
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile green synthesis of nanomagnets for modulating magnetohyperthermia: tailoring size, shape and phase
M. P. Silvaa,
A. L. Drummondb,
V. R. R. Aquinoc,
L. P. Silvad,
R. B. Azevedoe,
M. J. A. Salesb,
P. C. Moraisfg,
A. F. Bakuzisc and
M. H. Sousa *a
*a
aGreen Nanotechnology Group, University of Brasília, Brasília, DF 72220-900, Brazil. E-mail: mhsousa@unb.br
bInstitute of Chemistry, University of Brasília, Brasília, DF 70910-900, Brazil
cInstitute of Physics, Federal University of Goiás, Goiânia, GO 74690-900, Brazil
dLaboratory of Nanobiotechnology, Embrapa Genetic Resources and Biotechnology, Brasília, DF 70770-917, Brazil
eInstitute of Biological Sciences, University of Brasília, Brasília, DF 70919-900, Brazil
fInstitute of Physics, University of Brasília, Brasília, DF 70910-900, Brazil
gSchool of Chemistry and Chemical Engineering, Anhui University, Hefei 230601, China
First published on 11th October 2017
Abstract
This study reports the successful production of both isometric and anisometric iron oxide-based nanoparticles using, respectively, ammonia and urea for co-precipitating Fe2+/Fe3+ from aqueous solution. Spherical nanoparticles (SNPs) with 10–20 nm in diameter are obtained using ammonia under reflux from 1 h to 9 h, with their relative magnetite/maghemite content decreasing from 10 to 0.05. However, using a lower and higher concentration of urea under reflux from 1 h to 12 h results in rod-like nanoparticles (RNPs) with length/width varying from 40/16 to 80/20 nm and hexagonal nanoparticles (HNPs) with diagonal varying from 150 to 100 nm, respectively. For RNPs (HNPs) the relative magnetite/goethite content increases with refluxing time from 0.25 to 2 (1.25 to 3.75). Hysteresis cycles (300 K) show unblocked SNPs and blocked RNPs and HNPs with coercivity (remanence) increasing with refluxing time from 55 to 80 Oe (1 to 5 emu g−1) and 70 to 130 Oe (5 to 13 emu g−1), respectively. Saturation magnetization of SNPs, RNPs and HNPs spans from 50 to 65 emu g−1, 12 to 60 emu g−1 and 57 to 80 emu g−1, respectively. Under AC magnetic field (522 kHz), with amplitude ranging from 70 to 310 Oe, SNPs show a strong hyperthermia effect, following HNPs with mild and RNPs with weak effects.
Introduction
Shape-engineering is a key topic in nanoparticle (NP) synthesis with huge impacts in supercrystal construction, as it holds enormous potential to tailor new properties of nano- and micro-sized objects for practical applications. Actually, size and shape attributes can deeply affect the physicochemical properties (e.g. magnetic, optical, magneto-optical, and catalytic) of nanocrystalline materials through geometry and symmetry, and while exposing selected crystallographic faces to the surrounding medium. Moreover, the reciprocity between shape and property has been regularly shown by a diversity of compositions and shapes for different nanocrystals and supercrystals and may likewise increase the versatility of these structures in various applications1–3Regarding the increasing relevance of the theranostics applications of magnetic nanoparticles (MNPs) in the biomedical field, tailoring size, shape and understanding the role of composition and crystalline phase is fundamental for fine tuning the chemical and physical properties with the aim of developing highly efficient multitask-oriented nanoplatforms.4 Particularly, for such applications, spherical iron oxide nanoparticles, such as ∼20 nm size magnetite (Fe3O4) or maghemite (γ-Fe2O3) had been extensively investigated due to the excellent cross-relation between magnetic response, surface reactivity and biocompatibility, reflecting on easy manipulation, functionalization and interactivity with living tissues and cells.5 Although these isometric MNPs are widely employing, some works had already pointed out that not only particle size but also particle shape might play a dominant role on developing successful biocompatible platforms. For example, Champion and Mitragotri6,7 reported that phagocytosis of nanoparticles by macrophages depends on shape. Yue et al.8 demonstrated that, compared with spherical nanoparticles, nanorods could be internalized both quicker and to a higher extent, thus exhibiting a great potential for use in anticancer therapy. Additionally, Gratton et al.9 showed that anisometric NPs internalization strongly depends on the aspect ratio; keeping constant the volume, nanoparticles with aspect ratio equals three were internalized about four times faster than their spherical counterparts. Herd et al.10 showed that different geometries of silica nanoparticles, namely spheres, cylinders and worms exhibited quite different uptake profiles likely due to their orientation while interacting with the cell surface. Cribb et al.11 reported that under certain physiological conditions, especially in shear thinning fluids such as synovial fluid, mucus barriers and extracellular matrix, nanorods experienced higher velocity than spheres upon the same applied external stimuli. Moreover, the shape of nanoparticles might play a significant role in the immunological response. Lizotte et al.12 showed that virus-like nanoparticles from cowpea mosaic virus (CPMV) stimulated the systemic antitumor immunity to treat metastases. Niikura et al.13 demonstrated that shape and size of gold nanoparticles induced different immunological responses in vivo and in vitro while producing antibodies for West Nile virus and this can contribute to the future design of safe and effective nanoparticle-based vaccines through the activation of the desired immune response. Indeed, different uptake patterns triggered different downstream cellular pathways, which were dependent upon cell type and phenotype.
In addition to the influence on the way MNPs are taken up by cells, size and shape also affect the magnetic properties of an ensemble of NPs.14 For example, Guardia et al.15 reported high hyperthermia efficiency of magnetite nanocubes. The study concluded that cubic nanoparticles of size around 19 nm are the most efficient ones in magnetic hyperthermia. Boubeta et al.16 concluded that nanocubes heated better than spherical nanoparticles of similar size, probably due to the higher magnetic anisotropy. However, self-assembly due to chain formation had also been attributed to play a role in the hyperthermia efficiency. Indeed, in the low field regime,17 Branquinho et al.18 had also demonstrated that chain formation plays a role in magnetic hyperthermia. Chains of anisometric shaped iron oxide NPs are naturally formed in magnetotactic bacteria that had showed high heating efficiency at high field amplitude conditions.19,20
Shape-controlled synthesis of iron oxide-based MNPs such as magnetite is by far more conveniently achieved through thermal decomposition methods.21 However, the drawback of these procedures is their requirement for complex precursors, organic solvents, surfactants and reductants – normally toxic reagents – undesirable while addressing biomedical applications. Moreover, MNPs obtained from thermal decomposition routes are capped with non-polar and/or surfactant molecules, increasing their hydrophobicity and limiting the possibility of functionalization with biocompatible molecules. On the other hand, aqueous co-precipitation protocols, which are easier to be scaled-up while requiring milder and more eco-friendly conditions, offer superior synthesis routes for biocompatible nanomaterials.22 However, in conventional co-precipitation routes, reaction pathways leading to production of the magnetite phase are not fully understood and magnetite nanoparticles are crystallized in a quasi-immediate process at room temperature, normally as polydisperse and nearly spherical structures, making the shape control a very hard task.23 In this way, aiming to establish protocols for better controlling the nanoparticles' morphology, several adaptations to the conventional co-precipitation route had been proposed, such as magnetic field-assisted,24 ultrasound-assisted,25 microwave-assisted,26 hydrothermal,27,28 polyol-mediated,29,30 sol–gel31 and templated growth32 as well as reactions in constrained environments33 and seed-mediated (followed by calcination) processes.34 Among these adaptations, co-precipitation from homogeneous solutions using urea was utilized as an interesting tool for elaboration of shape-controlled magnetite nanoparticles.35 Actually, thermal decomposition of urea, which increases homogenously and continuously [OH−] in solution, can control the iron ions solvolysis/hydrolysis. Furthermore, the adsorption of ligands such as surfactants and molecules on specific surfaces of iron oxide/hydroxide precursors, during urea decomposition, was used to induce oriented growth and thus modulate nanoparticles shape in the final products. In this way, Lian et al.36 synthesized Fe3O4 nanorods, with average diameters of 40–50 nm and lengths of up to 1 mm, through hydrolysis of FeCl3 and FeSO4 solutions containing urea (at ∼95 °C) in reflux condition for 12 h. Yang et al.37 investigated the role of polymeric additives (polyvinyl alcohol and polyacrylic acid) on the shape and size during synthesis of magnetite nanoparticles obtained by heating aqueous solutions of Fe2+/Fe3+ and urea at 85 °C. Zhu et al.28 reported the building effect urea reveals during formation of hexagonal magnetite nanoplates precipitated in autoclave at 200 °C from aqueous solution containing NaOH and FeCl3 in the presence of β-cyclodextrin and urea as reducing agent and modifier, respectively. Li et al.38 demonstrated specific adsorption of urea inducing size and shape evolution of magnetite nanoparticles towards ellipsoidal geometry while co-precipitating a mixture of Fe2+/Fe3+ ions with NaOH in the presence of urea.
In this study we report findings on the production of iron oxide-based MNPs with spherical, rod-like and hexagonal geometries using a mild, efficient and environment-friendly synthesis route which involves the use of ammonium hydroxide or aqueous decomposition of urea in the presence of Fe2+/Fe3+ salts and sulfate anion, the latter well known by its building effect. The as-produced isometric and anisometric MNPs were morphologically, structurally and magnetically characterized using standard techniques. Additionally, aiming their possible application in magnetohyperthermia (MHT), the as-produced MNPs were comparatively characterized by their heating efficiency performance in terms of the amplitude and frequency of the applied AC magnetic field. More importantly, the role of phase and composition to the magneto thermal (hyperthermia) properties of the nanostructures were deeply investigated. This report shows the possibility to modulate size and shape of biocompatible isometric and anisometric MNPs with different magneto-thermal properties, from which we anticipate easy surface functionalization and different cell uptake profiles addressed to biomedical applications, such as for cancer treatment, magnetic contrast agent, and cell sorting.
Experimental
Synthesis
All chemical reagents were purchased with analytical quality and used without any further purification step. Water used to perform the experiments was purified by a Milli-Q water system (Millipore, USA). In a typical synthesis protocol, FeCl3·6H2O (3.0 mmol), FeSO4·7H2O (1.5 mmol) and a specified amount of OH−-source given by urea (CH4N2O) or ammonium hydroxide (NH4OH) were all dissolved in purified water (50 mL) in a flask with reflux condenser and constant magnetic stirring at 95 °C, for a specified refluxing time, using a Syrris Atlas Sodium reactor. After synthesis, each product was naturally cooling down to room temperature and separated by centrifugation or by magnetic decantation. Supernatant was disregarding and precipitate washed with water several times. Finally, samples were dried at 40 °C under vacuum.Characterization
The size and morphology of the as-produced MNPs were examined by high-resolution transmission electron microscopy (HRTEM) using a JEOL 1100 apparatus. X-ray diffraction (XRD) powder analyses of the samples were carried out in a Miniflex 600 diffractometer (Rigaku) over 2θ range of 20–70°, using Cu-Kα radiation (λ = 1.541 Å) and operating at 40 kV and 30 mA. Room temperature magnetization curves were recorded using a vibrating sample magnetometer (VSM) ADE model EV9. Hysteresis loops were obtained in the ±18 kOe range. The iron content of the as-produced samples39 was assessed by using an inductively coupled plasma optical emission spectrometer (ICP-OES) Perkin Elmer model Optima 8000, with radiofrequency power of 1400 W, 1.5 mL min−1 sample flux, 10 L min−1 argon plasma flux, 0.7 L min−1 nebulizer flux and 0.2 L min−1 flux of auxiliary argon gas. The [Fe3+]/[Fe2+] ratio was determined by titration with potassium dichromate, after dissolving the as-produced MNPs with concentrated HCl. Electrophoretic mobility of the MNPs was assessed from aqueous dispersions by dynamic light scattering (DLS) using a Zetasizer nano ZS system (Malvern Instrument). Magnetic hyperthermia data were obtained using a NanoTherics equipment model Magnetherm. The system operates from 170 to 990 kHz with a field amplitude ranging from 70 up to 310 Oe. All the measurements were performed on powder samples containing the same amount of magnetic material, i.e. 20 mg. The heating rate was obtained neglecting the first 5 s of hyperthermia data (after turning on the field) and doing a linear fit of the next 10 s of data. This parameter is the one chosen for comparison between samples that have a mixture of phases, namely magnetite/maghemite or goethite/magnetite. It is believed that the present analysis might avoid inconsistency procedures related to heat capacity determination of samples containing both phases. In addition, it allows us to easily identify the magnetic materials with higher heating efficiency, as a consequence better potential for MHT.Results and discussion
Different geometries of MNPs were obtained while applying the typical protocol described in experimental section and performing the co-precipitation synthesis via chemical routes 1 (R1), 2 (R2) or 3 (R3), as schematically shown in Fig. 1. Nearly spherical nanoparticles (SNP) were obtained while using 40 mmol of NH4OH under reflux for 6 h, as indicated by R1 (see Fig. 1). Different anisometric nanoparticles were obtaining while using increased content of CH4N2O under reflux. Using 20 mmol of CH4N2O rod nanoparticles (RNP) were formed whereas using 100 mmol of CH4N2O hexagonal nanoparticles (HNP) were produced, as schematically shown in Fig. 1 via R2 and R3, respectively. Fig. 2a, b and 2c show the typical morphologies resulting via R1 (SNP), R2 (RNP) and R3 (HNP) syntheses, respectively. Worth mentioning the key role the nature and concentration the OH− source plays while defining the end isometric or anisometric morphology. Nevertheless, as shown in the XRD patterns included in Fig. 2d, except for traces of goethite (γ-FeOOH) observed in the samples synthesized with urea (R2 and R3) the spinel structure (magnetite, maghemite) is the main crystalline phase observed in the samples produced via R1, R2 and R3 syntheses.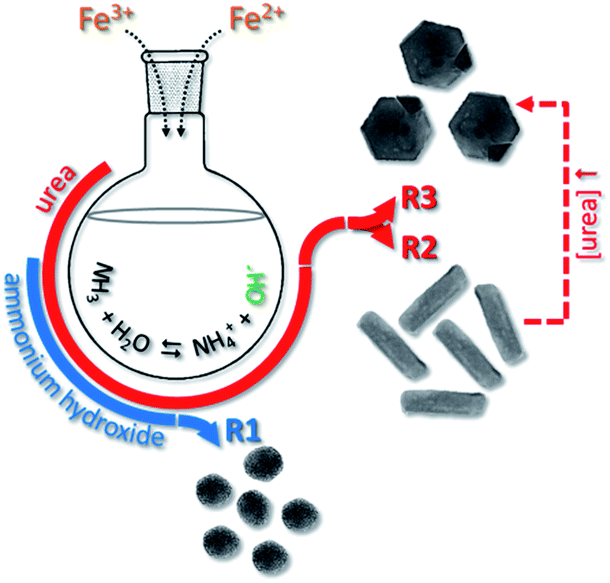 | ||
| Fig. 1 Schematic illustration of the formation of spherical (SNP), nanorod (RNP) and hexagonal nanoprism (HNP) nanoparticles in syntheses routes 1 (R1), 2 (R2) and 3 (R3). | ||
 | ||
| Fig. 2 TEM images of iron oxide nanoparticles obtained through syntheses R1 (a), R2 (b), and R3 (c). XRD patterns (d) and fittings of the XRD peak (511) performed in XRD data assessed from step scanning (between 2θ = 56° and 2θ = 59°) samples from R1, R2, and R3 (e). In (d) the main peaks of spinel were indexing while goethite is indicated by (g). In the bottom of the frame: (asterisk) goethite (JCPDS 29-0713), (double-cross) magnetite (JCPDS 19-0629), and (cross) maghemite (JCPDS 39-1346). | ||
However, it is important to take into account the limitation of the XRD technique in differentiating magnetite, maghemite or a solid solution of both once the two phases possess the same crystalline structure and almost identical lattice parameters. Actually, apart for few extra peaks, such as (210) and (211) solely observed in pure maghemite (not seen in Fig. 2d), the juncture of the XRD peaks in magnetite and maghemite is very analogous.
Nevertheless, the analogous XRD peaks slightly shift towards higher angles while changing the phase from magnetite to maghemite. Moreover, quantitative shift of the (511) XRD peak from lower (∼57.0°) towards higher (∼57.3°) angle indicates complete conversion of magnetite into maghemite.40
Therefore, considering dislocation of the (511) XRD peak as extracted from the fittings shown in Fig. 2e, which were performed in the XRD data recorded in a step scan pattern taken at angles (2θ) between 56° and 59°, it is possible to infer that maghemite predominates in the sample synthesized via R1 whereas magnetite is the dominant phase observed in the R2 and R3 syntheses, after refluxing for 6 h. Here, the upshift of XRD peaks (as in Fig. 2d) indicates that prolonged refluxing protocol under air condition (oxidative medium) while performing R1 synthesis prompts oxidation of magnetite into maghemite 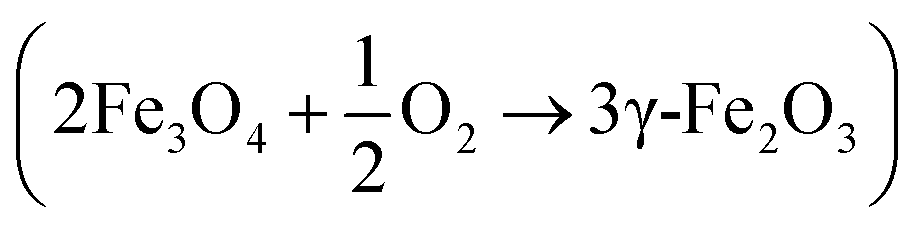 . Transmission electron microscopy (TEM) analyses of samples collected at different refluxing times show samples' aging process taking place, which increased the mean nanoparticles size while decreasing the polydispersity, probably due to a dissolution/recrystallization mechanism (ripening) triggered by refluxing. Nevertheless, no visible morphological alteration was noticed while refluxing via R1 synthesis.
. Transmission electron microscopy (TEM) analyses of samples collected at different refluxing times show samples' aging process taking place, which increased the mean nanoparticles size while decreasing the polydispersity, probably due to a dissolution/recrystallization mechanism (ripening) triggered by refluxing. Nevertheless, no visible morphological alteration was noticed while refluxing via R1 synthesis.
Fig. 3a and b show examples of TEM images of samples withdrawn during R1 synthesis at different refluxing times (1 h and 9 h). The second and third columns of Table 1 (upper panel) list both the mean TEM diameter and XRD crystalline size of samples collected after increasing reflux time (first column) in R1 synthesis. Samples were also collected at different refluxing times (1 h, 2 h, 4 h, 6 h, 8 h, and 12 h) for syntheses via R2 and R3. While Fig. 3c and d show typical TEM images of samples withdrawn (1 h and 12 h refluxing) from synthesis via R2, Fig. 3e and f show typical TEM images of samples collected from R3 synthesis after refluxing for 1 h and 12 h, respectively. As shown in Fig. 3c and d, morphology of the collected RNP is not noticeably altered as the refluxing time increases in R2 synthesis. Moreover, the acquired TEM images show that the RNP's length and width tend to increase as the refluxing time progresses, as listed in the second and third columns of Table 1 (middle panel). On the other hand, the TEM images of samples synthesized via R3 (Fig. 3e and f) reveal HNPs coexisting with RNPs at earlier refluxing times. However, as the refluxing time increases RNPs (R3 synthesis) tend to disappear giving place to well-defined HNPs, which are predominantly observed after refluxing for 12 h (see Fig. 3f).
 | ||
| Fig. 3 TEM images of samples withdrawn from syntheses via R1 at 1 h (a) and 9 h (b); R2 at 1 h (c) and 12 h (d), and R3 at 1 h (e) and 12 h (f). | ||
| R1 (SNP) | ||||||
|---|---|---|---|---|---|---|
| Refluxing time (h) | TEM diameter (nm) | XRD diameter (nm) | Fe3O4 (%) | Ms (emu g−1) | Hc (Oe) | Mr (emu g−1) |
| 1 | 11.5 ± 0.3 | 11.0 | 91 | 50.7 | ∼0.0 | ∼0.0 |
| 2 | 13.6 ± 0.4 | 11.9 | 11 | 57.0 | ∼0.0 | ∼0.0 |
| 4 | 12.8 ± 0.2 | 10.7 | 8 | 60.5 | ∼0.0 | ∼0.0 |
| 6 | 14.1 ± 0.1 | 13.1 | 7 | 63.7 | ∼0.0 | ∼0.0 |
| 9 | 19.4 ± 0.9 | 12.6 | 5 | 65.7 | ∼0.0 | ∼0.0 |
| R2 (RNP) | ||||||
|---|---|---|---|---|---|---|
| Refluxing time (h) | TEM length (nm) | TEM width (nm) | Fe3O4 (%) | Ms (emu g−1) | Hc (Oe) | Mr (emu g−1) |
| 1 | 41.3 ± 3 | 16 ± 0.2 | 19 | 12.3 | 79.6 | 0.7 |
| 2 | 57.9 ± 3 | 16.7 ± 0.9 | 43 | 37.8 | 93.4 | 3.2 |
| 4 | 49.7 ± 2 | 18.2 ± 0.4 | 52 | 50.8 | 57.0 | 3.0 |
| 6 | 50.1 ± 2 | 16.5 ± 0.3 | 61 | 56.9 | 71.5 | 3.1 |
| 8 | 55.4 ± 2 | 19.0 ± 0.3 | 65 | 55.5 | 82.1 | 4.1 |
| 12 | 79.0 ± 7 | 20.3 ± 0.4 | 68 | 60.9 | 79.5 | 4.6 |
| R3 (HNP) | |||||
|---|---|---|---|---|---|
| Refluxing time (h) | TEM diameter (nm) | Fe3O4 (%) | Ms (emu g−1) | Hc (Oe) | Mr (emu g−1) |
| 1 | — | 56 | 57.0 | 73.4 | 4.7 |
| 2 | — | 62 | 76.7 | 116.6 | 11.6 |
| 4 | 153.2 ± 7 | 66 | 79.0 | 138.0 | 13.7 |
| 6 | 146.4 ± 6 | 73 | 77.0 | 130.0 | 12.5 |
| 8 | 112.2 ± 6 | 74 | 79.7 | 120.0 | 12.1 |
| 12 | 105.1 ± 9 | 79 | 80.0 | 130.6 | 13.4 |
Differently from previous synthetic routes (R1 and R2), in the synthesis via R3 it was observed that as longer the refluxing time as shorter the diagonal (largest one) of the HNPs, as shown in the second column of Table 1 (lower panel). Notice that only hexagonal nanocrystal habit was computed in the TEM images for diagonal evaluation of HNPs.
HRTEM images obtained from samples synthesized via R2 (see Fig. 4a and b) showed that elongated nanoparticles seem to be morphologically organized from oriented aggregation of nanoclusters.41–43 Also, according to the lattice fringe profiles shown in Fig. 4a and b, only the intermediate goethite and the end product magnetite were detectable, in good agreement with XRD analyses discussed below. Besides, in R2 synthesis at early aging times (see Fig. 4a), goethite dominates the composition despite misorientation at the interface between nanocrystallites within RNPs. Nevertheless, as synthesis progresses via R2, goethite gives place to magnetite though still remaining in small amounts even after refluxing for 12 h, as shown in Fig. 4b. On the other hand, Fig. 3e and 4c show typical images of samples synthesized via R3, evidencing growth of HNPs by oriented aggregation of nanorods, where elongated structures and subsequently larger aggregates formed from them, align and fuse together to form a single crystal.44 Concomitantly, transformation of goethite into magnetite, though keeping the nanocluster's aggregate aspect, can also be seen in HNPs imaged in Fig. 4d.
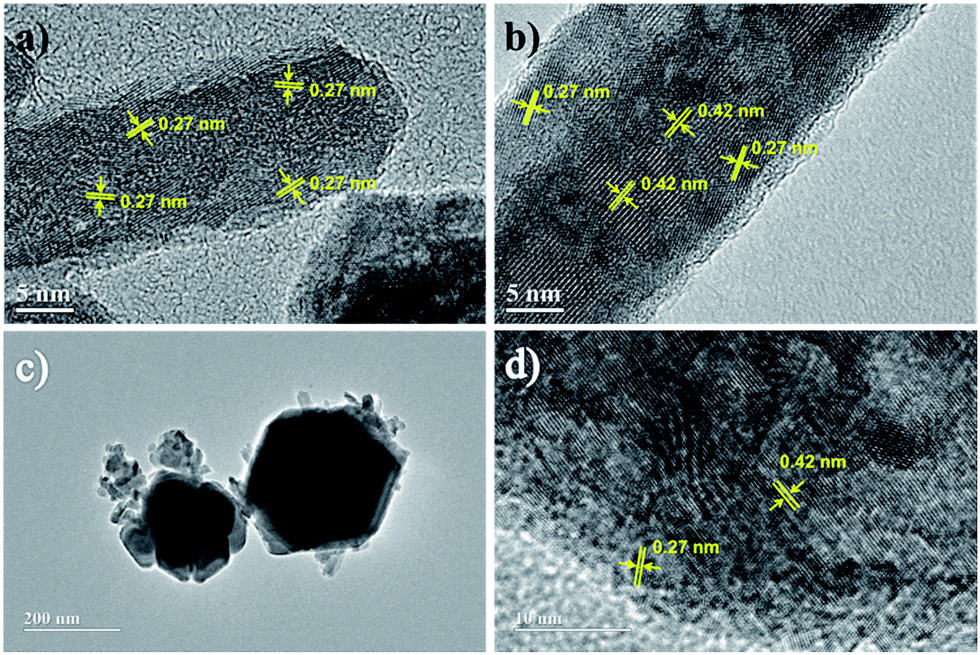 | ||
| Fig. 4 HRTEM images of samples withdrawn from syntheses via R2 at 1 h (a) and 12 h (b) and via R3 at 1 h (c) and 12 h (d). The lattice fringe of 0.42 nm corresponded to twice the (400) plane spacing of magnetite whereas the lattice fringe of 0.27 nm corresponded to the (130) plane spacing of goethite. | ||
Fig. 5 shows a set of XRD diffractograms of samples collected under increasing refluxing time (1 h, 2 h, 4 h, 6 h, 8 h, and 12 h) and synthesized via R2 and R3. Magnetite (indexed) and goethite (asterisks) crystalline phases have been identifyied in different contents depending on the synthesis route (R2 or R3) and time of refluxing. Moreover, as the refluxing time increases the XRD peaks assigned to goethite tend to disappear earlier (after ∼2 h) in R3 than in R2 synthesis. On the other hand, magnetite seems to be the major phase in R2 synthesis, but at a later stage, i.e. only after 6 h of refluxing.
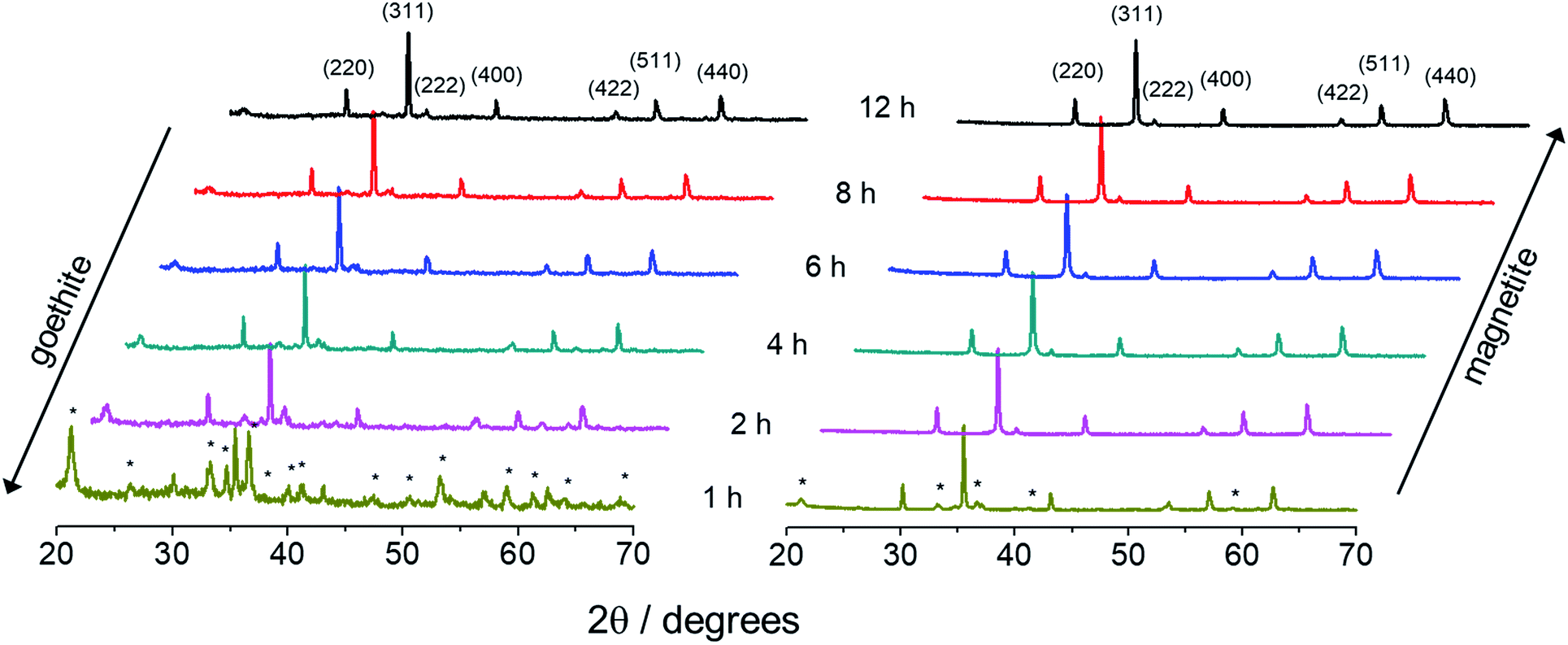 | ||
| Fig. 5 XRD patterns of nanoparticles obtained through syntheses R2 (left) and R3 (right) as a function of the refluxing time. Main peaks of the spinel phase are indexed while goethite is indicated by asterisks. | ||
Another important parameter investigated in the present study was the pH of the reaction medium, monitored as a function of the refluxing time for all syntheses routes. In the R1 synthesis, right after addition of the ammonium hydroxide we found pH ∼12 which linearly decreased down to pH ∼8 after ∼4 h, remaining afterwards nearly flat up to ∼9 h (see Fig. 6).
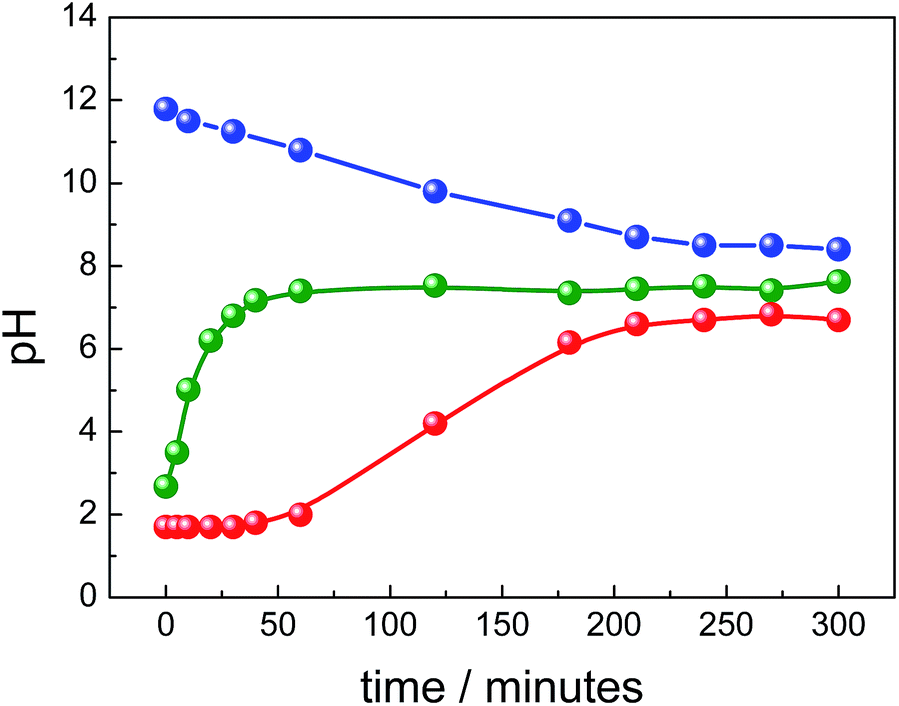 | ||
| Fig. 6 Variation of pH during syntheses via R1 (blue), R2 (red), and R3 (green). | ||
As previously reported,45 pH reduction in similar synthesis was assumed to be owing to hydroxide consumption (e.g. from NH4OH source) in the presence of Fe2+/Fe3+ due to the formation of magnetite (2Fe3+ + Fe2+ + 8OH− → Fe3O4 + 4H2O) and subsequently due to ammonia (NH3) volatilization from solution by heating. In this case, the high [OH−]/[Fe] ratio is supposed to favor nucleation and thus formation of isometric and nanosized particles. On the other hand, in the synthesis via R2 (see Fig. 6) using lower concentration of urea (20 mmol), the medium was initially acid (pH < 2) and the pH slowly increased as function of time in a sigmoid-like path to achieve a plateau at mild acid condition (pH slightly below 7) after ∼4 h of refluxing. Differently, in the synthesis via R3 (see Fig. 6), which started with higher concentration of urea (100 mmol), the pH upshift steeply and quickly achieved higher values, saturating at mild basic condition (pH slightly above 7) in ∼1 h. In both cases (R2 and R3), the thermal decomposition of urea, a well-known agent utilized in precipitation of nanoparticles from homogeneous solutions,46 generates OH− in solution in a rate strongly dependent upon the initial urea concentration (CO(NH2)2 + 3H2O → CO2 + 2NH4+ + 2OH−). This route provides an initial chemical environment with lower concentration of OH−, which could induce gradual precipitation and anisometric crystal growth of nanoparticles.47
In addition, in aqueous co-precipitation of Fe2+/Fe3+ aiming formation of magnetite, it is important to take into consideration the hydrolysis of Fe3+ and oxidation/hydrolysis of Fe2+ in the production of different iron oxides/hydroxides.
The actual chemical reaction is mainly governed by the [OH−]/[Fe] and [Fe3+]/[Fe2+] ratios plus the rate of oxidation and the temperature, and also influenced by foreign species in the system.48 As a result, it is a very difficult task controlling the chemical reaction conditions to obtain end monophasic products. Rather, formation of a mixture of oxides and even transient species not observed in the final precipitate is typical in this case. Since samples were collected at large intervals of time while carrying on R2 and R3 syntheses and ex situ methods were utilized to characterize the products, one assumes that only goethite appeared as intermediate for magnetite to tentatively explain the formation of the MNPs with different morphologies. Thus, once the solubility product of Fe(OH)2 is much higher than Fe(OH)3, in the starting mixed Fe3+/Fe2+ solution hydrolysis of Fe3+ is preferential. Therefore, as the [OH−] increased due to thermal decomposition of urea, the very low soluble goethite is formed from partial dehydroxylation of Fe3+ aquocations, as conveniently represented by the global equation Fe3+ + 3OH− → FeOOH + H2O, which is the sum of eqn (1)–(3) below:
| Fe3+ + 6H2O → [Fe(H2O)6]3+, | (1) |
| [Fe(H2O)6]3+ → FeOOH + 3H+ + 4H2O, | (2) |
| 3H+ + 3OH− → 3H2O. | (3) |
Therefore, as the pH of the medium monotonically rises in R2 and R3 syntheses (see Fig. 6) goethite progressively converts into magnetite. Moreover, once there is little morphological difference among nanorods as refluxing progresses it can be inferred that a topotactic conversion occurs, meaning transformation of the initial solid phase into the final solid phase without dissolution. Actually, several studies support this hypothesis while assuming that transformation is catalyzed by adsorption of Fe2+ onto the goethite surface and interfacial electron transfer takes place as [OH−] increases via dehydration, as represented by the chemical reaction 2FeOOH + Fe2+ + 2OH− → Fe3O4 + 2H2O. Ahn et al.23 reported that when NH4OH was slowly added to a Fe2+/Fe3+ solution described by R = [OH−]/([Fe3+] + [Fe2+]) akaganeite nucleated at very low pH, and was subsequently transformed through goethite into magnetite at pH ∼5 (R > 2).
Till et al.49 also investigated the role of goethite as a potential source of magnetite in sediments and argued that through dehydroxylation of poorly crystalline nanogoethite stoichiometric sub-micron magnetite was formed. Usman et al.50 showed that coordination of Fe2+ onto specific crystal faces of goethite's surface is a prerequisite to electron transfer and thus to promote topotactic transformation of goethite into magnetite in a system containing goethite seeds, Fe2+, and NaOH. Tronc et al.51 showed that alkalization and aging of aqueous solutions containing Fe2+/Fe3+, at various x = [Fe2+]/[Fe3+] ratios, led to transformation of quasi amorphous ferric hydroxide into spinel phase via Fe2+ adsorption and electron delocalization, which brings in local structural rearrangements and drives spinel ordering by means of topotactic crystallization or dissolution–recrystallization processes. The pathway depends on the [Fe2+] level in the system and, in particular, precursors were transformed into goethite at x = 0.05 and exclusively into spinel at x > 0.10. Briefly, the conditions for the predominance of one oxide phase in the pair goethite/magnetite are low pH for goethite or high pH, high temperature and high [Fe2+] for magnetite.48 Results of these studies corroborate with observations of the present report and help supporting the different pathways proposed for the formation of magnetic iron oxides. Shortly, in R1 synthesis the high pH, high [Fe2+] and high temperature (with nearly constant and high [OH−]/[Fe] ratio) benefit formation of magnetite and subsequently its oxidation into maghemite. Actually, in R1 synthesis goethite was not detected. However, in R2 and R3 syntheses temperature, [Fe3+]/[Fe2+] and [Fe2+] were the same as in R1 synthesis, but the pH and [OH−]/[Fe] strongly vary during synthesis (see Fig. 6). Indeed, in R2 synthesis the progressively rise of [OH−]/[Fe] and the low final pH reached have favored formation of goethite, which was slowly and partially transformed into magnetite under high [Fe3+]/[Fe2+] condition. On the other hand, in R3 synthesis, the sharp rise of [OH−]/[Fe] provided quick increasing in the content of magnetite by topotactic transformation of initially formed goethite.
Quantitative estimation of the magnetite content in the as-produced samples via R1, R2 and R3 syntheses was carrying out by assessing both the [Fe3+]/[Fe2+] ratio and the [Fe] using titration with potassium dichromate and ICP-OES, respectively. Additionally, for estimating the magnetite content it is assuming that Fe2+ is uniquely correlated with stoichiometric Fe3O4 whereas Fe3+ is associated with γ-Fe2O3 in R1 synthesis and with Fe3O4 and α-FeOOH in both R2 and R3 syntheses. Therefore, according to this approach the Fe3O4 content in all samples is given by [Fe2+] whereas the contents of stoichiometric γ-Fe2O3 (in R1) or stoichiometric α-FeOOH (in R2 and R3) are given by [Fe3+] − 2[Fe2+]. The fourth column of Table 1 collects the estimated content (w/w%) of magnetite in all samples. However, underestimated values of magnetite content could be obtained using this approach, once partial oxidation of Fe2+ into Fe3+ can occur in the spinel phase and may not change substantially the XRD pattern.
The electrophoretic mobility of the as-synthesized nanoparticles was measured as function of the pH and transformed into zeta potential (i.e. related to the nanoparticle surface charge density). From the assessed data, isoelectric points of charge at pH 7.1, 6.2 and 6.8 were determined for samples presenting the highest estimated values of magnetite content (see Table 1) and synthesized via R1, R2 and R3, respectively. Actually, the electrophoretic mobility depends not only on the nanoparticle composition, shape and size but also on both the ionization of the amphoteric surface sites and their further complexation with electrolyte ions.52 For bare magnetite, the isoelectric point of charge is around pH 7.0, and the presence of sulfate molecules in the as-synthesized samples could account for shifting the isoelectric point of charge to lower pH values.53 These surface characteristics suggest that oriented aggregation of nanoclusters in particle formation also seems to be governed by electrostatic forces arising from surface of nanoclusters. Actually, the oxide surface behaves as a diprotic acid, leading to three kinds of surface sites, most of them are positively charged (–OH2+) in acidic medium, negatively charged (–O−) in basic medium and neutral (–OH) around the isoelectric point.54 Thus, a faster growth under weak electrostatic repulsion forces at pH ∼7, nearby the isoelectric point of magnetite, during formation of samples in R2 and R3 is expected. On the other hand, an increase in pH above the isoelectric point, such as in R1, causes charging of primary particles preventing their aggregation and inducing a growth mechanism slower than R2 and R3 syntheses. Finally, it was reported that the presence of sulfate ions strongly complexes with Fe3+, resulting in precipitation at higher rate and lower [OH−]/[Fe] ratio.55 Also, at low precipitation rate the presence of sulfate ions can lead to the formation of anisometric crystals, due to the specific adsorption of SO42− to crystal faces parallel to the c-axis, retarding the growth of the particle in the direction normal to this axis.56
In short, one can infer that magnetite NPs obtained by route 1 are crystallized in a quasi-immediate process embedding both Fe3+ and Fe2+ into the crystal structure.
As it is well-known,48 due to the large surface area and once the synthesis is performed in oxidative atmosphere, magnetite is easily oxidized to maghemite by topotactical reaction. The reaction proceeds by outward migration of cations towards the surface of the crystal with simultaneous formation of cation vacancies and addition of oxygen atoms. On the other hand, in routes 2 and 3, goethite NP precursors are firstly formed, containing only Fe3+. Subsequently, magnetite NPs are slowly formed by transformation of goethite, catalyzed by adsorption of Fe2+ onto the crystal surface. Thus, the slow rate of magnetite formation, related to the slow rate of urea hydrolysis at lower pH, induces very slow oxidation of magnetite to maghemite.
The recorded (specific) saturation magnetization (Ms) of the samples collected from R1 (9 h), R2 (12 h) and R3 (12 h) syntheses were 51 emu g−1, 55 emu g−1 and 59 emu g−1, respectively. Additionally, analysis of the hysteresis cycles at low field region (see inset of Fig. 7) shows that SNPs are superparamagnetic-like whereas RNPs and HNPs are at the blocked state. Actually, bulk maghemite and bulk magnetite present saturation magnetization of 60–80 emu g−1 and 90–100 emu g−1, respectively.48 Therefore, higher Ms values are expected for the anisometric samples (RNP and HNP at 12 h refluxing) in which magnetite dominates whereas lower value of Ms accounts for the isometric sample (SNP at 9 h refluxing) with prevalence of maghemite. Besides, in the nanosized range the larger the volume the higher the saturation magnetization, in good agreement with the data collected in Table 1: upper panel for SNPs (second column, last row), middle panel for RNPs (second and third columns, last row) and lower panel for HNPs (second column, last row).
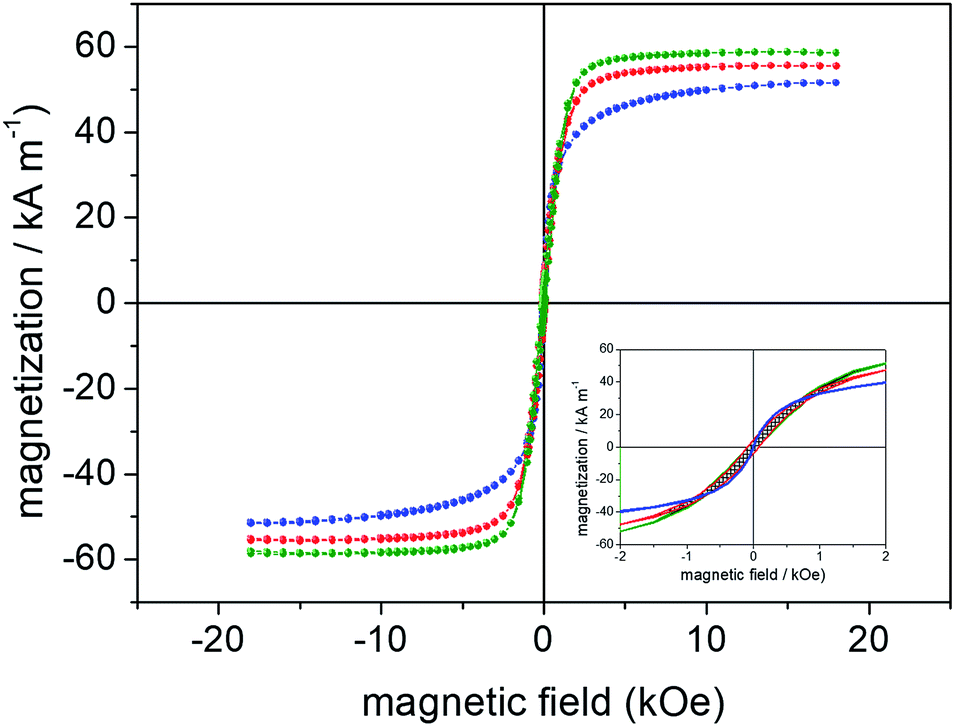 | ||
| Fig. 7 Magnetization curves of samples obtained via R1 (blue), R2 (red) and R3 (green) syntheses and collected after 9 h, 12 h and 12 h of refluxing, respectively. The inset shows the recorded hysteresis cycles in the low field range (±2 kOe). | ||
Moreover, the presence and relative content of antiferromagnetic and/or poorly magnetically-ordered secondary phase, as in the case of antiferromagnetic goethite, should decrease the Ms value of the samples synthesized via R2 and R3. Indeed, the strong implications of the nanometric scale, such as cationic redistribution57 plus surface and finite size effects58 may also change the end magnetic characteristics of nanosized materials.
Fig. 8 shows the extracted values of the room temperature saturation magnetization of all synthesized samples as function of the refluxing time. Regardless the synthesis route employed (R1, R2, or R3), the saturation magnetization shown in Fig. 8 increases with increasing refluxing time, though different contributions are claimed to play key roles, such as phase composition and mean crystallite size. In addition to saturation magnetization (Ms), coercive field (Hc) and magnetic remanence (Mr) were also extracted from the room temperature magnetic hysteresis cycles of all samples and collected in Table 1 (see last three columns).
 | ||
| Fig. 8 Variation of room temperature saturation magnetization as a function of refluxing time for R1 (blue), R2 (red), and R3 (green) syntheses. The insets showed the variation of Mr and Hc as a function of refluxing time for R2 and R3 syntheses. | ||
Values of the coercive field (Hc) and magnetic remanence (Mr) are also plotted in the insets of Fig. 8. As observed from the hysteresis cycles and the data collected in Table 1, all samples obtained from R1 present superparamagnetic-like behavior, with negligible Mr and Hc. Moreover, samples synthesized via R1 present Ms slightly increasing with refluxing time, despite the expected reduction of Ms arising from oxidation of magnetite into maghemite. However, the SNP growth observed from both XRD and TEM data (see upper panel in Table 1) and its contribution to the enhancement of Ms clearly overcomes the reduction of the saturation magnetization due to phase transformation with increasing refluxing time. Samples obtained from syntheses R2 and R3 present room temperature blocked state with significant Hc and Mr values, which also vary as a function of the refluxing time. For these samples, increasing of Ms as the refluxing time increases is a combination of both transformation of goethite into magnetite and variation in the mean crystallite size. While in samples synthesized via R2 both tendencies (phase transformation and size as the refluxing time increases) favor enhancement of Ms; in samples obtained via R3 the phase transformation dominates over reduction in the mean crystallite size, resulting in enhancement of Ms upon refluxing time (see data in Table 1). These tendencies provide a clear picture for the total enhancement (from 1 h up to 12 h of refluxing) of Ms by a factor of ∼5 in R2 against a mild enhancement of Ms by a factor of ∼1.4 in R3.
The magnetothermal properties of the as-produced and shape-modulated MNPs were also studied here. Fig. 9a–c show the time profile of temperature increase of samples from R1, R2 and R3 syntheses obtained after 1 h of refluxing for distinct magnetic field amplitudes (70 to 310 Oe) at an AC frequency of 522 kHz. In all samples one observes an increase of temperature the higher the field amplitude. According to the recorded data, one can also conclude that the best morphology for strong hyperthermia applications are the SNPs (R1), followed by the HNPs (R3) with mild heating, while the RNPs showed weak heating at this experimental condition.
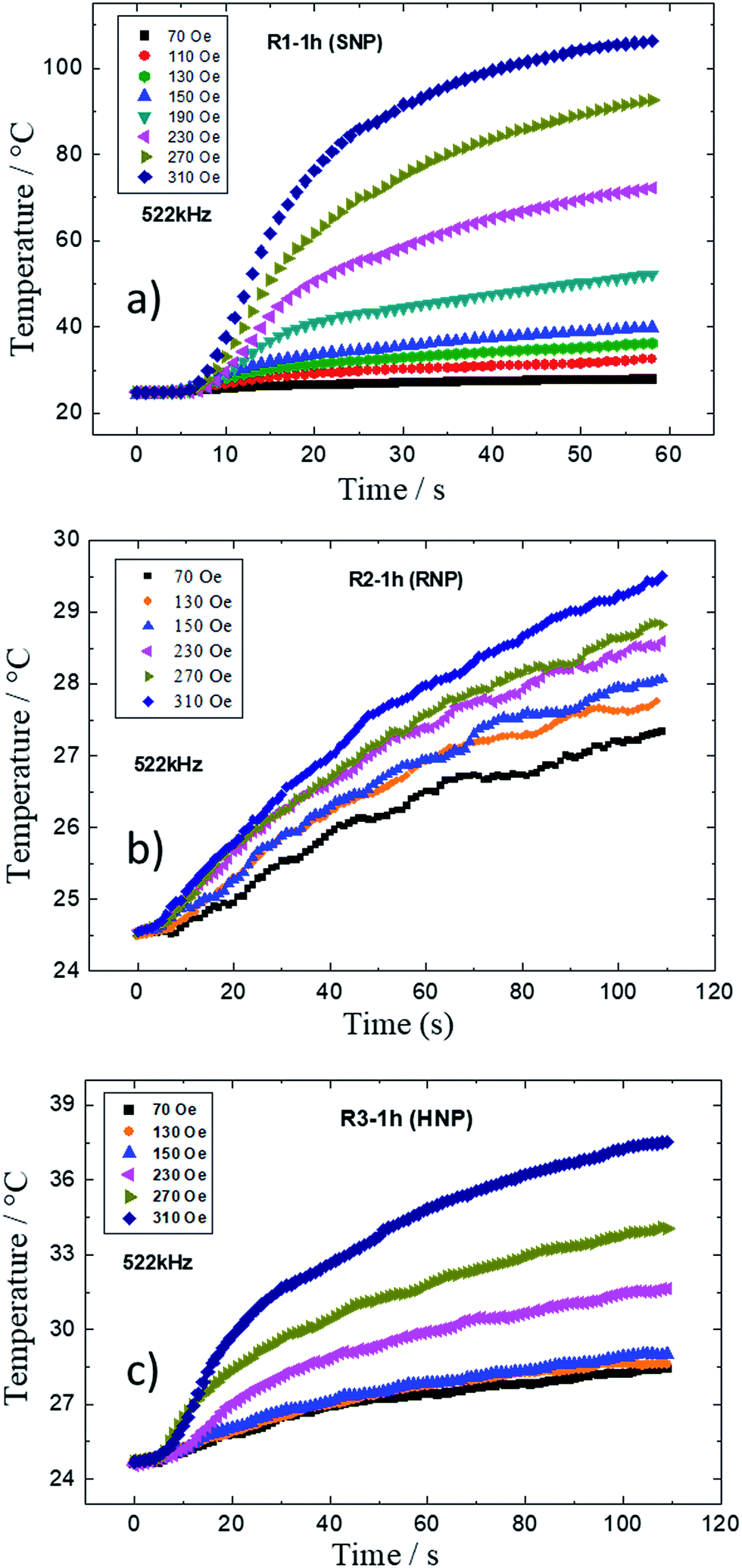 | ||
| Fig. 9 Time profile of temperature increase of samples from R1 (a), R2 (b) and R3 (c) obtained after 1 h of refluxing for distinct magnetic field amplitudes. Data at 130 Oe were omitted from the graphs to avoid superposition of curves. | ||
The same conclusion can be obtained from the analysis of the heating rate (that is proportional to the heating efficiency, namely specific loss power – SLP – or also the commonly used name specific absorption rate – SAR) as function of the square of the applied field, as shown in Fig. 10a. Symbols represent experimental data whereas the dash lines are just guides to the eye. The lines are included aiming to compare data with theoretical predictions from the linear response theory (LRT), which predicts square field dependence.17 According to this approach only at high fields, for samples synthesized via R1, deviations are observed at this frequency. The approach of analyzing the heating rate performance allowed one to compare several samples without having to make a step forward towards stabilizing them by adequately surface coating the nanoparticles to avoid particle agglomeration and guarantee a highly stable magnetic colloid. Although this is an important step forward, firstly it is important to check, as accurate as possible, which distinct shapes have potential for hyperthermia therapy.
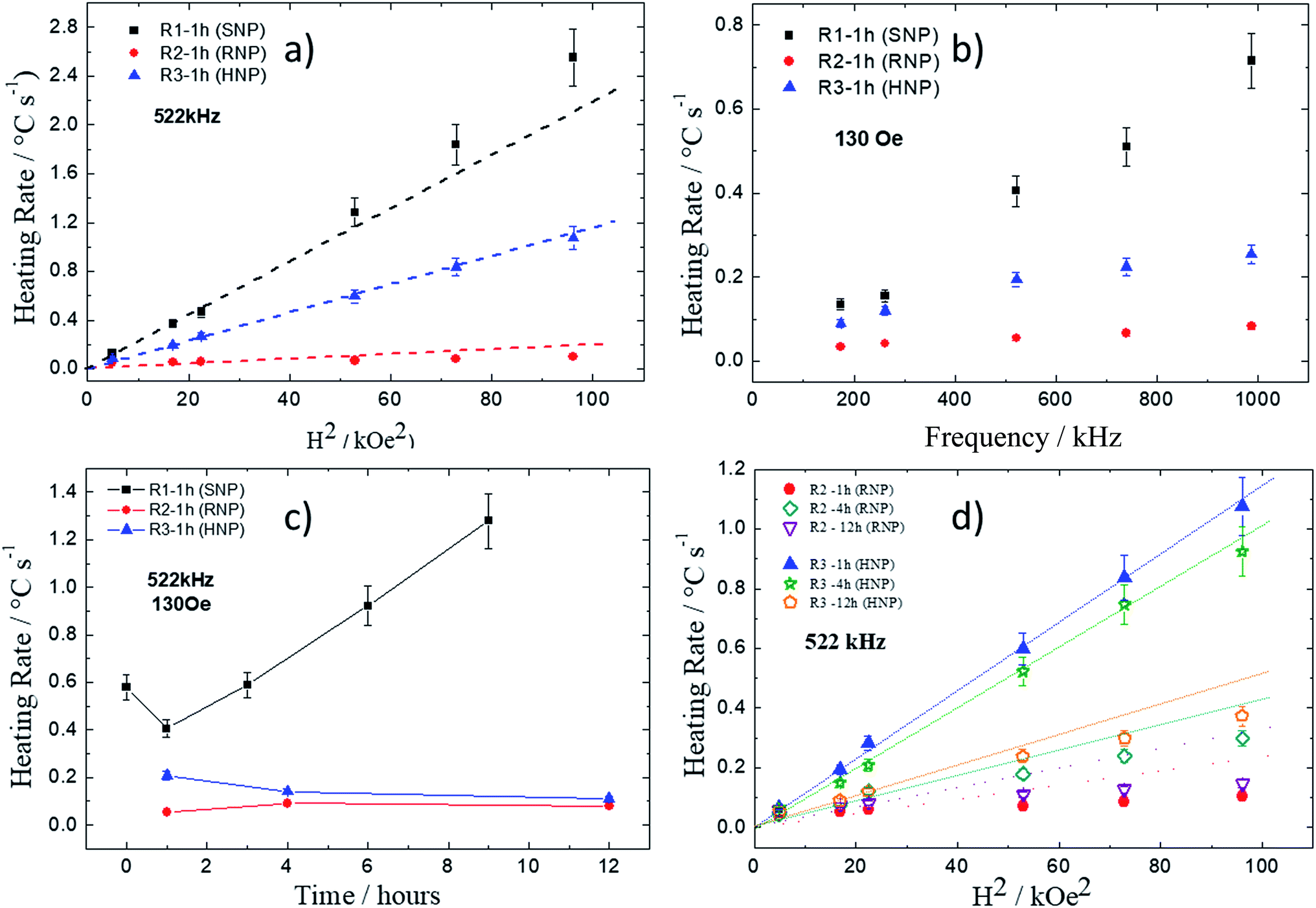 | ||
| Fig. 10 Heating rate as function of the: square of the applied field (a); field frequency (b); refluxing time (c) for R1, R2 and R3. Heating rate as function of the square field for samples from R2 and R3 at different refluxing time (d). | ||
Nevertheless, relative comparison between samples by performing measurements with powder can still be made and allowed one to identify the most promising materials for such specific applications, once higher heating rate values mean more efficient heat delivery.
Fig. 10b shows heating rate as function of field frequency for the magnetic field amplitude of 130 Oe. Square symbols represent samples from R1 (SNP), triangles represent samples from R2 (RNP) and circles represent samples from R3 (HNP). Dashed lines are the expected linear frequency dependence for NPs that does not show significant dynamic hysteresis effect.59 Note that deviation from this behavior occurs for samples from R3 and R2 (although in a smaller content), which seems to tend to saturation the higher the frequency values.
Fig. 10c shows heating rate as function of refluxing time for all three synthetic routes at 522 kHz and 130 Oe. This again confirms the result that SNPs heat more efficiently than HNPs or RNPs. Indeed, one can observe that samples from R1 showed higher heating rate values the longer the refluxing time. According to TEM data this happened because of an increase of the mean particle size, which varied from around 11 nm (1 h) to 19 nm (9 h). Curiously, neither coercive field nor remanence was found in all the samples prepared from R1, indicating a quasi-static superparamagnetic-like behavior for SNPs at this dimension range. On the other hand, samples synthesized from both R2 and R3, showed non-zero coercive field and remanence. Moreover, samples from R3 synthesis showed a decrease of heating rate the longer the refluxing time, whereas samples from R2 synthesis showed a maximum of heating rate at around 4 h of refluxing with a slight decrease the longer the refluxing time. Fig. 10d shows heating rate as function of the square of field amplitude for samples from R2 and R3 syntheses at distinct refluxing times, namely 1 h, 4 h, and 12 h. As shown in the other figure, it is clear for the RNPs that a maximum of SLP is occurring around 4 h of refluxing. On the contrary, HNPs showed higher heating rate for 1 h of refluxing and a decrease of heating efficiency the longer the refluxing time. Comparison between samples from R2 (1 h) and R3 (1 h) suggests that the better heating performance of the sample from R3 might be related to higher magnetization values, which also correlates with its lower goethite content, due to the fast pH increase while using a higher amount of urea in the synthesis protocol. On the other hand, the lower heating rate value for the R3 (12 h) sample in comparison to the R3 (1 h) one might be related to the increase of coercive field the longer the refluxing time. R3 (12 h) presented Hc = 131 Oe whereas R3 (1 h) sample had Hc = 73 Oe.
As far as the hyperthermia efficiency is concerned it is clear from all the assessed data that the spheres are better than any anisometric nanoparticle investigated in this study. However, it is important to point out that the size/length of the structures are not in the same range, so effects due mainly to dimension are relevant to the present discussion. Nevertheless, it is fair to comment that the SNPs showed no coercivity at quasi-static conditions (VSM data), in contrast to the other samples (RNPs and HNPs). Indeed, this suggests that dynamic hysteresis60 played a key role in the heating efficiency of the SNPs, which means that at higher frequencies a dynamic hysteresis appeared in those samples increasing the heating efficiency. On the other hand, the HNPs showed better heating efficiency than RNPs. It is curious that HNPs showed higher coercivity values in comparison to the RNPs. Therefore, one might expect other contributions for this type of NPs due to its shape characteristics. The first hypothesis could be some multi-domain nanoparticles that are expected to show Rayleigh loss.61 However, the HNPs had not showed any evidence of such contribution once the heating rate did not scale with the cubic of the field amplitude. On the other hand, one may not rule out the vortex-like spin rotation as a possible explanation for better magneto-thermal properties of HNPs in comparison to RNPs. Independent of that, none of those materials were able to heat better than the SNPs, suggesting that the spherical shape might be the best at these experimental conditions. Indeed, from the hyperthermia experiments one can conclude that the best NP for heating performance are the SNPs, followed by HNPs. However, in the latter the best sample corresponds to a short time of synthesis, which suggests that one should not increase significantly the coercive field value if one is interested in strong hyperthermia applications, i.e. at low field amplitude conditions where Atkinson's criteria for clinical application stands. In addition, although it was observed a lower efficiency as magnetic heating sources for the anisometric nanostructures, both shapes (HNPs and RNPs) might be still interesting for mild and weak efficiency once the control of particle shape might have several impacts on the biomedical field spanning from more efficient cell uptake up to the activation of the systemic immune system, which for instance could impact the development of vaccines for several diseases. Notwithstanding, this could still be an important contribution to the non-conventional applications of MHT, such as in the remote controlled recycling of magnetic nanosorbents during the treatment of water containing pollutants.62
Conclusion
The eco-friendly chemical co-precipitation protocol herein described was successfully used to produce iron oxide-based nanoparticles with modulated size and shape in the nanoscale range and modulated phase between magnetite/maghemite and magnetite/goethite. In contrast to the commonly employed thermal decomposition methods, the green synthesis protocol herein described used ammonia to precipitate spherical magnetite/maghemite nanoparticles (SNPs) from aqueous solution of Fe2+/Fe3+ whereas magnetite/goethite rod/hexagonal nanoparticles (RNPs/HNPs) were precipitated from aqueous solution of Fe2+/Fe3+ using urea at 1/5 concentration ratio. Increasing refluxing time at 95 °C, from 1 h to 9 h with ammonia and from 1 h to 12 h with urea was successfully used to modulate the mean size of the SNPs from 10 to 20 nm. Likewise, modulation of the length/width (diagonal) of the RNPs (HNPs) in the range of 40/16 nm (150 nm) to 80/20 nm (100 nm) was achieved by varying the refluxing time from 1 h to 12 h in the presence of urea. Moreover, modulation of the relative magnetite/maghemite content in the range of about 0.05 to 10 was accomplished while using ammonia whereas the use of urea produced RNPs (HNPs) with magnetite/goethite relative content varying in the range of about 0.25 to 2 (1.25 to 3.75). As for the room-temperature magnetic characteristics the SNPs showed superparamagnetic behavior whereas both the RNPs and HNPs were in the blocked state; the anisometric NPs presenting coercivity (remanence) varying in the range of about 55 to 80 Oe (1 to 5 emu g−1) and 70 to 130 Oe (5 to 13 emu g−1), respectively. Additionally, the saturation magnetization of SNPs, RNPs and HNPs varied in the range of about 50 to 65 emu g−1, 12 to 60 emu g−1 and 57 to 80 emu g−1, respectively. Finally, the as-produced samples were tested in regard to their heating efficiency for magnetic hyperthermia applications. In this regard, strong hyperthermia effect was found for the SNPs, while HNPs presented mild hyperthermia effect, followed by weak heating efficacy with RNPs. Indeed, the green synthesis route herein employed allowed successful modulation of size, shape and iron oxide-based magnetic phase in the nanosized range, thus covering a wide range of biological applications using strong, mild and weak hyperthermia effect.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors gratefully acknowledge financial support from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), Fundação de Amparo à Pesquisa do Estado de Goiás (FAPEG), Decanato de Pesquisa e Inovação (DPI-UnB) and Fundação de Empreendimentos Científicos e Tecnológicos (FINATEC). The authors also thank LabMic-UFG for TEM measurements.Notes and references
- Y. Yu, Q. B. Zhang, J. P. Xie and J. Y. Lee, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- J. P. Xie, J. Y. Lee, D. I. C. Wang and Y. P. Ting, ACS Nano, 2007, 1, 429–439 CrossRef CAS PubMed.
- Q. F. Yao, Y. Yu, X. Yuan, D. Zhao, J. P. Xie and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 184–189 CrossRef CAS PubMed.
- D. Yoo, J. H. Lee, T. H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863 CrossRef CAS PubMed.
- N. Tran and T. J. Webster, J. Mater. Chem., 2010, 20, 8760–8767 RSC.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
- J. A. Champion and S. Mitragotri, Pharm. Res., 2009, 26, 244–249 CrossRef CAS PubMed.
- Z. G. Yue, W. Wei, Z. X. You, Q. Z. Yang, H. Yue, Z. G. Su and G. H. Ma, Adv. Funct. Mater., 2011, 21, 3446–3453 CrossRef CAS.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- H. Herd, N. Daum, A. T. Jones, H. Huwer, H. Ghandehari and C. M. Lehr, ACS Nano, 2013, 7, 1961–1973 CrossRef CAS PubMed.
- J. A. Cribb, T. D. Meehan, S. M. Shah, K. Skinner and R. Superfine, Ann. Biomed. Eng., 2010, 38, 3311–3322 CrossRef PubMed.
- P. H. Lizotte, A. M. Wen, M. R. Sheen, J. Fields, P. Rojanasopondist, N. F. Steinmetz and S. Fiering, Nat. Nanotechnol., 2016, 11, 295–302 CrossRef CAS PubMed.
- K. Niikura, T. Matsunaga, T. Suzuki, S. Kobayashi, H. Yamaguchi, Y. Orba, A. Kawaguchi, H. Hasegawa, K. Kajino, T. Ninomiya, K. Ijiro and H. Sawa, ACS Nano, 2013, 7, 3926–3938 CrossRef CAS PubMed.
- E. A. Perigo, G. Hemery, O. Sandre, D. Ortega, E. Garaio, F. Plazaola and F. J. Teran, Appl. Phys. Rev., 2015, 2, 35 Search PubMed.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080–3091 CrossRef CAS PubMed.
- C. Martinez-Boubeta, K. Simeonidis, A. Makridis, M. Angelakeris, O. Iglesias, P. Guardia, A. Cabot, L. Yedra, S. Estrade, F. Peiro, Z. Saghi, P. A. Midgley, I. Conde-Leboran, D. Serantes and D. Baldomir, Sci. Rep., 2013, 3, 8 Search PubMed.
- M. S. Carriao, V. R. R. Aquino, G. T. Landi, E. L. Verde, M. H. Sousa and A. F. Bakuzis, J. Appl. Phys., 2017, 121, 13 CrossRef.
- L. C. Branquinho, M. S. Carriao, A. S. Costa, N. Zufelato, M. H. Sousa, R. Miotto, R. Ivkov and A. F. Bakuzis, Sci. Rep., 2013, 3, 10 Search PubMed.
- E. Alphandery, Y. Ding, A. T. Ngo, Z. L. Wang, L. F. Wu and M. P. Pileni, ACS Nano, 2009, 3, 1539–1547 CrossRef CAS PubMed.
- E. Alphandery, I. Chebbi, F. Guyot and M. Durand-Dubief, Int. J. Hyperthermia, 2013, 29, 801–809 CrossRef PubMed.
- H. Zeng, P. M. Rice, S. X. Wang and S. H. Sun, J. Am. Chem. Soc., 2004, 126, 11458–11459 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- T. Ahn, J. H. Kim, H. M. Yang, J. W. Lee and J. D. Kim, J. Phys. Chem. C, 2012, 116, 6069–6076 CAS.
- W. Zhang, S. Y. Jia, Q. Wu, J. Y. Ran, S. H. Wu and Y. Liu, Mater. Lett., 2011, 65, 1973–1975 CrossRef CAS.
- R. V. Kumar, Y. Koltypin, X. N. Xu, Y. Yeshurun, A. Gedanken and I. Felner, J. Appl. Phys., 2001, 89, 6324–6328 CrossRef CAS.
- H. F. Zhou, R. Yi, J. H. Li, Y. Su and X. H. Liu, Solid State Sci., 2010, 12, 99–104 CrossRef CAS.
- L. Gu and H. X. Shen, J. Alloys Compd., 2009, 472, 50–54 CrossRef CAS.
- J. J. Zhu, D. Li, D. L. Jiang and M. Chen, Micro Nano Lett., 2013, 8, 383–385 CAS.
- C. M. Cheng, F. J. Xu and H. C. Gu, New J. Chem., 2011, 35, 1072–1079 RSC.
- J. Wang, Z. M. Peng, Y. J. Huang and Q. W. Chen, J. Cryst. Growth, 2004, 263, 616–619 CrossRef CAS.
- K. Woo and H. J. Lee, J. Magn. Magn. Mater., 2004, 272, E1155–E1156 CrossRef.
- M. Mahmoudi, A. Simchi, M. Imani, P. Stroeve and A. Sohrabi, Thin Solid Films, 2010, 518, 4281–4289 CrossRef CAS.
- O. Bomati-Miguel, A. F. Rebolledo and P. Tartaj, Chem. Commun., 2008, 4168–4170 RSC.
- H. Itoh and T. Sugimoto, J. Colloid Interface Sci., 2003, 265, 283–295 CrossRef CAS PubMed.
- M. Ocana, M. P. Morales and C. J. Serna, J. Colloid Interface Sci., 1999, 212, 317–323 CrossRef CAS PubMed.
- S. Y. Lian, E. Wang, Z. H. Kang, Y. P. Bai, L. Gao, M. Jiang, C. W. Hu and L. Xu, Solid State Commun., 2004, 129, 485–490 CrossRef CAS.
- C. Yang and H. S. Yan, Mater. Lett., 2012, 73, 129–132 CrossRef CAS.
- S. Li, G. W. Qin, T. W. L. Pei, Y. P. Ren, Y. D. Zhang, C. Esling and L. Zuo, J. Am. Ceram. Soc., 2009, 92, 631–635 CrossRef CAS.
- M. H. Sousa, G. J. da Silva, J. Depeyrot, F. A. Tourinho and L. F. Zara, Microchem. J., 2011, 97, 182–187 CrossRef CAS.
- W. Kim, C. Y. Suh, S. W. Cho, K. M. Roh, H. Kwon, K. Song and I. J. Shon, Talanta, 2012, 94, 348–352 CrossRef CAS PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- D. S. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS PubMed.
- V. M. Yuwono, N. D. Burrows, J. A. Soltis and R. L. Penn, J. Am. Chem. Soc., 2010, 132, 2163–2165 CrossRef CAS PubMed.
- C. Frandsen, B. A. Legg, L. R. Comolli, H. Z. Zhang, B. Gilbert, E. Johnson and J. F. Banfield, CrystEngComm, 2014, 16, 1451–1458 RSC.
- A. L. Drummond, N. C. Feitoza, G. C. Duarte, M. J. A. Sales, L. P. Silva, J. A. Chaker, A. F. Bakuzis and M. H. Sousa, J. Nanosci. Nanotechnol., 2012, 12, 8061–8066 CrossRef CAS PubMed.
- E. Matijevic, Chem. Mater., 1993, 5, 412–426 CrossRef CAS.
- J. Park, V. Privman and E. Matijevic, J. Phys. Chem. B, 2001, 105, 11630–11635 CrossRef CAS.
- R. M. Cornell and U. Schertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrence and Uses, VCH Publishers, Weinheim, 2003 Search PubMed.
- J. L. Till, Y. Guyodo, F. Lagroix, G. Morin and G. Ona-Nguema, Geology, 2015, 43, 75–78 CrossRef.
- M. Usman, M. Abdelmoula, P. Faure, C. Ruby and K. Hanna, Geoderma, 2013, 197, 9–16 CrossRef.
- E. Tronc, P. Belleville, J. P. Jolivet and J. Livage, Langmuir, 1992, 8, 313–319 CrossRef CAS.
- L. Garcell, M. P. Morales, M. Andres-Verges, P. Tartaj and C. J. Serna, J. Colloid Interface Sci., 1998, 205, 470–475 CrossRef CAS PubMed.
- M. A. Verges, R. Costo, A. G. Roca, J. F. Marco, G. F. Goya, C. J. Serna and M. P. Morales, J. Phys. D: Appl. Phys., 2008, 41, 10 Search PubMed.
- N. C. Feitoza, T. D. Goncalves, J. J. Mesquita, J. S. Menegucci, M. Santos, J. A. Chaker, R. B. Cunha, A. M. M. Medeiros, J. C. Rubim and M. H. Sousa, J. Hazard. Mater., 2014, 264, 153–160 CrossRef CAS PubMed.
- C. M. Flynn, Chem. Rev., 1984, 84, 31–41 CrossRef CAS.
- T. Sugimoto and Y. S. Wang, J. Colloid Interface Sci., 1998, 207, 137–149 CrossRef CAS PubMed.
- J. A. Gomes, M. H. Sousa, G. J. da Silva, F. A. Tourinho, J. Mestnik, R. Itri, G. D. Azevedo and J. Depeyrot, J. Magn. Magn. Mater., 2006, 300, E213–E216 CrossRef CAS.
- E. C. Sousa, C. R. Alves, R. Aquino, M. H. Sousa, G. F. Goya, H. R. Rechenberg, F. A. Tourinho and J. Depeyrot, J. Magn. Magn. Mater., 2005, 289, 118–121 CrossRef CAS.
- E. L. Verde, G. T. Landi, M. S. Carriao, A. L. Drummond, J. A. Gomes, E. D. Vieira, M. H. Sousa and A. F. Bakuzis, AIP Adv., 2012, 2, 23 Search PubMed.
- E. L. Verde, G. T. Landi, J. A. Gomes, M. H. Sousa and A. F. Bakuzis, J. Appl. Phys., 2012, 111, 8 CrossRef.
- R. Hiergeist, W. Andra, N. Buske, R. Hergt, I. Hilger, U. Richter and W. Kaiser, J. Magn. Magn. Mater., 1999, 201, 420–422 CrossRef CAS.
- F. L. Rodovalho, G. Capistrano, J. A. Gomes, F. F. Sodre, J. A. Chaker, A. F. C. Campos, A. F. Bakuzis and M. H. Sousa, Chem. Eng. J., 2016, 302, 725 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |