
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the toxicity of silicon dioxide nanoparticles on neural stem cells using RNA-Seq
Dayu Sun†
 abc,
Linji Gong†de,
Jing Xieac,
Xiao Heac,
Siyu Chenac,
Luodan Aac,
Qiyou Liac,
Zhanjun Gu*de and
Haiwei Xu*ac
abc,
Linji Gong†de,
Jing Xieac,
Xiao Heac,
Siyu Chenac,
Luodan Aac,
Qiyou Liac,
Zhanjun Gu*de and
Haiwei Xu*ac
aSouthwest Hospital/Southwest Eye Hospital, Third Military Medical University, Chongqing 400038, China. E-mail: haiweixu2001@163.com
bDepartment of Physiology, Third Military Medical University, Chongqing 400038, China
cKey Lab of Visual Damage and Regeneration & Restoration of Chongqing, Chongqing 400038, China
dKey Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China. E-mail: zjgu@ihep.ac.cn
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 9th October 2017
Abstract
Neural stem cells are characterized by self-renewal and multipotency, and a capacity to regenerate in response to brain injury or neurodegenerative disease. Silicon dioxide nanoparticles (SiO2 NPs) are novel materials, which enable the delivery of specific payloads to stem cells; for example, genes or proteins, to enable cell-fate manipulation, or tracer materials, to enable in vivo tracing. However, little is known about the dose-dependent cytotoxicity of SiO2 NPs, and how exposure to SiO2 NPs changes mRNA expression profiles in neural stem cells. In this study, a mouse C17.2 neural stem cell line was treated with 90 nm monodisperse fluorescein isothiocyanate-SiO2 NPs at 0, 100, 200 and 400 μg mL−1 for 48 hours. Internalization of SiO2 NPs was observed in C17.2 cells in a dose-dependent manner. SiO2 NP exposure induced apoptosis and inhibited cell proliferation in the C17.2 cell line at dosage levels of 200 μg mL−1 and above. Microscopically, mitochondrial swelling and cristae fracture were observed. Furthermore, next generation RNA sequencing (RNA-Seq) indicated that high-dose SiO2 NP exposure specifically inhibited transcription of glutathione-S-transferase (GST) genes, including GSTM1, GSTM7 and GSTT1. These results suggest that application of high-dose SiO2 NPs to the nervous system may cause neurotoxicity, induce apoptosis and reduce neural stem cell proliferation by inhibiting GST gene expression.
1. Introduction
Nanoparticles (NPs) are a new technology that is increasingly finding applications in industry and everyday life, including in electronics,1,2 food production,3,4 cosmetics5 and pharmaceutical products.6,7 Silicon dioxide nanoparticles (SiO2 NPs) are one of the most widely used inorganic NPs. Monodisperse SiO2 NPs are considered to have good biocompatibility,8 and thus have great potential for applications in the biomedical and biotechnological fields. A number of recent studies have revealed that some SiO2 NPs have the ability to cross biological barriers, including the blood–brain barrier,9–11 opening up the possibility of applications in the central nervous system (CNS).SiO2 NPs can bind a DNA payload, through modification of specific surface amino groups, and have been successfully used as a non-viral vector for gene delivery.12–14 For example, a DNA plasmid of fibroblast growth factor receptor type 1, carried by organically modified silica (ORMOSIL) nanoparticles, has been demonstrated to inhibit the proliferation of neural stem/progenitor cells in the mouse subventricular zone and the adjacent rostral migratory stream.12 SiO2 NPs have also been used as carriers of proteins to manipulate cell function. For example, delivery of SiO2 NP-anti-pAkt induced cell death in rat neural stem cells (NSCs),13 and uptake of GFP-FRATtide-SiO2 NPs led to inhibition of GSK-3β and elevation of the β-catenin pathway in rat NSCs, thus maintaining the self-renewal and undifferentiated state of the NSCs.14
Besides acting as a delivery vector for biological molecules, SiO2 NPs have also been used for tracing of NSCs in vivo. For example, through doping with magnetic iron, SiO2 NPs can be used as magnetic resonance imaging (MRI) probes for stem-cell tracking.15 Also, Gd3+-incorporated mesoporous SiO2 (Gd2O3@SiO2) nanoparticles have been used to label transplanted NSCs, allowing them to be monitored as a hyperintense signal by 3T MRI from 4 to 14 days in the living brain.16 Finally, Fe3O4@SiO2-(fluorescein isothiocyanate)-CD133 nanocomposites, a four-in-one system, have recently been used in magnetic cell collection, magnetic and optical imaging, and specific tracking of NSCs.17
Considering the wide range of possible applications of SiO2 NPs, from lab research to industry, to medical therapy, it is very important to study their potential adverse effects on human health and the environment. Resolving the safety and toxicity issues associated with this kind of nanomaterial will not only be beneficial to industry, but will also facilitate development of biomedical applications, for drug delivery, bioimaging and biosensors.
Numerous studies have been carried out into the toxicity of SiO2 NPs. However, most research has focused on cytotoxicity in the respiratory system. For example, it has been demonstrated that SiO2 NPs trigger anti-oxidative gene expression in the RAW 267.4 macrophage cell line, but not in the A549 lung epithelial cell, but they induce an inflammatory response in both cell-types.18 In human lung cancer cells, SiO2 NP exposure causes oxidative stress, with increased levels of reactive oxygen species (ROS) and lactate dehydrogenase (LDH), and a reduced glutathione (GSH) level.19
However, very few toxicity studies have been performed in the neurological system. They are urgently needed, given the large demand for safe therapies that are able to cross the blood–brain barrier (BBB). One candidate target cell in the brain is the neural stem cell (NSC), an endogenous, CNS stem cell (mainly found in the hippocampus and subventricular zone of the adult brain), which is capable of self-renewal, and differentiation, for repair and regeneration of the nervous system damage.20–22 Manipulation of the fate-determination of NSCs (e.g. using SiO2 NPs) could lead to treatments for diseases such as Alzheimer's disease and Parkinson's disease.
Several studies have exposed NSCs to SiO2 NPs, to test internalization, cell viability and mitochondrial activity.23–25 However, the effect of SiO2 NPs on NSC proliferation and death, and the underlying mechanisms, are still unclear. The recent development of next-generation RNA sequencing (so-called RNA-Seq) allows for high-throughput mapping and quantification of gene transcripts,26,27 and therefore enables us to study these effects more closely. RNA-Seq has already shown great potential in studying the biomedical compatibility of nanomaterials, and environmental risk assessment.3,28
In this study, we used a C17.2 mouse NSC line as a model to explore the potential harmful effect of SiO2 NPs on the CNS. To do this, we fabricated a fluorescein isothiocyanate (FITC)-functionalized SiO2 with ∼90 nm diameter. Fluorescence-activated cell sorting, immunocytochemistry staining and confocal microscopy were used to detect the effects of SiO2 NPs on apoptosis and proliferation of NSCs.
Furthermore, previous reports have focused on very specific cellular effects of SiO2 NPs, rather than on the overall internal state of the cell, thus limiting the scope and generalizability of their findings. To try and address these limitations, we here present the results of RNA-Seq analysis of NSC gene expression profiles following exposure to SiO2 NPs. This allows us to identify all genes whose expression is affected by SiO2 NPs, and specifically those which may be associated with cytotoxicity, to provide deeper insights into the underlying molecular mechanisms of toxicity.
Here we show that high-dose SiO2 NPs can down-regulate the expression of GSTM1, GSTM7 and GSTT1, leading to mitochondrial damage in NSCs, which suggests that SiO2 NPs cytotoxicity is caused by induced oxidative stress. This study therefore provides a unique and novel insight into the cytotoxicity of SiO2 NPs, and illustrates the value of using RNA-Seq in future studies, in order to better understand and hopefully mitigate the toxicity of SiO2 nanoparticles, so they can be safely used for new therapies.
2. Experimental section
2.1 Materials
3-Aminopropyltriethoxysilane (APTES, 98%), fluorescein isothiocyanate (FITC, 95%), ammonium hydroxide (NH4OH, 28–30% as NH3) and tetraethyl orthosilicate (TEOS, 98%) were purchased from Alfa Aesar Ltd (Shanghai, China). Absolute ethyl alcohol was acquired from Beijing Chemical Reagent Company (Beijing, China). All materials were used as received and without further purification. Deionized water was used in all experiments.2.2 Preparation of FITC precursor solution
FITC-APTES stock solution was prepared as follows. 20 mg FITC was stirred with 12 μL APTES in 10 mL ethanol overnight, in the dark, at room temperature, to achieve APTES-modified FITC. The resulting FITC-APTES solution was stored in the dark at −20 °C for later use.2.3 Synthesis and characterization of SiO2 NPs
To prepare green fluorescein-doped silica nanoparticles (FITC-SiO2 NPs) for visible cellular internalization, we used a modified Stober process, as described in a previous report.29,30 Briefly, an excess of FITC-APTES solution was slowly added to a mixture containing 150 mL ethanol, 15 mL TEOS and 15 mL deionized water. Next, 1 mL ammonia solution dispersed in ethanol was added dropwise, and the mixture was stirred at room temperature in a dark place. Finally, the mixed solution was centrifuged and washed with ethanol and water several times, to remove free FITC-APTES and ammonium ions.To reduce the cytotoxicity of the synthesized SiO2 NPs, the SiO2 NP mixture was dialyzed against purified water for 1 day using a dialysis tube (3.0 kDa molecular cut-off), to remove impurities, especially ammonium ions. The synthesized FITC-SiO2 NPs were then re-dispersed in sterile deionized water, or penicillin–streptomycin solution, and stored in a dark place for future use.
The size and morphology of the as-synthesized SiO2 NPs were observed using a scanning electron microscope (SEM, S-4800, Hitachi) and a field emission transmission electron microscope (TEM, JEM-2100PLUS, JEOL Ltd.). The hydrodynamic diameter distributions and zeta potential of the SiO2 NPs were measured by dynamic light scattering (DLS) on a NanoBrook ZetaPlus (Brookhaven). The green fluorescence of the as-synthesized SiO2 NPs was observed under ultraviolet illumination (365 nm wavelength).
2.4 Cell culture and SiO2 NP treatment
C17.2 NSC line was purchased form CellBank Australia, which was originally supplied by European Collection of Authenticated Cell Cultures (ECACC). As previously described, the C17.2 NSC line was cultured in Dulbecco's Modified Eagle's Medium (DMEM, Hyclone), supplemented with 10% fetal bovine serum (FBS, Gibco BRL), 100 units per mL penicillin and 100 μg mL−1 streptomycin (Invitrogen), and kept at 37 °C, 95% humidity and 5% CO2 atmosphere.31,32 In this work, all the biological samples were from the mouse C17.2 NSC line.For sample preparation, SiO2 NPs were dispersed for 10 minutes using a sonicator (Qsonica Q700) to prevent aggregation. C17.2 NSCs were then seeded in six-well plates, and cultured for 5 hours, before exposing to SiO2 NPs at doses of 0 (vehicle control), 100, 200 and 400 μg mL−1. To evaluate the long-term influence on stem cell proliferation, the C17.2 NSCs were cultured with SiO2 NPs for 48 hours.23
2.5 Cultured cell imaging
After treatment with FITC-SiO2 NPs for 48 hours, cell images were obtained using a Leica inverted microscope (Leica DMI3000B). The fluorophore of SiO2 NPs was excited with a mercury lamp with a 482 nm bandpass filter. Both phase-contrast images and fluorescence images were captured in the same field for all four groups. Image analysis was performed by overlying fluorescence and phase contrast images.2.6 Immunocytochemical staining
For immunocytochemical staining, coverslips seeded with C17.2 cells were rinsed once in 0.01 M PBS, fixed with 4% paraformaldehyde (PFA) for 30 minutes, and then washed three times with 0.01 M PBS for 10 minutes. Fixed cells were permeated for 15 minutes in 0.3% Triton-X 100 and blocked in 3% bovine serum albumin (BSA) for 20 minutes, to reduce non-specific binding.Coverslips were then incubated with the primary antibody, rabbit anti-Ki67 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500, CST) in 1% BSA overnight at 4 °C, then washed three times with 0.01 M PBS, and incubated with Alexa Fluor 568-conjugated secondary antibody (1:500; goat anti-rabbit, Invitrogen) for 1 hour at 37 °C. After washing three times with 0.01 M PBS, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich).
:500, CST) in 1% BSA overnight at 4 °C, then washed three times with 0.01 M PBS, and incubated with Alexa Fluor 568-conjugated secondary antibody (1:500; goat anti-rabbit, Invitrogen) for 1 hour at 37 °C. After washing three times with 0.01 M PBS, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich).
Staining was visualized and quantified using a confocal laser-scanning microscope (Zeiss LSM 800, Carl Zeiss) and analyzed using Zeiss imaging software. For viewing the distribution of FITC-SiO2 NPs, orthogonal projections of the confocal Z-stack were analyzed.
2.7 Flow cytometry
For analysis of the cell cycle, C17.2 cells, which had been exposed to SiO2 NPs for 48 hours, were dissociated using TryPLE (Gibco) and fixed in 70% ethanol. A FACS cell cycle kit (Yueya Institute of Biotechnology) was used according to the manufacturer's instructions. The cell cycle of C17.2 cells was detected using flow cytometry (FACS Calibur, BD Biosciences) and analyzed with FlowJo 7.2 software.An apoptosis assay was performed with propidium iodide/APC-Annexin V (BD Pharmingen™) according to the manufacturer's instructions. Briefly, cells were harvested and re-suspended with 400 μL Annexin V binding buffer at a concentration of 106 cells per mL. Then, 5 μL propidium iodide and 5 μL APC (allophycocyanin)-conjugated Annexin V were added to 100 μL aliquots of cell solution, and incubated at room temperature for 15 minutes in the dark. The cell apoptosis assay was performed within 1 hour post-staining on the flow cytometer.
2.8 Transmission electron microscopy (TEM)
TEM was performed to verify SiO2 NP internalization and detect the subcellular effects, as previously described.33 Briefly, after 48 hour of SiO2 NP exposure, C17.2 cells were harvested, centrifuged at 200 g for 10 min and fixed in 3% glutaraldehyde. Then the pellets were post-fixed in 1% osmium tetroxide, dehydrated with an ascending acetone series, ending with propylene oxide, and embedded in epoxy resin 618. Ultra-thin sections were cut with a diamond knife (diatome) and post-stained with uranyl acetate and lead citrate. Sections on copper grids were analyzed with a JEM-1400 Plus (JEOL) electron microscope at an operating voltage of 120 kV.2.9 RNA-Seq and bioinformatic analysis
Three samples of the C17.2 NSC line, treated with 200 μg mL−1 SiO2 NPs, were collected for RNA extraction with TRIzol reagent (Sigma Aldrich). For convenience, this is referred to in the Results section as the “SiO2 NP-200” group. A second group, consisting of three vehicle control (0 μg mL−1 SiO2 NPs) samples, underwent the same analysis. This is referred to in the Results section as simply the “control” group.mRNA was enriched with oligo (dT) magnetic beads and fragmented into short fragments using fragmentation buffer. The fragments were enriched by PCR amplification to construct transcriptome libraries. Primary raw reads, produced by Illumina HiSeq 4000 (Illumina), were qualified and filtered to obtain clean reads.
Gene expression levels were calculated using FPKM (fragments per kilobase of transcript per million fragments mapped) methods. Differentially Expressed Genes (DEGs) between the two groups were analyzed using the NOISeq method. Genes with a fold-change greater than 1.5-fold and a diverge probability more than 0.8 were considered to be DEGs.34
The KEGG (Kyoto Encyclopedia of Genes and Genomes) database was used to perform pathway analysis of these DEGs. KEGG analysis identifies significantly-enriched metabolic pathways or signal transduction pathways in DEGs, compared to the whole-genome background.35 Based on the KEGG database, “pathway-act-network” analysis was used to construct an interactive network for graphical representation of central pathways, using Cytoscape (open source, http://www.cytoscape.org).36
Next, we constructed a co-expression network based on DEGs. Pairs of genes with Pearson's correlation coefficients >0.99 were used to construct the network. k-Core scoring was used to infer the location of a gene in the network, and the k-Core difference (difk-Core) between the control and SiO2 NP-200 groups was used to identify core regulatory genes.37
2.10 Real-time quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as previously described.38 Briefly, total RNA was extracted from the cell pellets using TRIzol reagent (Sigma Aldrich), according to manufacturer's instructions. RNA samples were quantified and qualified using a spectrophotometer (NanoDrop-2000) and then reverse-transcribed into cDNA using a PrimeScript® RT Reagent Kit (Takara). Quantitative PCR was then performed using a CFX96 Real-time PCR System (Bio-Rad) using SYBR Green qPCR Mix (Takara), according to the manufacturer's instructions.Relative expression levels were normalized to GAPDH and were calculated using the 2−ΔΔC(t) method. RT-PCR was performed with the following primers (Table 1). Amplification was performed under the following conditions: 5 min at 94 °C, 35 cycles of 30 s at 94 °C, 30 s at 65 °C, 30 s at 72 °C, 10 min at 72 °C and storage at 4 °C.
| Gene specificity | Primer |
|---|---|
| GSTM1 | Forward, 5′-CCTATGATACTGGGATACTGGAACG-3′ reverse, 5′-GGAGCGTCACCCATGGTG-3′ |
| GSTM7 | Forward, 5′-TCCGTGTGGATATTCTGGAG-3′ reverse, 5′-CCTCATCATTCCAGGGAGTT-3′ |
| GSTT1 | Forward, 5′-GTTCTGGAGCTGTACCTGGATC-3′ reverse, 5′-AGGAACCTTATACTTGTGTGCC-3′ |
| GSTP1 | Forward, 5′-TTGCTCAAGCCCACTTGTCTGT-3′ reverse, 5′-CAGGGCCTTCACGTAGTCATTC-3′ |
| GAPDH | Forward, 5′-AATCTCCACTTTGCCACTG-3′ reverse, 5′-CCTCGTCCCGTAGACAAAA-3′ |
2.11 Statistical analysis
All data are presented in this manuscript as mean ± SEM. Statistical comparison was performed using SPSS (Statistical Package for the Social Sciences), version 19 (IBM corporation, Armonk, NY, USA). The effect of SiO2 NP dose (factor: dose; levels: 4) was analyzed using one-way ANOVA. Tukey's or Tamhane's T2 post hoc tests were used for pairwise comparisons. Statistical significance was established with a threshold p-value <0.05.3. Results and discussion
3.1 Synthesis and characterization of SiO2 NPs
We successfully synthesized SiO2 NPs, which had an average diameter of around 90 nm under SEM (e.g. Fig. 1A and B). The hydrodynamic diameter was larger (∼150 nm), due to superior dispersibility in water (Fig. 1C). Dispersion of SiO2 NPs was also achieved in penicillin–streptomycin solution (e.g. Fig. 1D). We successfully achieved conjugation of FITC onto the surface of SiO2 NPs, as evidenced by bright green fluorescence of the final FITC-SiO2 NPs under UV illumination (e.g. Fig. 1E). | ||
| Fig. 1 Characterization of FITC-SiO2 NPs. Representative images of FITC-SiO2 NPs under (A) SEM and (B) TEM. (C) Diameter distribution of FITC-SiO2 NPs, assessed by dynamic light scattering (DLS). Blue line shows the cumulative distribution. (D) Representative example of dispersion in penicillin–streptomycin solution, (E) fluorescent photograph of 90 nm FITC-SiO2 NPs under 365 nm UV light. | ||
3.2 Phagocytosis of FITC-SiO2 NPs by C17.2 neural stem cells
After being exposed to SiO2 NPs for 48 hours, C17.2 NSCs engulfed SiO2 NPs in a dose-dependent manner. Compared with the vehicle control group (e.g. Fig. 2A), SiO2 NP-treated groups showed obvious phagocytosis of FITC-SiO2 NPs. Orthogonal projections of confocal microscopy indicated that the FITC-SiO2 NPs were localized on the nuclear surface, but not inside the cell nucleus (e.g. Fig. 2B–D). | ||
| Fig. 2 C17.2 neural stem cell line engulfs FITC-SiO2 NPs in a dose-dependent manner. (A–D) Representative merged images from fluorescent confocal microscopy of C17.2 cells, following treatment with FITC-SiO2 NPs at doses of 0 (control), 100, 200 and 400 μg mL−1, respectively. Green channel, FITC; blue channel, DAPI nuclear counterstain; scale bar = 20 μm (E) representative flow cytometry analysis of phagocytosis by NSCs of FITC-SiO2 NPs at 0, 100, 200 and 400 μg mL−1. (F) Quantitative group data for relative fluorescence intensity indicating the degree of engulfment of FITC-SiO2 NPs by C17.2 NSCs. Data are expressed as a percentage, normalized such that the value for the 100 μg mL−1 group was equal to 100% (n = 3). Bars show mean ± SEM. *p < 0.05, **p < 0.01. | ||
Flow cytometry was used to quantify the level of phagocytosis by the NSCs after exposure to different concentrations of SiO2 NPs (Fig. 2E). The relative fluorescent intensities were 3.9 ± 0.3%, 100.0 ± 7.2%, 505.3 ± 24.1% and 731.0 ± 52.7% following treatment with 0 (control), 100, 200 and 400 μg mL−1 doses of FITC-SiO2 NPs, respectively. Phagocytized FITC fluorescence intensities from flow cytometry clearly and significantly increased (compared to control) following treatment with 100, 200 and 400 μg mL−1 FITC-SiO2 NPs (Fig. 2F; 100 μg mL−1 vs. control, p < 0.05; 200 μg mL−1 vs. control, p < 0.01; 400 μg mL−1 vs. control, p < 0.05).
Engulfment of SiO2 NPs has been previously demonstrated in both mouse and human NSCs. Further to these reports, our results confirm phagocytosis of SiO2 NPs by NSCs using confocal scanning microscopy, and flow cytometry. This is essential for the future development of nanoparticles for biomedical applications, such as in therapy-delivery or cell-tracing.
3.3 Effect of FITC-SiO2 NPs on apoptosis and proliferation of C17.2 neural stem cells
NSCs contribute to brain repair by cell replacement, trophic support and immunomodulation.24,39,40 The homeostasis of stem cell pools in the brain mainly depends on the balance between proliferation and cell death of NSCs.41 Though SiO2 NPs are useful as biomaterial carriers and nervous system tracing agents, their adverse effects on NSCs have not been sufficiently evaluated. Therefore, we next studied the influence of SiO2 NPs on apoptosis and proliferation of NSCs.FITC fluorescence was observed clearly in cells treated with 200 and 400 μg mL−1 FITC-SiO2 NPs. However, we found that cell numbers reduced in these two groups, compared with the control group (Fig. 3C–D vs. Fig. 3A). Flow cytometry (FACS) analysis was performed for quantitative evaluation of the number of NSCs present in each of the SiO2 NP treatment groups. The proportion of live cells significantly decreased following treatment with 200 μg mL−1 (20.8% reduction compared to control; p < 0.001) and 400 μg mL−1 (38.6% reduction; p < 0.001) FITC-SiO2 NPs (Fig. 3E–H and M), indicating increased cell death in these doses. No significant difference was observed in proportion of live cells between the 100 μg mL−1 group and control group (p > 0.05).
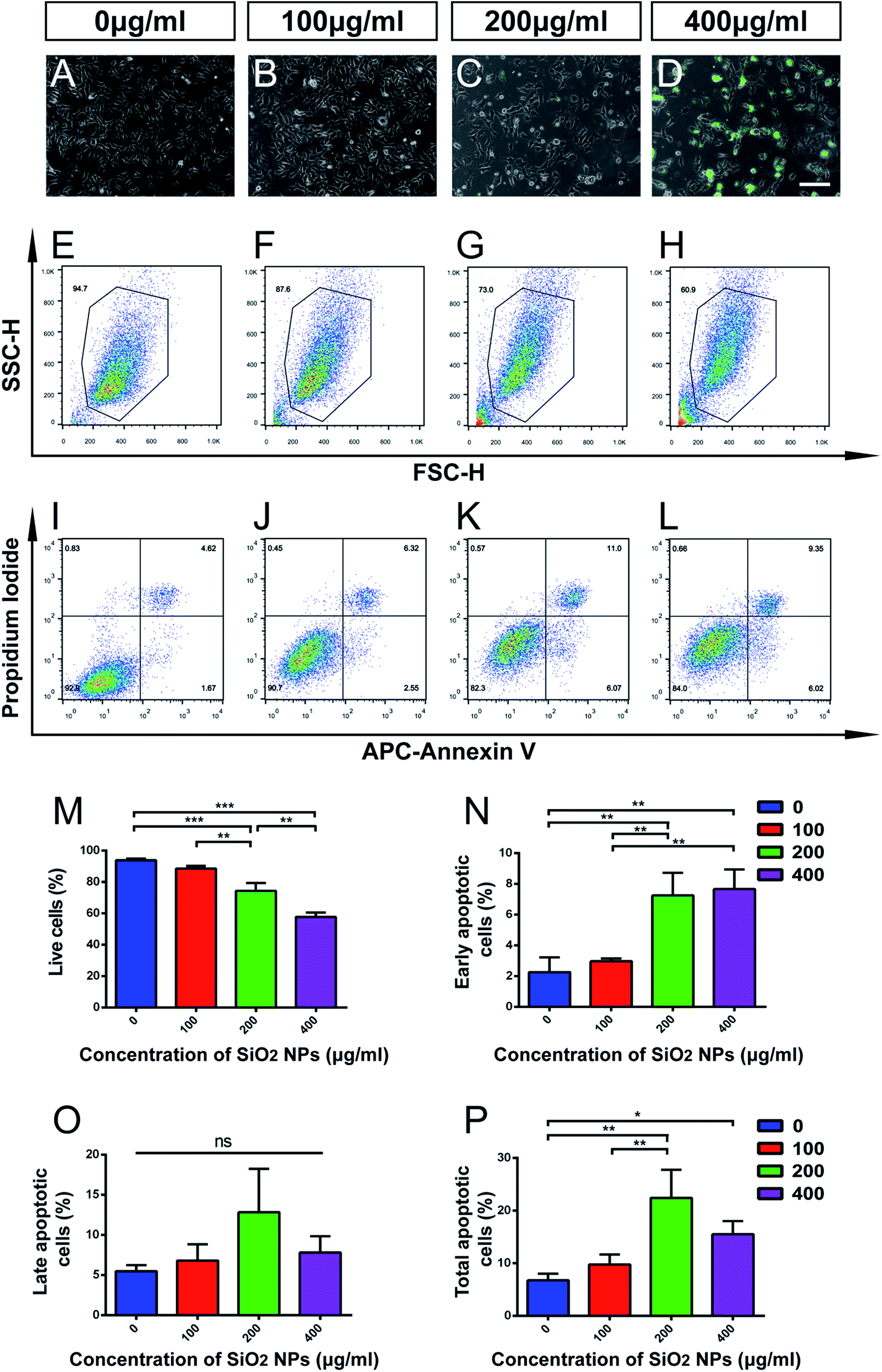 | ||
| Fig. 3 High-dosage SiO2 NPs induces cell death and apoptosis in C17.2 neural stem cells. (A–D) Representative phase contrast images of C17.2 NSCs, showing morphological changes following treatment with FITC-SiO2 NPs at doses of 0 (control), 100, 200 and 400 μg mL−1, for 48 hours. Scale bar = 200 μm. (E–H) Representative FACS output of live cell proportions following SiO2 NP treatment. (I–L) Quantification of apoptosis by dual-staining with Annexin V and propidium iodide. (M) Quantitative group data of proportion (%) of live cells in C17.2 cell sample following SiO2 NP treatment (n = 3). (N) Proportion (%) of cells in C17.2 cell sample showing early apoptosis (Annexin V+/propidium iodide−), following SiO2 NP treatment (n = 3). (O) Proportion (%) of cells in C17.2 cell sample showing late apoptosis (Annexin V+/propidium iodide+), following SiO2 NP treatment (n = 3). (P) Proportion (%) of cells in C17.2 cell sample showing early or late apoptosis, following SiO2 NP treatment (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Apoptosis is a form of programmed cell death (PCD) that occurs during nervous system development, and also following cell damage under pathological conditions.42,43 To specifically explore the effect of FITC-SiO2 NPs on NSC apoptosis, we used propidium iodide/APC-Annexin V with FACS to detect apoptotic cells.
We found that proportions of early apoptotic cells increased after exposure to 200 and 400 μg mL−1 FITC-SiO2 NPs, relative to the control group (Fig. 3I–L, N and O). In contrast, we saw no significant difference in proportions of late apoptotic cells between any of the groups. However, SiO2 NP treatment did markedly increase the proportion of total apoptotic cells at doses of 200 (3.3-fold increase vs. control; p < 0.01) and 400 μg mL−1 (2.3-fold increase vs. control; p < 0.05) (Fig. 3P; control, 6.7 ± 1.3; 200 μg mL−1, 22.4 ± 5.3; 400 μg mL−1, 15.5 ± 2.5). However, there was no significant effect of the 100 μg mL−1 SiO2 NP dose (p > 0.05 vs. control) on the number of early, late or total apoptotic cells.
Self-renewal is one of the classical properties of stem cells. It is the ability to go through numerous cycles of cell division without differentiation. Ki67 is a protein that is expressed during all active phases of cell division, and is associated with cell self-renewal.44
Using immunocytochemical staining, we found that FITC-SiO2 NPs reduced the proportion of Ki67-positive C17.2 NSCs. FITC-SiO2 NP treatment for 48 hours at 200 μg mL−1 reduced the proportion of Ki67-positive cells by 27.0%, relative to control (p < 0.05) and 400 μg mL−1 reduced the proportion by 33.0% (p < 0.01 vs. control) (Fig. 4I). No significant difference was observed between the 100 μg mL−1 group and control group.
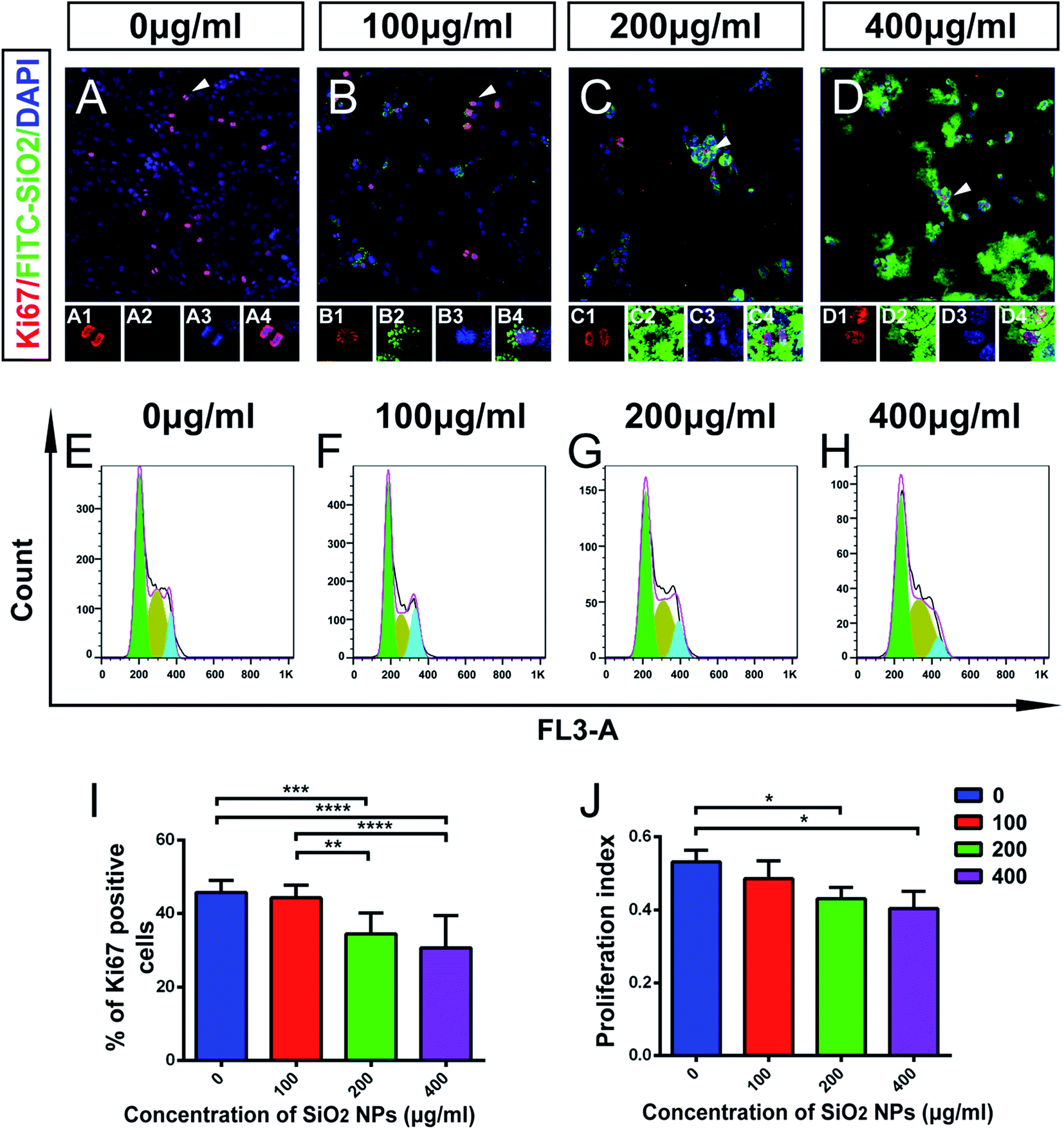 | ||
| Fig. 4 Effect of SiO2 NPs on proliferation of C17.2 neural stem cells. (A–D) Representative merged images from fluorescent confocal microscopy of C17.2 cells, following treatment with FITC-SiO2 NPs at doses of 0 (control), 100, 200 and 400 μg mL−1, respectively. Scale bar = 100 μm; red, Ki67; green, FITC; blue, DAPI. Insets show high-power views of the arrowheads. (E–H) Cell cycle analysis of C17.2 NSCs line, treated as per (A–D). (I) Quantitative group data of Ki67 expression (% positive cells) by NSCs after SiO2 NPs treatment, as per (A–D) (n = 3). (J) Quantitative group data of proliferation index of NSCs after SiO2 NP treatment, as per (A–D) (n = 3). *p < 0.05, **p < 0.01. | ||
To further explore this effect, we next detected the phases of the cell cycle in NSCs by FACS (Fig. 4E–H). A proliferation index (PI) was calculated as the proportion of the total cell cycle spent in either S-phase or G2/M-phase. The PI was 0.53 ± 0.03 for the control group and 0.49 ± 0.06 for 100 μg mL−1 group (p > 0.05). The PI decreased significantly in 200 μg mL−1 treated group (22.9% lower than control; p < 0.05) and 400 μg mL−1 treated group (24.1% lower than control; p < 0.05) (Fig. 4J). These results support the hypothesis that FITC-SiO2 NPs interfere with NSC proliferation.
Taken together, these data suggest that biomedical usage of SiO2 NPs, at doses over 200 μg mL−1, might damage NSCs, by inducing apoptosis and inhibiting their proliferation. However, no significant effect was detected following a dose of 100 μg mL−1 of SiO2 NPs.
3.4 Influence of FITC-SiO2 NP exposure on the subcellular characteristics of C17.2 neural stem cells
The main mechanisms of nanotoxicity are induction of reactive oxygen species (ROS) and mitochondrial damage.45 It has been shown, in a PC12 neuronal cell line, that nano-size silicon dioxide exposure increases ROS levels, and correspondingly decreases the level of glutathione, suggesting that SiO2 NPs cause a ROS-related neurotoxicity.46Our TEM studies of NSCs treated with FITC-SiO2 NPs revealed SiO2 NPs aggregated in intracellular vesicles, as well as deformed nuclei and condensed chromatin (Fig. 5B–D, F and H) that were not seen in control experiments (Fig. 5A). We also observed SiO2 NP phagocytosis at all doses of FITC-SiO2 NPs.
 | ||
| Fig. 5 TEM images of cell membranes and mitochondria of C17.2 neural stem cells, following incubation with SiO2 NPs at various doses. (A–D) Representative TEM images, showing the internalization of SiO2 NPs at all three active doses. Arrowhead, internalized SiO2 NPs. Scale bar = 2 μm. (E–H) High-power views of boxed areas in (A–D), to show mitochondria. Scale bar = 0.5 μm. | ||
Mitochondria of C17.2 NSCs in the control group exhibited well-defined double membranes and well-organized cristae (Fig. 5E). The same was also seen in the NSCs treated with 100 μg mL−1 FITC-SiO2 NP (Fig. 5F). However, the mitochondria of NSCs treated with 200 μg mL−1 SiO2 NPs were swollen, and mitochondrial cristae were ruptured or absent (Fig. 5G). After exposure to 400 μg mL−1 SiO2 NPs for 48 hours, the cell membrane of NSCs was completely disrupted, forming vacuolated cytoplasm and organelles (Fig. 5H). TEM confirmed that SiO2 NPs entered the cytoplasm but not the nuclei of the NSCs.
Mitochondrial damage was thus observed in NSCs with SiO2 NP at dosage levels of 200 μg mL−1 and above, which is consistent with the toxicity suggested by the other experiments presented here. Mitochondria are the major organelles that clear ROS from the cell, and it is likely that mitochondrial damage caused by SiO2 NPs leads to the accumulation of ROS, leading to broader cytotoxicity.
3.5 Changes in gene expression profile induced by FITC-SiO2 NPs
mRNA sequencing was used to analyze the impact of SiO2 NPs on the whole gene expression profile of C17.2 NSCs. A total of more than 6.8 billion clean reads were generated from all six cDNA libraries, using the Illumina HiSeq 4000 platform. Gene expression in NSCs exposed to exposing to 200 μg mL−1 was compared to vehicle controls. For convenience, we hereafter refer to this SiO2 NP treatment group as “SiO2 NP-200”, and the vehicle control (0 μg mL−1) group simply as “control”. Fold-changes greater than 1.5 and diverge probability values of more than 0.8 were set as the threshold for defining differentially expressed genes (DEGs).47,48We found 42 up-regulated and 77 down-regulated DEGs in NSCs in the SiO2 NP-200 group (Fig. 6A and B). DEG expression levels in the control group and SiO2 NP-200 group were plotted in a heat-map (Fig. 6C), and KEGG analysis was used to further examine the underlying mechanistic effects of SiO2 NPs on NSCs. Ten particularly important pathways in NSCs were tested, including metabolism of xenobiotics by cytochrome P450, leishmaniasis, pertussis, drug metabolism by cytochrome P450, platinum drug resistance, chemical carcinogenesis, pathways in cancer, prostate cancer, glutathione metabolism and HTLV-I infection (Fig. 6D).
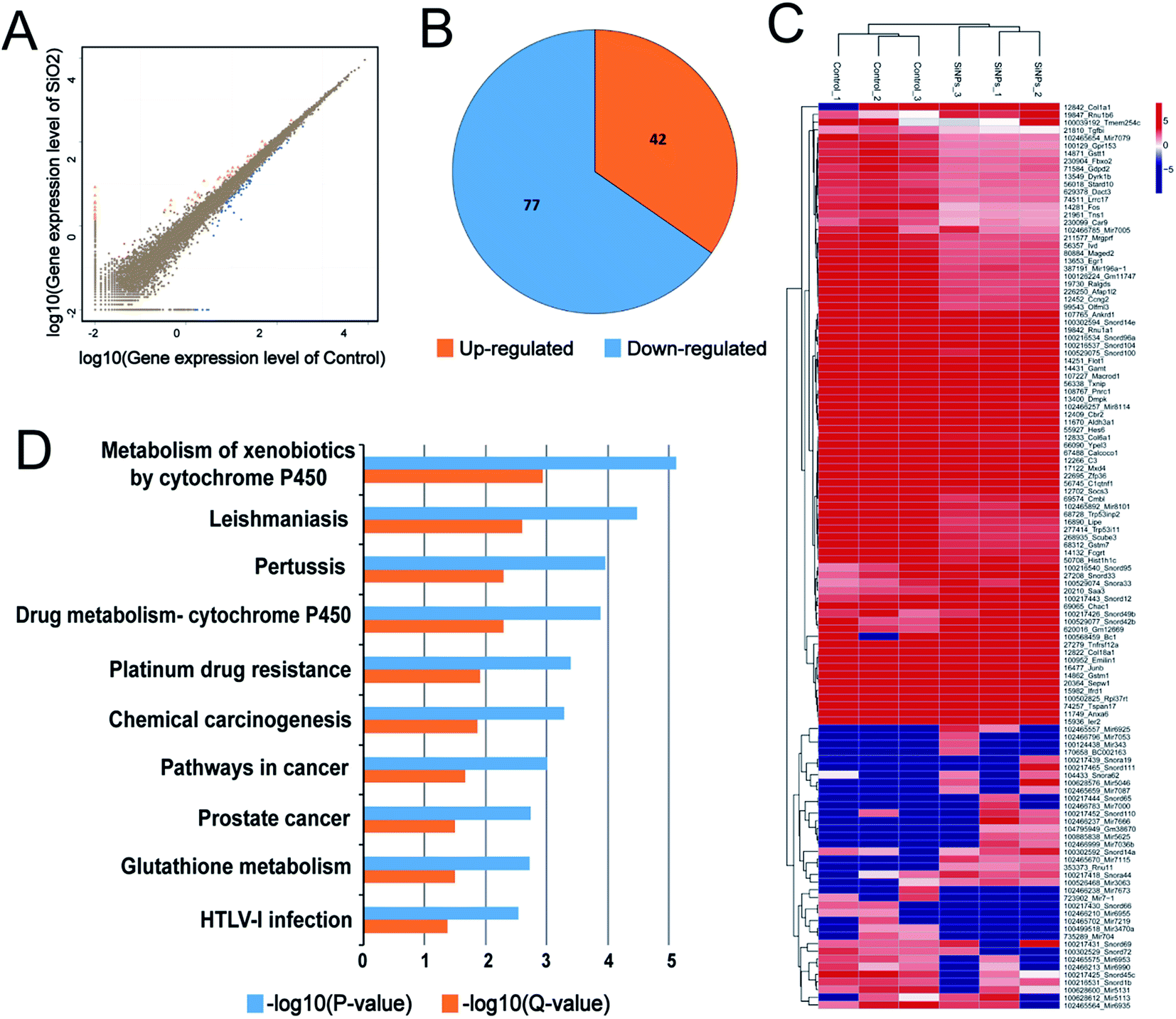 | ||
| Fig. 6 Distribution of DEGs in and KEGG pathway analysis of C17.2 neural stem cells treated with SiO2 NPs. (A) Scatter-plots showing all expressed genes: control group expression level vs. SiO2 NP-200 group expression level. Orange, gene up-regulated by SiO2 NP; blue, gene down-regulated; brown, gene not regulated. (B) Pie chart, showing the distribution of dysregulated mRNAs. (C) Hierarchical clustering of differently expressed mRNAs in control and SiO2 NP-200 NSCs. Each row represents a single mRNA and each column represents one tissue sample. Red, high relative expression; blue, low relative expression. (D) KEGG pathway analysis for differently expressed mRNAs. | ||
A pathway-act-network was constructed according to the interactions assigned between pathways in the KEGG database, to further screen core pathways of NSCs exposed to SiO2 NPs (Fig. 7). We identified three main pathways as being particularly important in FITC-SiO2 NP-induced gene expression in NSCs: the apoptosis pathway, the MAPK signaling pathway, and the PI3K-Akt signaling pathway. MAPK signaling is related to stem cell maintenance in NSCs,49 and PI3K-Akt signaling regulates proliferation of NSCs.50,51 The pathway-act-network indicated that apoptosis and proliferation were both involved following SiO2 NP treatment, which supports the findings presented in previous sections of this report.
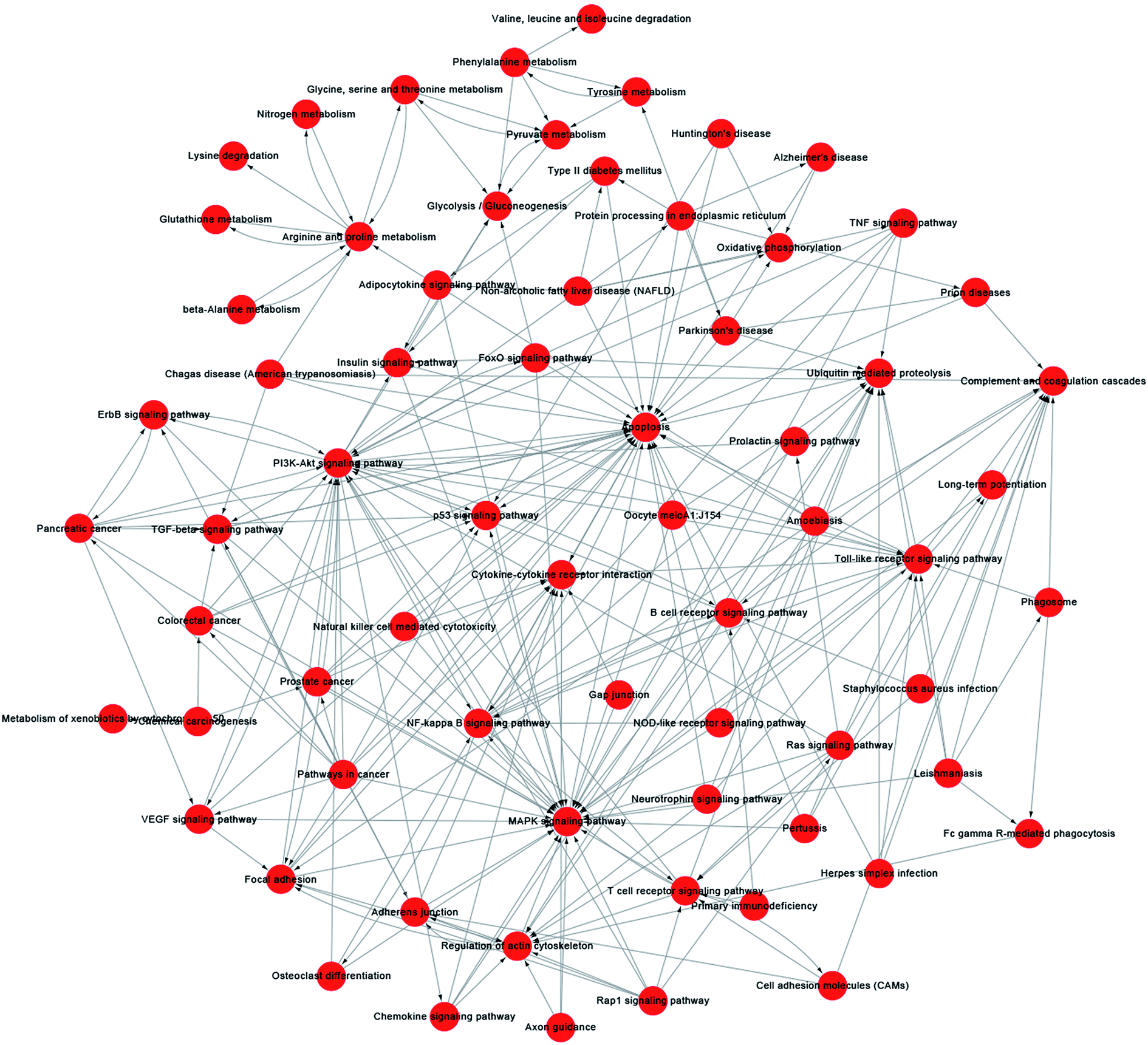 | ||
| Fig. 7 Pathway-act-network analysis of C17.2 neural stem cells treated with 200 μg mL−1 SiO2 NPs. A pathway-act-network was constructed according to the interactions within pathways identified in the KEGG database. Each node (red circle) represents a signaling pathway. Arrows represents interactive relationships between two signaling pathways. | ||
We also constructed co-expression networks for the control group and SiO2 NP-200 group, based on the DEG data. Here, the co-expression network in the control group contained 104 network nodes and 348 connections. Of these, 272 were positive connections and 76 were negative connections (Fig. 8A). The network in the SiO2 NP-200 group comprised 110 network nodes and 337 connections. 196 were positive and 141 negative (Fig. 8B). In this case, these were three genes from the glutathione-S-transferase (GST) gene family, namely GSTM1, GSTM7 and GSTT1 (Fig. 8C).
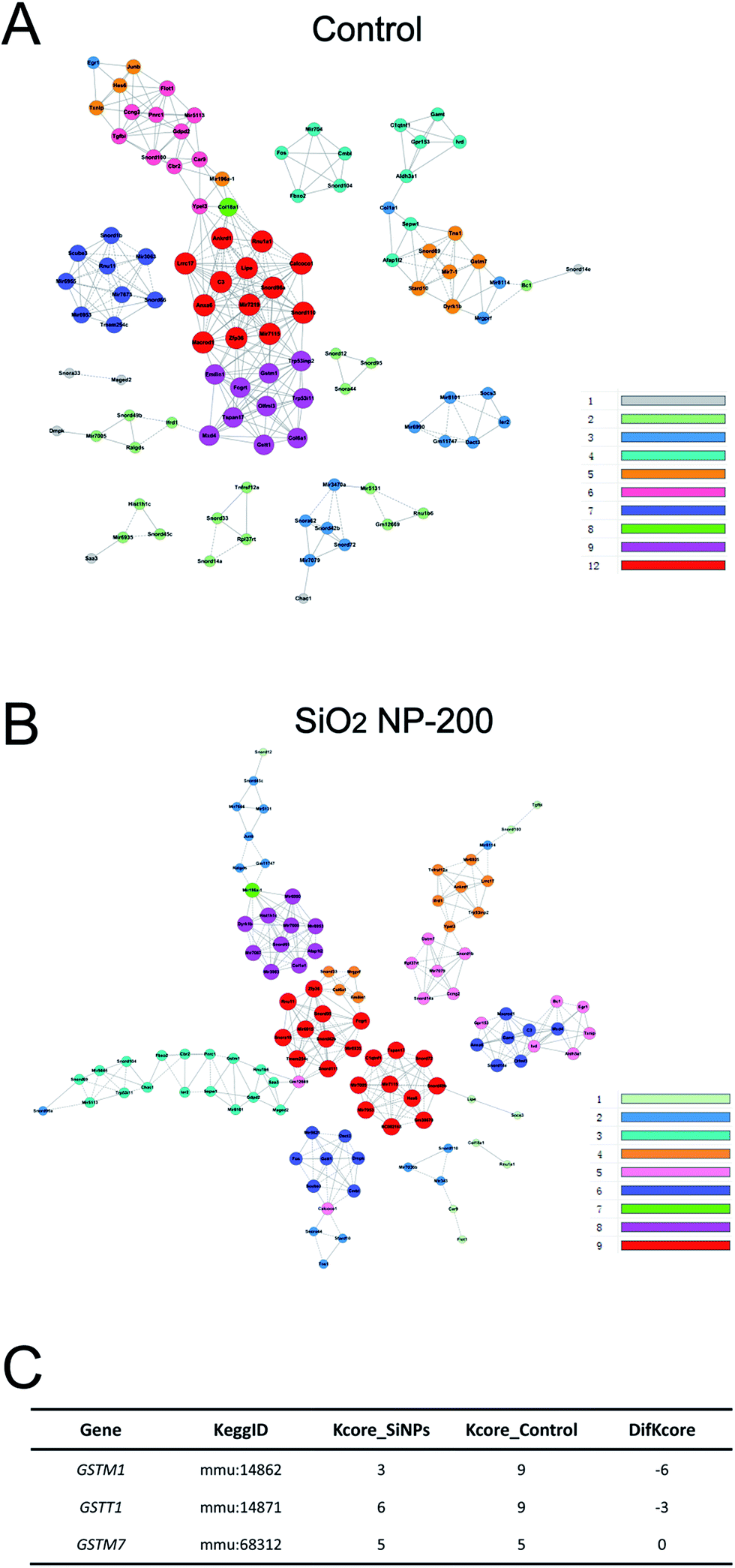 | ||
| Fig. 8 Co-expression networks of DEGs in C17.2 NSCs treated with SiO2 NPs. (A) Co-expression network of DEGs expressed in NSCs in the control group. Colored nodes indicate mRNA. Solid/dashed lines represent positive/negative regulatory relationships, respectively. (B) Same as (A), but for the SiO2 NP-200 group. (C) Potential core regulatory factors for SiO2 NPs exposing in NSCs as determined by the k-Core difference (difk-Core) of control and SiO2 NPs networks. | ||
To validate the results of our mRNA-sequencing analysis, we also performed RT-qPCR of these core genes. We found that GSTM1, GSTM7 and GSTT1, but not GSTP1 (used as a negative control), were down-regulated in the SiO2 NP-200 group, compared to the control group (Fig. 9A–D). We therefore confirmed specific GSTs as core genes, underlying the mechanism by which SiO2 NPs cause cytotoxicity in NSCs.
 | ||
| Fig. 9 Quantitative RT-PCR validation of core genes from RNA-Seq analysis. Real-time PCR analysis of the expression of (A) GSTM1, (B) GSTM7, (C) GSTT1 and (D) GSTP1 (n = 3 per bar). ***p < 0.001, ****p < 0.0001. | ||
GSTs are a family of phase II detoxification enzymes, which catalyze the conjugation of a reduced form of glutathione to xenobiotic substrates.52 Previously, Shi and colleagues have found that SiO2 nanoparticles induce cytotoxicity in the MCF-7 cell line (a human breast carcinoma line), and overexpression of microsomal glutathione transferase 1 reverses this toxicity.53 Our results demonstrate similar findings in NSCs, and go beyond previous work, by confirming specific GST gene expression changes, using RT-qPCR.
Taken together, these results suggest that drugs targeting the GST genes should be investigated for their potential to mitigate the adverse toxicity associated with SiO2 NPs. This may facilitate development of SiO2 NP therapies that can be safely given to human patients.
4. Conclusions
We have shown that SiO2 NPs exert cytotoxicity on NSCs at dosage levels of 200 μg mL−1 and above. In this series of experiments, SiO2 NPs reduced the proportion of live cells, increased the apoptosis level and inhibited proliferation of C17.2 NSCs. Our results suggest that the toxicity of SiO2 NPs is induced via mitochondrial damage. RNA-Seq analysis indicated that metabolism of xenobiotics by cytochrome P450, drug metabolism by cytochrome P450 and glutathione metabolism were involved in the toxicity of SiO2 NPs on the C17.2 cell line. Specific subtypes of the glutathione-S-transferase gene, namely GSTM1, GSTM7 and GSTT1, were inhibited in the C17.2 cell line after SiO2 NP treatment. Our findings demonstrate the cytotoxicity of SiO2 NPs on NSCs, help elucidate the underlying mechanism or toxicity, and suggest the GST pathways as possible therapeutic targets for future study.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 31271051, 31571015 and 81570890), the National Basic Research Programs of China (No. 2016YFA0201600) and the Foundation of Southwest Hospital (No. SWH2016LHYS-03).References
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS PubMed.
- W. Shen, X. Zhang, Q. Huang, Q. Xu and W. Song, Nanoscale, 2014, 6, 1622–1628 RSC.
- N. Feliu, P. Kohonen, J. Ji, Y. Zhang, H. L. Karlsson, L. Palmberg, A. Nystrom and B. Fadeel, ACS Nano, 2015, 9, 146–163 CrossRef CAS PubMed.
- A. Weir, P. Westerhoff, L. Fabricius, K. Hristovski and N. von Goetz, Environ. Sci. Technol., 2012, 46, 2242–2250 CrossRef CAS PubMed.
- J. W. Wiechers and N. Musee, J. Biomed. Nanotechnol., 2010, 6, 408–431 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- M. A. Malvindi, V. Brunetti, G. Vecchio, A. Galeone, R. Cingolani and P. P. Pompa, Nanoscale, 2012, 4, 486–495 RSC.
- S. D. Kong, J. Lee, S. Ramachandran, B. P. Eliceiri, V. I. Shubayev, R. Lal and S. Jin, J. Controlled Release, 2012, 164, 49–57 CrossRef CAS PubMed.
- C. Saraiva, C. Praca, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS PubMed.
- S. Ku, F. Yan, Y. Wang, Y. Sun, N. Yang and L. Ye, Biochem. Biophys. Res. Commun., 2010, 394, 871–876 CrossRef CAS PubMed.
- D. J. Bharali, I. Klejbor, E. K. Stachowiak, P. Dutta, I. Roy, N. Kaur, E. J. Bergey, P. N. Prasad and M. K. Stachowiak, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11539–11544 CrossRef CAS PubMed.
- S. S. Bale, S. J. Kwon, D. A. Shah, A. Banerjee, J. S. Dordick and R. S. Kane, ACS Nano, 2010, 4, 1493–1500 CrossRef CAS PubMed.
- D. A. Shah, S. J. Kwon, S. S. Bale, A. Banerjee, J. S. Dordick and R. S. Kane, Biomaterials, 2011, 32, 3210–3219 CrossRef CAS PubMed.
- M. Mahmoudi, H. Hosseinkhani, M. Hosseinkhani, S. Boutry, A. Simchi, W. S. Journeay, K. Subramani and S. Laurent, Chem. Rev., 2011, 111, 253–280 CrossRef CAS PubMed.
- Y. Shen, Y. Shao, H. He, Y. Tan, X. Tian, F. Xie and L. Li, Int. J. Nanomed., 2013, 8, 119–127 Search PubMed.
- Y. K. Peng, C. N. Lui, T. H. Lin, C. Chang, P. T. Chou, K. K. Yung and S. C. Tsang, Faraday Discuss., 2014, 175, 13–26 RSC.
- A. Panas, C. Marquardt, O. Nalcaci, H. Bockhorn, W. Baumann, H. R. Paur, S. Mulhopt, S. Diabate and C. Weiss, Nanotoxicology, 2013, 7, 259–273 CrossRef CAS PubMed.
- W. Lin, Y. W. Huang, X. D. Zhou and Y. Ma, Toxicol. Appl. Pharmacol., 2006, 217, 252–259 CrossRef CAS PubMed.
- F. H. Gage and S. Temple, Neuron, 2013, 80, 588–601 CrossRef CAS PubMed.
- E. Llorens-Bobadilla, S. Zhao, A. Baser, G. Saiz-Castro, K. Zwadlo and A. Martin-Villalba, Cell Stem Cell, 2015, 17, 329–340 CrossRef CAS PubMed.
- Y. Mu and F. H. Gage, Mol. Neurodegener., 2011, 6, 85 CrossRef PubMed.
- Y. Wang, J. Wang, M. Wu, X. Deng, T. Wen, C. Chen, H. Zhang, Y. Liu and Z. Jiao, J. Nanosci. Nanotechnol., 2010, 10, 7121–7125 CrossRef CAS PubMed.
- K. Fujioka, S. Hanada, Y. Inoue, K. Sato, K. Hirakuri, K. Shiraishi, F. Kanaya, K. Ikeda, R. Usui, K. Yamamoto, S. U. Kim and Y. Manome, Int. J. Mol. Sci., 2014, 15, 11742–11759 CrossRef PubMed.
- E. Izak-Nau, K. Kenesei, K. Murali, M. Voetz, S. Eiden, V. F. Puntes, A. Duschl and E. Madarasz, Nanotoxicology, 2014, 8(suppl. 1), 138–148 CrossRef CAS PubMed.
- Z. Wang, M. Gerstein and M. Snyder, Nat. Rev. Genet., 2009, 10, 57–63 CrossRef CAS PubMed.
- A. Mortazavi, B. A. Williams, K. McCue, L. Schaeffer and B. Wold, Nat. Methods, 2008, 5, 621–628 CrossRef CAS PubMed.
- D. F. Simon, R. F. Domingos, C. Hauser, C. M. Hutchins, W. Zerges and K. J. Wilkinson, Appl. Environ. Microbiol., 2013, 79, 4774–4785 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- L. Zhou, W. Yin, W. Ren, Z. Gu, W. Li, S. Jin, L. Yan, G. Tian, Z. Hu and Y. Zhao, New J. Chem., 2012, 36, 2599–2606 RSC.
- S. J. Wang, C. H. Weng, H. W. Xu, C. J. Zhao and Z. Q. Yin, J. Membr. Biol., 2014, 247, 493–500 CrossRef CAS PubMed.
- J. Huang, S. Jing, X. Chen, X. Bao, Z. Du, H. Li, T. Yang and X. Fan, Mol. Neurobiol., 2016, 53, 1031–1044 CrossRef CAS PubMed.
- W. Wu, L. Yan, Q. Wu, Y. Li, Q. Li, S. Chen, Y. Yang, Z. Gu, H. Xu and Z. Q. Yin, Nanotoxicology, 2016, 10, 1329–1340 CrossRef CAS PubMed.
- S. Tarazona, F. Garcia-Alcalde, J. Dopazo, A. Ferrer and A. Conesa, Genome Res., 2011, 21, 2213–2223 CrossRef CAS PubMed.
- M. Kanehisa, M. Araki, S. Goto, M. Hattori, M. Hirakawa, M. Itoh, T. Katayama, S. Kawashima, S. Okuda, T. Tokimatsu and Y. Yamanishi, Nucleic Acids Res., 2008, 36, D480–D484 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- S. Hu, G. Yao, Y. Wang, H. Xu, X. Ji, Y. He, Q. Zhu, Z. Chen and Y. Sun, J. Clin. Endocrinol. Metab., 2014, 99, E2744–E2753 CrossRef CAS PubMed.
- Z. Li, Y. Zeng, X. Chen, Q. Li, W. Wu, L. Xue, H. Xu and Z. Q. Yin, Cytotherapy, 2016, 18, 771–784 CrossRef CAS PubMed.
- G. Gincberg, H. Arien-Zakay, P. Lazarovici and P. I. Lelkes, Br. Med. Bull., 2012, 104, 7–19 CrossRef CAS PubMed.
- V. Bonnamain, I. Neveu and P. Naveilhan, Front. Cell. Neurosci., 2012, 6, 17 Search PubMed.
- C. C. Homem, M. Repic and J. A. Knoblich, Nat. Rev. Neurosci., 2015, 16, 647–659 CrossRef CAS PubMed.
- M. P. Mattson, Nat. Rev. Mol. Cell Biol., 2000, 1, 120–129 CrossRef CAS PubMed.
- J. Yuan and B. A. Yankner, Nature, 2000, 407, 802–809 CrossRef CAS PubMed.
- T. Scholzen and J. Gerdes, J. Cell. Physiol., 2000, 182, 311–322 CrossRef CAS PubMed.
- F. Joris, B. B. Manshian, K. Peynshaert, S. C. De Smedt, K. Braeckmans and S. J. Soenen, Chem. Soc. Rev., 2013, 42, 8339–8359 RSC.
- F. Wang, C. Jiao, J. Liu, H. Yuan, M. Lan and F. Gao, Toxicol. In Vitro, 2011, 25, 1548–1556 CrossRef CAS PubMed.
- C. Wu, X. Li, S. Guo and S. M. Wong, Sci. Rep., 2016, 6, 36007 CrossRef CAS PubMed.
- M. Pescatori, D. Bedognetti, E. Venturelli, C. Menard-Moyon, C. Bernardini, E. Muresu, A. Piana, G. Maida, R. Manetti, F. Sgarrella, A. Bianco and L. G. Delogu, Biomaterials, 2013, 34, 4395–4403 CrossRef CAS PubMed.
- L. S. Campos, D. P. Leone, J. B. Relvas, C. Brakebusch, R. Fassler, U. Suter and C. ffrench-Constant, Development, 2004, 131, 3433–3444 CrossRef CAS PubMed.
- J. Peltier, A. O'Neill and D. V. Schaffer, Dev. Neurobiol., 2007, 67, 1348–1361 CrossRef CAS PubMed.
- J. E. Le Belle, N. M. Orozco, A. A. Paucar, J. P. Saxe, J. Mottahedeh, A. D. Pyle, H. Wu and H. I. Kornblum, Cell Stem Cell, 2011, 8, 59–71 CrossRef CAS PubMed.
- D. Sheehan, G. Meade, V. M. Foley and C. A. Dowd, Biochem. J., 2001, 360, 1–16 CrossRef CAS PubMed.
- J. Shi, H. L. Karlsson, K. Johansson, V. Gogvadze, L. Xiao, J. Li, T. Burks, A. Garcia-Bennett, A. Uheida, M. Muhammed, S. Mathur, R. Morgenstern, V. E. Kagan and B. Fadeel, ACS Nano, 2012, 6, 1925–1938 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |