troitskaia, Inorg. Mater., 2008, 44, 622 CrossRef CAS.
- A. D. Merg, J. C. Boatz, A. Mandal, G. Zhao, S. Mokashi-Punekar, C. Liu, X. Wang, P. Zhang, P. C. A. van der Wel and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 13655–13663 CrossRef CAS PubMed.
- L. Ziserman, H.-Y. Lee, S. R. Raghavan, A. Mor and D. Danino, J. Am. Chem. Soc., 2011, 133, 2511–2517 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- H. Choi, M. T. Jeena, L. Palanikumar, Y. Jeong, S. Park, E. Lee and J.-H. Ryu, Chem. Commun., 2016, 52, 5637–5640 RSC.
- D. Mumcuoglu, M. Sardan Ekiz, G. Gunay, T. Tekinay, A. B. Tekinay and M. O. Guler, ACS Appl. Mater. Interfaces, 2016, 8, 11280–11287 CAS.
- P. K. Sukul, P. Bose, T. Takei, O. M. Yaghi, Y. He, M. Lee and K. Tashiro, Chem. Commun., 2016, 52, 1579–1581 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- C. Guo, Z. A. Arnon, R. Qi, Q. Zhang, L. Adler-Abramovich, E. Gazit and G. Wei, ACS Nano, 2016, 10, 8316–8324 CrossRef CAS PubMed.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- N. Baccile, A.-S. Cuvier, S. Prévost, C. V. Stevens, E. Delbeke, J. Berton, W. Soetaert, I. N. A. Van Bogaert and S. Roelants, Langmuir, 2016, 32, 10881–10894 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T.-C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- T. Shimizu, N. Kameta, W. Ding and M. Masuda, Langmuir, 2016, 32, 12242–12264 CrossRef CAS PubMed.
- J. B. Matson, Y. Navon, R. Bitton and S. I. Stupp, ACS Macro Lett., 2015, 4, 43–47 CrossRef CAS.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- K. Ma, R. Xing, T. Jiao, G. Shen, C. Chen, J. Li and X. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 30759–30767 CAS.
- C. E. Morgan, A. W. Dombrowski, C. M. Rubert Pérez, E. S. M. Bahnson, N. D. Tsihlis, W. Jiang, Q. Jiang, J. M. Vercammen, V. S. Prakash, T. A. Pritts, S. I. Stupp and M. R. Kibbe, ACS Nano, 2016, 10, 899–909 CrossRef CAS PubMed.
- A. C. Eldredge, M. E. Johnson, N. J. Oldenhuis and Z. Guan, Biomacromolecules, 2016, 17, 3138–3144 CrossRef CAS PubMed.
- S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC.
- M. Konda, B. Kauffmann, D. B. Rasale and A. K. Das, Org. Biomol. Chem., 2016, 14, 4089–4102 CAS.
- E. R. da Silva, M. N. M. Walter, M. Reza, V. Castelletto, J. Ruokolainen, C. J. Connon, W. A. Alves and I. W. Hamley, Biomacromolecules, 2015, 16, 3180–3190 CrossRef CAS PubMed.
- J. Jaworski, K. Yokoyama, C. Zueger, W.-J. Chung, S.-W. Lee and A. Majumdar, Langmuir, 2011, 27, 3180–3187 CrossRef CAS PubMed.
- J. J. Bang, K. K. Rupp, S. R. Russell, S. W. Choong and S. A. Claridge, J. Am. Chem. Soc., 2016, 138, 4448–4457 CrossRef CAS PubMed.
- S. Wang and K. S. Schanze, ACS Appl. Mater. Interfaces, 2013, 5, 4487 CAS.
- B. E. I. Ramakers, S. A. Bode, A. R. Killaars, J. C. M. van Hest and D. W. P. M. Löwik, J. Mater. Chem. B, 2015, 3, 2954–2961 RSC.
- K. Maeda, L. Hong, T. Nishihara, Y. Nakanishi, Y. Miyauchi, R. Kitaura, N. Ousaka, E. Yashima, H. Ito and K. Itami, J. Am. Chem. Soc., 2016, 138, 11001–11008 CrossRef CAS PubMed.
- M. Suzuki, J. F. K. Kotyk, S. I. Khan and Y. Rubin, J. Am. Chem. Soc., 2016, 138, 5939–5956 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- S. R. Diegelmann, N. Hartman, N. Markovic and J. D. Tovar, J. Am. Chem. Soc., 2012, 134, 2028–2031 CrossRef CAS PubMed.
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC.
- I. Maity, H. S. Parmar, D. B. Rasale and A. K. Das, J. Mater. Chem. B, 2014, 2, 5272–5279 RSC.
- A. K. Das, I. Maity, H. S. Parmar, T. O. McDonald and M. Konda, Biomacromolecules, 2015, 16, 1157–1168 CrossRef CAS PubMed.
- J. Kwak and S.-Y. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 6461–6468 CAS.
- I. W. Hamley, Angew. Chem., Int. Ed., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- Q. Liu, S. Chen, J. Chen and J. Du, Macromolecules, 2015, 48, 739–749 CrossRef CAS.
- G. W. Coates, A. R. Dunn, L. M. Henling, D. A. Dougherty and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 248–251 CrossRef CAS.
- L. Zhu, H. Tran, F. L. Beyer, S. D. Walck, X. Li, H. Ågren, K. L. Killops and L. M. Campos, J. Am. Chem. Soc., 2014, 136, 13381–13387 CrossRef CAS PubMed.
- J. R. Néabo, K. I. S. Tohoundjona and J.-F. Morin, Org. Lett., 2011, 13, 1358–1361 CrossRef PubMed.
- A. Potisatityuenyong, R. Rojanathanes, G. Tumcharern and M. Sukwattanasinitt, Langmuir, 2008, 24, 4461–4463 CrossRef CAS PubMed.
- E. Jahnke, J. Weiss, S. Neuhaus, T. N. Hoheisel and H. Frauenrath, Chem.–Eur. J., 2009, 15, 388–404 CrossRef CAS PubMed.
- S. Biswas, D. B. Rasale and A. K. Das, RSC Adv., 2016, 6, 54793–54800 RSC.
- M. Shirakawa, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164–4165 CrossRef CAS PubMed.
- K. Lv, L. Zhang and M. Liu, Langmuir, 2014, 30, 9295–9302 CrossRef CAS PubMed.
- J.-M. Kim, Y. B. Lee, S. K. Chae and D. J. Ahn, Adv. Funct. Mater., 2006, 16, 2103–2109 CrossRef CAS.
- J.-M. Heo, Y. Kim, S. Han, J. F. Joung, S. Lee, S. Han, J. Noh, J. Kim, S. Park, H. Lee, Y. M. Choi, Y.-S. Jung and J.-M. Kim, Macromolecules, 2017, 50, 900–913 CrossRef CAS.
- K. Morrisa and L. Serpel, Chem. Soc. Rev., 2010, 39, 3445–3453 RSC.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- L. Wang, C. Pan, A. Liang, X. Zhou, W. Zhou, T. Wan and L. Wang, Polym. Chem., 2017, 8, 4644–4650 RSC.
- K. Zhang, A. Nalaparaju and J. Jiang, J. Mater. Chem. A, 2015, 3, 16327–16336 CAS.
- J. Hotz and W. Meier, Langmuir, 1998, 14, 1031–1036 CrossRef CAS.
- S.-L. Zhong, R. Xu, L.-F. Zhang, W.-G. Qu, G.-Q. Gao, X.-L. Wu and A.-W. Xu, J. Mater. Chem., 2011, 21, 16574–16580 RSC.
- S. Ren, R. Dawson, A. Laybourn, J. Jiang, Y. Khimyak, D. J. Adams and A. I. Cooper, Polym. Chem., 2012, 3, 928–934 RSC.
- S. K. Gupta, D. Kaleeswaran, S. Nandi, R. Vaidhyanathan and R. Murugavel, ACS Omega, 2017, 2, 3572–3582 CrossRef CAS.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- A. Bhunia, V. Vasylyeva and C. Janiak, Chem. Commun., 2013, 49, 3961–3963 RSC.
- Q. Xiao, J. Wen, Y. Guo, J. Hu, J. Wang, F. Zhang, G. Tu, Y. Zhong and W. Zhu, Ind. Eng. Chem. Res., 2016, 55, 12667–12674 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details, spectroscopic data for all synthesized compounds, SEM image, TGA data and gas adsorption data. See DOI: 10.1039/c7ra09538d |
| This journal is © The Royal Society of Chemistry 2017 |