
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular packing and morphological stability of dihydro-indeno[1,2-b]fluorenes in the context of their substitution pattern†
M. Hempe and
M. Reggelin *
*
Clemens-Schöpf-Institut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Alarich-Weiss-Straße 4, 64287 Darmstadt, Germany. E-mail: re@chemie.tu-darmstadt.de
First published on 6th October 2017
Abstract
The synthesis of a series of dihydroindeno[1,2-b]fluorene (IF) derivatives with various side chain substituents is reported, these have been investigated by single-crystal X-ray analysis in terms of their molecular packings and the thermal stability of these morphologies observed by DSC measurements. It is shown that symmetrically substituted IFs bearing longer linear aliphatic side chains tend to crystallize in thermolabile coplanar layers, in which the aliphatic and dihydroindeno[1,2-b]fluorene core segments are spatially segregated. In contrast to that, the attachment of aryl side chains to the dihydroindenofluorene core results in a stabilization of the cofacial morphology, which can be observed by the absence of thermal phase transitions. In addition to this, asymmetrically substituted derivatives, so called “mixed indenofluorenes” (MIFs), bearing pairwise linear aliphatic side chains of variable length, as well as aryl substituents have been synthesized. These derivatives tend to exhibit thermostable morphologies with an enhanced tendency to form edge-to-face contacts of the dihydroindenofluorene structures.
Introduction
One of the most important correlations in the field of optoelectronic organic materials is the interplay between the optical properties and the morphology of the molecules in the bulk of a solid state film.1 It is well known that the morphology of the molecules has an impact on the charge carrier and exciton dynamics2–5 and can lead to excimers, which may influence the pristine emission color and intensity upon excitation.6A powerful strategy to utilize this behavior for the rational design of high-performance materials has been termed sidechain engineering and was successfully applied in the field of organic photovoltaic devices.7–10 Furthermore, the influence of side chain substituents on the emission properties of polyfluorenes has been intensively investigated. Besides the stabilization of the deep blue emission color by use of aryl substituents,11 it has been shown that thin films of alkylated polyfluorenes are prone to the formation of polymorphs with distinct optical properties.12–16
Shifting the blue emission color into a spectral region in which the human eye is more sensitive, higher homologs of bridged poly-para-phenylene derivatives have been developed. The scaffold of dihydroindeno[1,2-b]fluorenes was used for the design of both, small molecule and polymer light-emitting materials.17–19 It has been shown by Müllen et al., that there is a connection between the molecular structure, the microscopic morphology and the optical properties of poly(dihydroindenofluorenes) (PIFs).20 In addition to this, theoretical studies demonstrated that the energy transfer between the polymer chains of PIFs is dependent on the interchain distance of the polymers and can be very efficient.21,22 Besides the intermolecular distance between chromophores the relative orientation of the emitting molecules is of paramount importance. It has been shown, that a cofacial arrangement of the emitting aromatic motifs in combination with an intermolecular face-to-face distance of 3–4 Å can lead to the formation of aggregates, which may determine and alter the emission properties of their thin films.6,23 Therefore, efficient and color pure emitting materials should provide a high morphological stability of their amorphous solid state, which can be observed by the absence of thermal phase transitions.24
To learn more about the molecular packing and the morphological stability of dihydroindeno[1,2-b]fluorene-based materials with respect to their substitution pattern, a series of monomeric derivatives with different side chain substituents has been synthesized in this work. The set of molecules covers symmetrically substituted dihydroindenofluorenes (IFs) with linear aliphatic side chains of variable length, as well as molecules bearing sterically demanding tert-butylphenyl substituents. In addition, asymmetrically substituted derivatives, so called “mixed indenofluorenes” (MIFs), compromising pairwise linear aliphatic side chains of variable length, as well as aryl substituents have been synthesized. The molecular packing behavior of these derivatives was investigated by single-crystal X-ray analysis and was correlated to their thermal behavior observed by DSC measurements.
Results & discussion
Synthesis
The synthesis of the symmetrically substituted dihydroindenofluorenes was achieved starting from the 2,5-diphenylterephthalic ester 1, following two strategies based on reaction sequences reported in the literature (Scheme 1).17,18,25 Along route A, the terephthalic ester 1 was transformed to the biscarbinols 2 by the attack of metallated carbon nucleophiles. Without further purification, the biscarbinols 2 were subsequently cyclized to the dihydroindeno[1,2-b]fluorenes. It is noteworthy that the cyclization reaction using hydrochloric acetic acid (method 1) was only feasible in the case of the aryl substituted IFs. The synthesis of alkylated IFs was achieved by using methanesulfonic acid and poly phosphoric acid (method 2) under mild reaction conditions.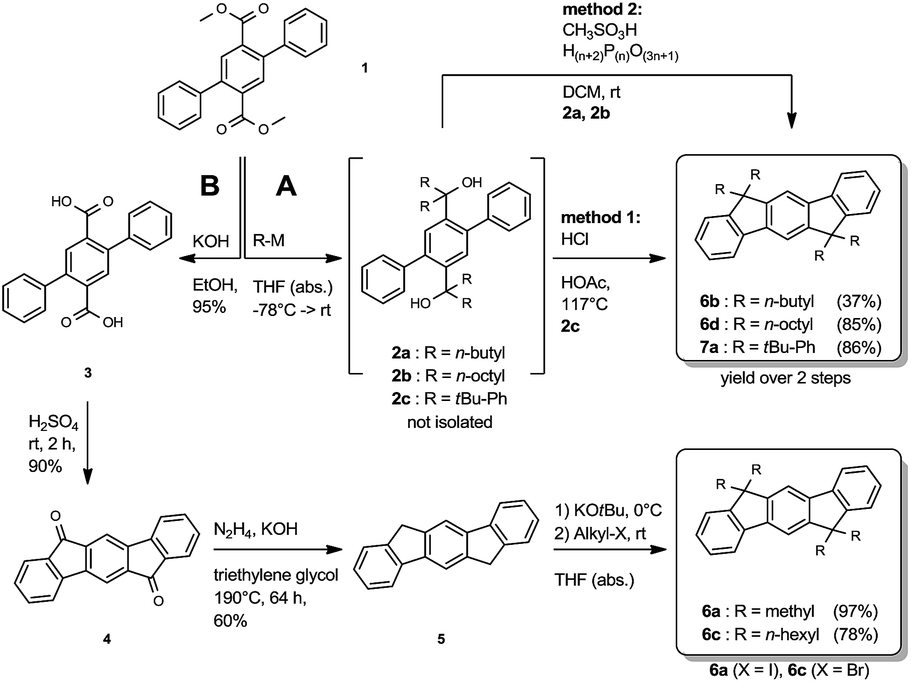 | ||
| Scheme 1 Reaction sequence of the symmetrically substituted dihydroindeo[1,2-b]fluorenes. | ||
Alternatively, alkylated IFs have been synthesized using a different strategy based on the work of Deuschel and Chardonnens (Route B).26,27 Following this reaction sequence, the terephthalic ester 1 was saponified and the resulting terephthalic acid 3 subsequently cyclized to the diketone 4 using sulfuric acid.28 The diketone 4 was reduced to the dihydroindenofluorene 5 in a Wolff-Kishner reaction28 and afterwards alkylated using a reaction protocol by Lardíes et al.29
The synthesis of the mixed substituted dihydroindenofluorene derivatives was achieved starting from 2-bromo fluorene 8 (Scheme 2). In a sequenced one-pot Miyaura borylation/Suzuki–Miyaura cross-coupling reaction cascade the corresponding fluorenyl pinacole boronate ester was generated in situ, which afterwards was reacted with 2-bromo-methylbenzoate 9 leading to 2-fluorenyl-methylbenzoate 10.
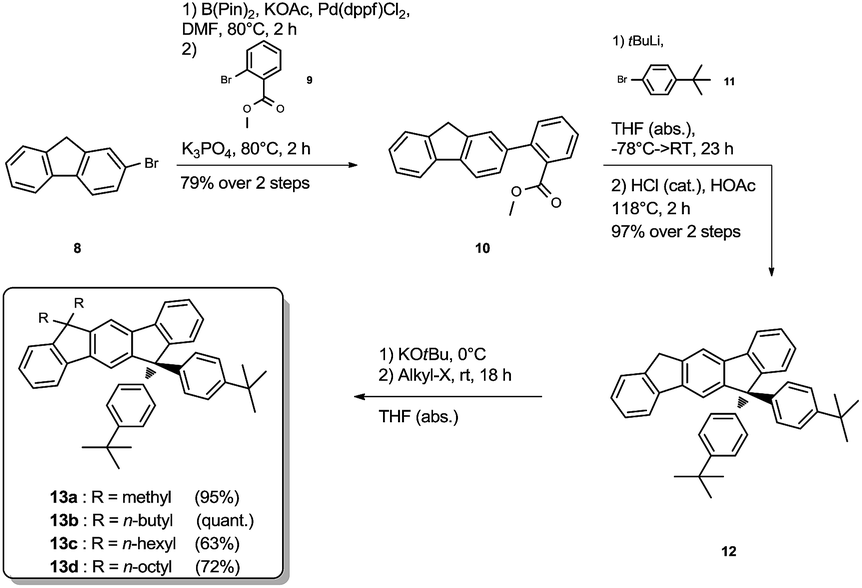 | ||
| Scheme 2 Synthesis of the mixed substituted derivatives 13a–d. | ||
Here, the use of the stronger base K3PO4 upon the addition of the electrophile 9 promotes the Suzuki–Miyaura cross-coupling reaction, leading to the benzoate 10 in good yield over two steps. Using an excess amount of the metallated species of 4-bromo-tert-butylbenzene 11, compound 10 was converted to the unisolated carbinol intermediate, which was subsequently cyclized to the dihydroindeno[1,2-b]fluorene scaffold 12. The derivatization of compound 12 was achieved in analogy to the unsubstituted dihydroindenofluorene 5, yielding the homologous series of methyl-MIF 13a, butyl-MIF 13b, hexyl-MIF 13c, and octyl-MIF 13d.
Single-crystal X-ray analysis
The influence of the geminal substituents on the molecular geometry (Fig. 1) and packing behavior (Fig. 2) of the symmetrically substituted derivatives (6a–d and 7a), as well as of the mixed substituted compounds methyl-MIF 13a and hexyl-MIF 13c were investigated using single-crystal X-ray analysis (for details, see the ESI† and Table 1). Compounds butyl-MIF 13b and octyl-MIF 13d were obtained as fine powders only.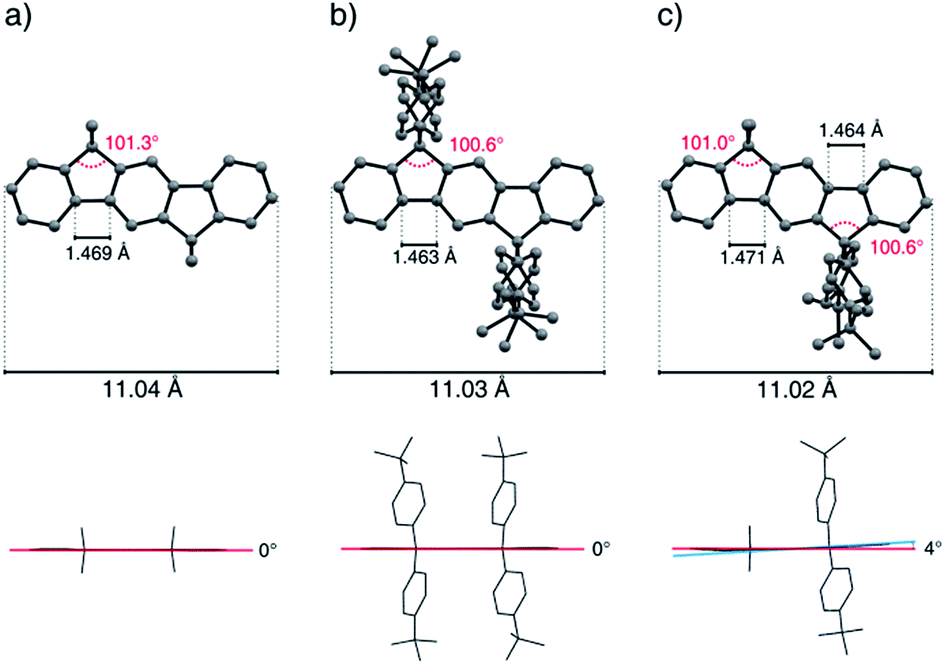 | ||
| Fig. 1 Exemplary dihydroindeno[1,2-b]fluorene geometries as a function of the geminal substitution pattern. (a) Methyl-IF 6a, (b) tBuPh-IF 7a, (c) methyl-MIF 13a (H atoms have been omitted for clarity). | ||
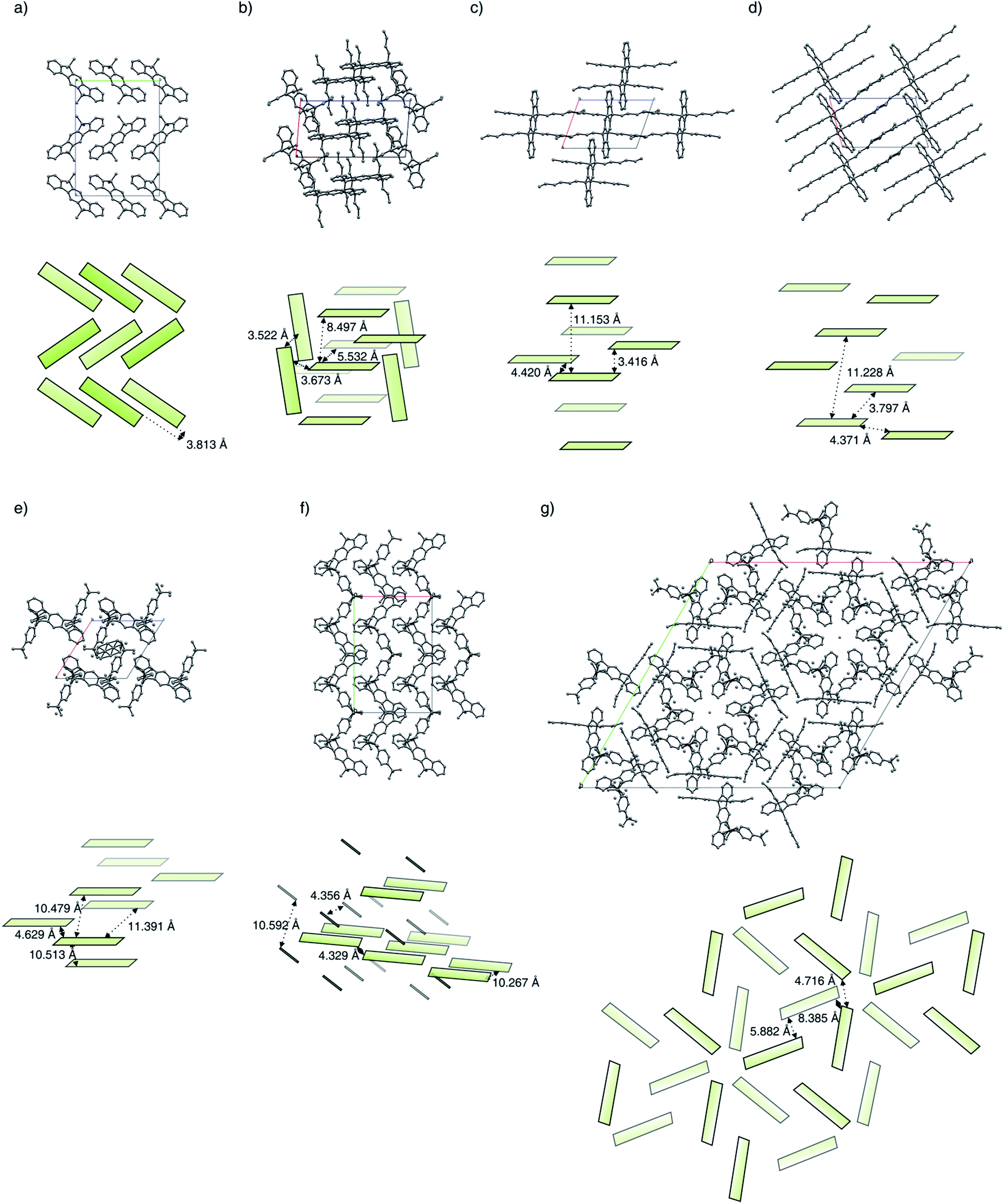 | ||
| Fig. 2 Molecular packings obtained by single-crystal X-ray analysis and illustrations with intermolecular distances (H atoms have been omitted for clarity). (a) Methyl-IF 6a, (b) butyl-IF 6b, (c) hexyl-IF 6c, (d) octyl-IF 6d, (e) tBuPh-IF 7a, (f) methyl-MIF 13a, (g) hexyl-IF 13c. | ||
| Methyl-IF 6a | Butyl-IF 6b | Hexyl-IF 6c | Octyl-IF 6d | tBuPh-IF 7a | Methyl-MIF 13a | Hexyl-MIF 13c | |
|---|---|---|---|---|---|---|---|
| Formula | C24H22 | C36H46 | C44H26 | C52H78 | C60H62 | C42H42 | C52H62 |
| MW [g mol−1] | 310.41 | 478.73 | 590.93 | 703.14 | 903.19 | 546.75 | 687.05 |
| Crystal system | Orthorhombic | Triclinic | Triclinic | Triclinic | Triclinic | Monoclinic | Trigonal |
| Space group | Pbca | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
Cc | R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| a [Å] | 6.7189(8) | 9.350(3) | 8.453(1) | 8.502(1) | 11.660(4) | 15.416(2) | 43.677(2) |
| b [Å] | 13.926(1) | 13.930(4) | 10.868(1) | 10.722(2) | 11.794(3) | 20.078(2) | 43.677(2) |
| c [Å] | 19.061(2) | 19.084(4) | 11.903(2) | 14.069(2) | 11.944(3) | 12.162(2) | 12.140(1) |
| α [deg] | 90 | 109.98(3) | 92.85(1) | 82.38(2) | 102.39(2) | 90 | 90 |
| β [deg] | 90 | 91.28(2) | 108.07(1) | 73.88(2) | 115.41(3) | 118.63(2) | 90 |
| γ [deg] | 90 | 98.95(2) | 104.44(1) | 75.30(2) | 107.53(3) | 90 | 120 |
| V [Å3] | 1783.49 | 2299.92 | 997.087 | 1189.9 | 1297.12 | 3304.15 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 056.5 056.5 |
| Z | 4 | 3 | 1 | 1 | 1 | 4 | 3 |
| ρcalc [g cm−3] | 1.156 | 1.037 | 0.984 | 0.982 | 1.156 | 1.099 | 1.028 |
| μ [mm−1] | 0.065 | 0.058 | 0.055 | 0.054 | 0.065 | 0.062 | 0.058 |
| T [K] | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) |
| Θmin–Θmax [deg} | 2.925–25.184 | 2.570–25.183 | 2.639–25.186 | 2.560–25.184 | 2.941–25.349 | 2.785–25.185 | 2.468–25.187 |
| R/wR [I > 2σ(I)] | 0.0691/0.1233 | 0.0769/0.1227 | 0.0608/0.1356 | 0.1538/0.2016 | 0.1147/0.2738 | 0.0478/0.0957 | 0.0910/0.2535 |
The total length (C2–C12 distance) of the indeno[1,2-b]fluorene scaffold was determined to be 11.05(3) Å and displays only minor variations by deformations resulting from packing effects. The bond angle of the sp3-bridging carbon atoms to the carbon atoms of the aromatic rings shows a slightly distorted tetrahedral geometry and has been determined in the range of 100.4° and 101.6°. In the case of the fourfold alkylated derivatives, the length of the terphenyl C–C-bond axis of the aromatic backbone was measured to be 1.469 Å to 1.473 Å, while this bond was shortened to 1.463 Å in the case of the aryl substituted compound tBuPh-IF 7a. Depending on the kind of substituents, the mixed substituted derivatives show bond length patterns which match those of the corresponding symmetrically substituted compounds.
Because of packing effects, the compounds methyl-MIF 13a and hexyl-MIF 13c (see ESI†) show a deformation of the dihydroindeno[1,2-b]fluorene backbone, which is almost perfectly planar in the case of the symmetric substituted derivatives (6a–6d and 7a). The tetra-methylated dihydroindeno[1,2-b]fluorene 6a crystallizes orthorhombically in the space group Pbca. The molecules within a unit cell arrange in a “herringbone”-type structure, exhibiting a C–C-distance of 3.813 Å as the shortest intermolecular contact. In this arrangement, the molecular packing might be stabilized by C–H⋯π-interactions.
The derivative butyl-IF 6b crystallizes triclinically in the space group P, as already reported by Zhang et al.30 Within the molecular packing, two coplanar arranged molecules are surrounded by four molecules in an almost orthogonal but among themselves cofacial alignment. These molecules are slightly shifted towards each other with an intermolecular C–C-distance of 3.522 Å (π–π-stacking).
The derivative hexyl-IF 6c crystallizes triclinically in the space groupe P. In an unit cell, two coplanar arranged molecules form layers in which the molecules are slightly shifted towards each other. The aromatic systems within the molecular packing exhibit a minimal intermolecular edge-to-edge-distance of 4.420 Å and a minimal interlayer distance of 3.416 Å.
Similarly to the hexyl-substituted compound hexyl-IF 6c, the derivative octyl-IF 6d crystallizes in the space group P (triclinic) as well. The molecules within the elementary cell arrange in the same orientation as in the case of the hexyl-IF 6c, whereas the interlayer distance expands from 11.153 Å to 11.228 Å. Within the molecular packing, the closest intermolecular edge-to-edge contacts are identified to be 3.797 Å, while the minimal interlayer distance is 3.390 Å.
As is the case for the aliphatic substituted derivatives, the tetraaryl substituted compound tBuPh-IF 7a crystallizes triclinically in the space group P. Within the molecular packing, the dihydroindeno[1,2-b]fluorene moieties arrange in a coplanar orientation, in which the sterically demanding tert-butylphenyl side chains form a cavity that hosts a solvent molecule within the crystals analyzed. The minimal intermolecular edge-to-edge distance was identified to be 4.629 Å, while the interlayer distances were determined to 10.479 Å and 10.513 Å. Comparing the molecular arrangement of tBuPh-IF 7a with the dispirofluorene-substituted dihydroindeno[1,2-b]fluorene DSF-IF reported by Poriel et al.,19 it can be observed that the fluorene substituents of DSF-IF lead to a edge-to-face orientation of the dihydro-indenofluorene core structures, while in the case of the tetraaryl-substituted derivative 7a a preservation of the strictly coplanar orientation can be observed. This behaviour is in good agreement with the molecular packing of the tetra-(4-tolyl)-substituted derivative 3Ph reported by Wong et al., adopting a intermolecular edge-to-edge distance of 3.58 Å.31
The mixed substituted derivative methyl-MIF 13a crystallizes monoclinically in the space group Cc. The four dihydroindeno[1,2-b]fluorenes of one unit cell arrange in pairs of lamellas, where the two layers are rotated towards each other. The intermolecular distances between the molecules of the two kinds of lamellas have been measured to be 10.267 Å and 10.592 Å, while the minimal edge-to-edge distance is 4.329 Å.
The molecules of the derivative hexyl-MIF 13c crystallize trigonally in the space group R. The molecules within a unit cell arrange in a layered and stacked radial orientation of three dihydroindeno[1,2-b]fluorene molecules per layer. The minimal intermolecular distance within one layer is 4.716 Å, while the intermolecular distance between two layers expands to 8.385 Å.
Along the investigated series of molecules it can be observed that by the extension of the alkyl side chains, a widening of the molecular packing takes place. In the case of the symmetrically substituted derivatives, the close “herringbone”-style packing mode of the methyl-IF 6a changes to a strictly coplanar orientation of the dihydroindenofluorene segments for compounds 6b–6d. This orientation can also be observed in the case of the tetraaryl substituted derivative tBuPh-IF 7a, while the mixed substituted derivatives methyl-MIF 13a and hexyl-MIF 13c tend to exhibit intercalated structures.
Thermal behavior (DSC)
To investigate the influence of the geminal substituents on the thermal behavior of the dihydroindeno[1,2-b]fluorenes synthesized, differential scanning calorimetry (DSC) measurements have been performed (Fig. 3).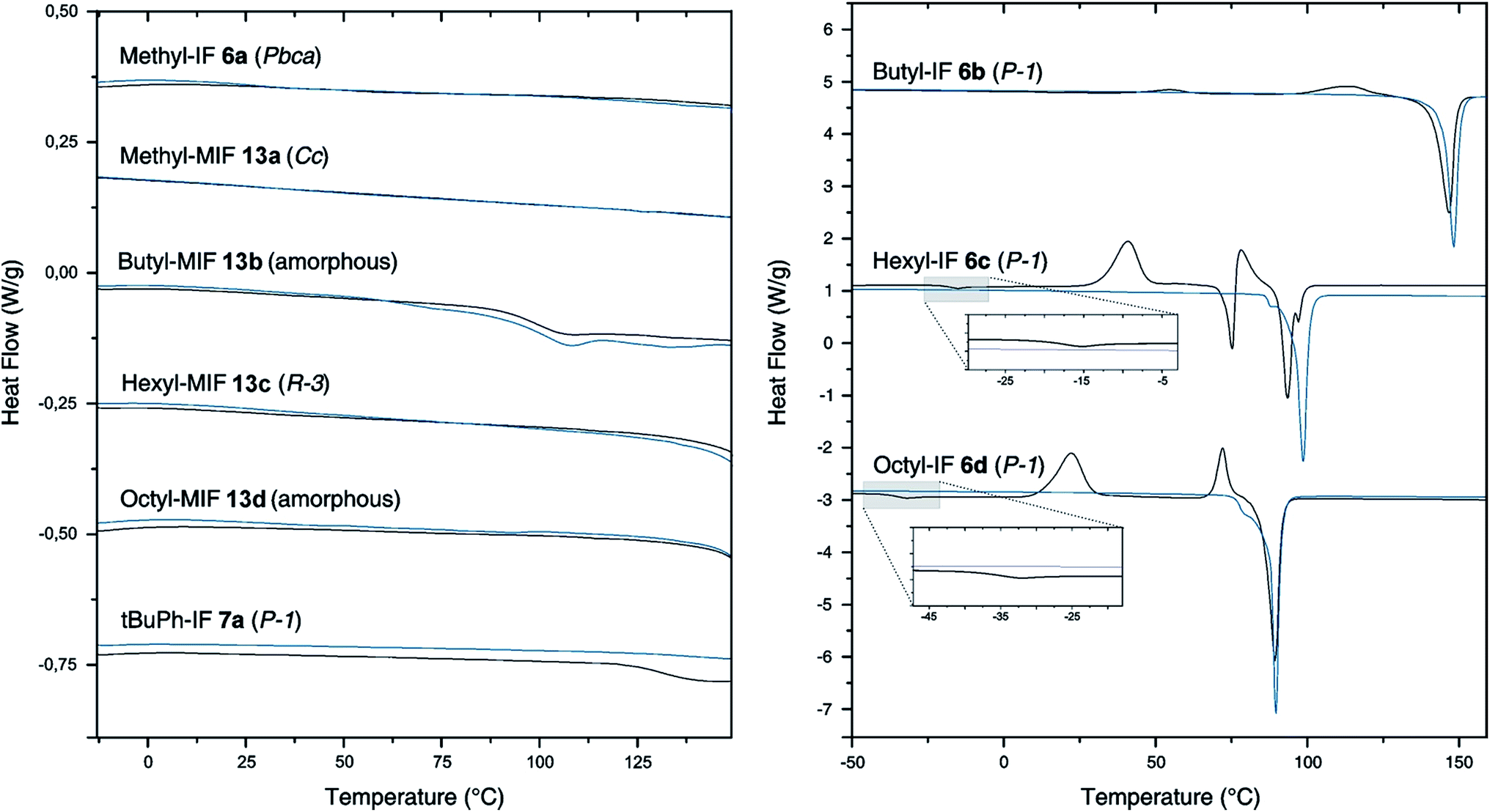 | ||
| Fig. 3 Differential scanning calorimetry (DSC) of the derivatives methyl-IF 6a, butyl-IF 6b, hexyl-IF 6c, octyl-IF 6d, tBuPh-IF 7a, methyl-MIF 13a, butyl-MIF 13b, hexyl-MIF 13c and octyl-MIF 13d (N2-atmosphere, 10 K min−1, black line: first heating cycle, blue line: second heating cycle). The traces have been shifted vertically for clarity. | ||
The thermogram of the symmetrically substituted derivative methyl-IF 6a shows no thermal phase transitions in the range between −10 °C and 200 °C. In sharp contrast to this, the thermograms of the symmetrically substituted derivatives with longer linear aliphatic side chains (butyl-IF, hexyl-IF, octyl-IF) exhibit several signals in their first heating cycle. The measurements reveal weak endothermic signals at −18 °C and −36 °C, which can be correlated to glass transitions of the hexyl-IF 6c and octyl-IF 6d derivatives. In addition to this, the thermograms of the three derivatives 6b–d show further exothermic signals below their melting temperature, which can be assigned to recrystallization processes and the formation of mesophases that already have been reported by Elmahdy et al. in the case of octyl-substituted dihydroindenofluorene mono- and oligomers.32 Furthermore, the DSC curve of the hexyl-IF 6c exhibits one additional endothermic signal at a temperature of 73 °C, which merges into the second exothermic recrystallization signal. The thermograms of the second heating cycle of the tetraalkylated dihydroindenofluorenes butyl-IF 6b, hexyl-IF 6c and octyl-IF 6d only exhibit endothermic signals, which have been assigned to the melting points of the compounds at 143 °C (butyl-IF), 95 °C (hexyl-IF) and 87 °C (octyl-IF).
In comparison to the tetraalkylated derivatives, a morphological stabilization can be observed in the case of the tert-butylphenyl substituted compounds. The thermogram of the first heating cycle of the derivative tBuPh-IF 7a only exhibits a weak endothermic signal, which was assigned to the glass transition of the material. The stabilization effect of the aryl side chains can be observed in the case of the mixed dihydroindenofluorenes as well. The derivatives methyl-MIF 13a, butyl-MIF 13b, hexyl-MIF 13c and octyl-MIFs 13d did not show any significant thermal transitions in the temperature range between −10 °C and 150 °C. Note that only the butyl-substituted compound butyl-MIF 13b shows a weak endothermic signal at 89 °C, which could not be assigned.
Discussion
We have observed that the tetramethyl-substituted derivative methyl-IF 6a crystallizes in a “herringbone”-type structure, while longer linear aliphatic substituents lead to a widening of this close packing mode and result in a coplanar arrangement of the terphenylic systems. In the course of that, a spatial segregation of aliphatic and aromatic segments takes place, which was already reported by Elmahdy et al. in the case of oligoindenofluorenes.32 The interlayer distance of this alignment increases along the homologous series of the derivatives butyl-IF 6b, hexyl-IF 6c and octyl-IF 6d. The substitution of the dihydroindeno[1,2-b]fluorene scaffold with four tert-butylphenyl groups also results in the orientation of the molecules in parallel layers, while the intermolecular edge-to-edge-distance within a layer further increases in comparison to the aliphatic-substituted derivatives.In contrast to the symmetric substitution pattern, the mixed substituted dihydroindenofluorenes methyl-MIF 13a and hexyl-MIF 13c tend to crystallize in lamellar, intercalating structures with an increased number of edge-to-face contacts of the dihydroindenofluorene core structures.
The widening of the molecular packing with increasing linear aliphatic side chains directly corresponds to the thermal behavior of the derivatives, which has been examined by DSC measurements. For the compound methyl-IF 6a, which crystallizes in a “herringbone”-type structure, no thermal phase transition could be observed up to a temperature of 150 °C. In contrast to this, the coplanar arrangement of the symmetrically substituted molecules of longer aliphatic side chain lengths results in a thermolabile morphological behavior, which leads to the existence of polymorphs and rather low melting points.
In accordance with the observations of the symmetrically aliphatic substituted derivatives, the attachment of four tert-butylphenyl groups to the dihydroindenofluorene core leads to coplanar molecular packing. Unlike the thermolabile character of the aliphatic substituents, the rigid nature of the aryl side chains results in a stabilization of the morphology of the material.
The stabilizing effect of the aryl side chains can be observed in the case of the mixed substituted derivatives, as well. The thermograms of the compounds methyl-MIF 13a, butyl-MIF 13b, hexyl-MIF 13c and octyl-MIF 13d lack any significant features up to a temperature of 150 °C. A possible explanation of this behavior can be found in the enhanced tendency of the molecules to form edge-to-face contacts of the dihydroindenofluorene core structures, which could be observed in the solid-state structures of the derivatives methyl-MIF 13a and hexyl-MIF 13c. In that case, the aryl side chains disturb the spatial segregation between the aliphatic and aromatic segments and result in an interlocked arrangement of the molecules within the molecular packing. The rigidity of the aryl side chains as well as the enlargement of the intermolecular contact surface of the aromatic systems could be the reason for the higher morphological stability and the absence of thermolabile mesophases.
Conclusions
This work offers insights into the molecular packing and the morphological stability of dihydroindeno[1,2-b]fluorenes in dependence of their substitution pattern.A series of dihydroindeno[1,2-b]fluorenes with various substituents has been synthesized and was investigated in terms of the thermal stability of their morphology. A correlation of the geometry and molecular packing with the thermal behavior of the derivatives has been observed. It was found that symmetrically substituted IFs bearing longer linear aliphatic side chains tend to crystallize in coplanar layers, in which the aliphatic side chains and aryl core segments spatially segregate. This segregation results in the formation of thermolabile mesophases, which have been detected by DSC measurements. In contrast to aliphatic substituents, the attachment of aryl side chains results in a stabilization of the morphology, which can be observed by the absence of thermal phase transitions in the temperature range investigated. This kind of behavior has previously been identified as one important material property for the improvement of OLED lifetimes.24,33–35 Therefore, the results obtained will be valuable for the development of future dihydroindeno[1,2-b]fluorene-based materials and may be transferable to related structures and applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to thank Sabine Foro (Technical University of Darmstadt) for performing the single-crystal X-ray analysis and Christian Rüttiger (Rehahn group, Technical University of Darmstadt) for conducting the DSC measurements. Generous support by Merck KGaA (Darmstadt) is gratefully acknowledged.Notes and references
- P. E. Keivanidis, J. Jacob, L. Oldridge, P. Sonar, B. Carbonnier, S. Baluschev, A. C. Grimsdale, K. Müllen and G. Wegner, ChemPhysChem, 2005, 6, 1650–1660 CrossRef CAS PubMed.
- C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef CAS.
- M. Surin, E. Hennebicq, C. Ego, D. Marsitzky, A. C. Grimsdale, K. Müllen, J.-L. Brédas, R. Lazzaroni and P. Leclère, Chem. Mater., 2004, 16, 994–1001 CrossRef CAS.
- B.-G. Kim, E. J. Jeong, J. W. Chung, S. Seo, B. Koo and J. Kim, Nat. Mater., 2013, 12, 659–664 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CAS.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- X. Xu, Y. Wu, J. Fang, Z. Li, Z. Wang, Y. Li and Q. Peng, Chem.–Eur. J., 2014, 20, 13259–13271 CrossRef CAS PubMed.
- X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2013, 25, 5467–5472 CrossRef CAS PubMed.
- T. W. Holcombe, J. E. Norton, J. Rivnay, C. H. Woo, L. Goris, C. Piliego, G. Griffini, A. Sellinger, J.-L. Brédas, A. Salleo and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 12106–12114 CrossRef CAS PubMed.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946–953 CrossRef CAS PubMed.
- S. H. Chen, A. C. Su and S. A. Chen, J. Phys. Chem. B, 2005, 109, 10067–10072 CrossRef CAS PubMed.
- M. Grell, D. D. C. Bradley, G. Ungar, J. Hill and K. S. Whitehead, Macromolecules, 1999, 32, 5810–5817 CrossRef CAS.
- M. Knaapila, F. B. Dias, V. M. Garamus, L. Almásy, M. Torkkeli, K. Leppänen, F. Galbrecht, E. Preis, H. D. Burrows, U. Scherf and A. P. Monkman, Macromolecules, 2007, 40, 9398–9405 CrossRef CAS.
- M. Knaapila, M. Torkkeli, F. Galbrecht and U. Scherf, Macromolecules, 2013, 46, 836–843 CrossRef CAS.
- D. W. Bright, F. B. Dias, F. Galbrecht, U. Scherf and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 67–73 CrossRef CAS.
- S. Setayesh, D. Marsitzky and K. Müllen, Macromolecules, 2000, 33, 2016–2020 CrossRef CAS.
- J. Jacob, J. Zhang, A. C. Grimsdale, K. Müllen, M. Gaal and E. J. W. List, Macromolecules, 2003, 36, 8240–8245 CrossRef CAS.
- C. Poriel, J.-J. Liang, J. Rault-Berthelot, F. Barrière, N. Cocherel, A. M. Z. Slawin, D. Horhant, M. Virboul, G. Alcaraz, N. Audebrand, L. Vignau, N. Huby, G. Wantz and L. Hirsch, Chem.–Eur. J., 2007, 13, 10055–10069 CrossRef CAS PubMed.
- A. C. Grimsdale, P. Leclère, R. Lazzaroni, J. D. MacKenzie, C. Murphy, S. Setayesh, C. Silva, R. H. Friend and K. Müllen, Adv. Funct. Mater., 2002, 12, 729–733 CrossRef CAS.
- D. Beljonne, G. Pourtois, Z. Shuai, E. Hennebicq, G. D. Scholes and J. L. Brédas, Synth. Met., 2003, 137, 1369–1371 CrossRef CAS.
- T. A. Papadopoulos, L. Muccioli, S. Athanasopoulos, A. B. Walker, C. Zannoni and D. Beljonne, Chem. Sci., 2011, 2, 1025–1032 RSC.
- G. Zeng, W.-L. Yu, S.-J. Chua and W. Huang, Macromolecules, 2002, 35, 6907–6914 CrossRef CAS.
- K. Naito and A. Miura, J. Phys. Chem., 1993, 97, 6240–6248 CrossRef CAS.
- S. Y. Cho, A. C. Grimsdale, D. J. Jones, S. E. Watkins and A. B. Holmes, J. Am. Chem. Soc., 2007, 129, 11910–11911 CrossRef CAS PubMed.
- W. Deuschel, Helv. Chim. Acta, 1951, 34, 2403–2416 CrossRef CAS.
- L. Chardonnens and L. Salamin, Helv. Chim. Acta, 1968, 51, 1095–1102 CrossRef.
- S. Merlet, M. Birau and Z. Y. Wang, Org. Lett., 2002, 4, 2157–2159 CrossRef CAS PubMed.
- E. Cerrada, M. Laguna and N. Lardíes, Eur. J. Inorg. Chem., 2009, 2009, 137–146 CrossRef.
- G.-H. Zhang, D.-X. Cao, H.-Y. Chen and Z.-Q. Liu, Z. Kristallogr. – New Cryst. Struct., 2007, 310–312, DOI:10.1524/ncrs.2007.0131.
- K.-T. Wong, L.-C. Chi, S.-C. Huang, Y.-L. Liao, Y.-H. Liu and Y. Wang, Org. Lett., 2006, 8, 5029–5032 CrossRef CAS PubMed.
- M. M. Elmahdy, G. Floudas, L. Oldridge, A. C. Grimsdale and K. Müllen, ChemPhysChem, 2006, 7, 1431–1441 CrossRef CAS PubMed.
- E. Ettedgui, G. T. Davis, B. Hu and F. E. Karasz, Synth. Met., 1997, 90, 73–76 CrossRef CAS.
- H. Aziz, Z. Popovic, S. Xie, A.-M. Hor, N.-X. Hu, C. Tripp and G. Xu, Appl. Phys. Lett., 1998, 72, 756–758 CrossRef CAS.
- H. Spreitzer, H. W. Schenk, J. Salbeck, F. Weissörtel, H. Reil and W. Riess, in Proc. SPIE 3797, 1999, vol. 3797, pp. 316–324 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Seven X-ray crystallographic files of compounds 6a–d, 7a, 13a and 13c, experimental procedures and characterization data. CCDC 1568960–1568966. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09401a |
| This journal is © The Royal Society of Chemistry 2017 |