
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-locking strategy facilitating determination of absolute optical purity of 2-amino-1-butanol by reverse-phase high-performance liquid chromatography†
Bo Li‡
,
Jie Zhang‡,
Bei-Bei Yang,
Li Li * and
Xiao-Xiao Yang*
* and
Xiao-Xiao Yang*
Beijing Key Laboratory of Active Substances Discovery and Druggability Evaluation, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. E-mail: annaleelin@imm.ac.cn; Fax: +86-10-63165247; Tel: +86-10-63165247 Tel: +86-10-63165251
First published on 25th September 2017
Abstract
A concise and efficient reverse-phase high-performance liquid chromatography (RPLC) method has been established for absolute optical purity assay of 2-amino-1-butanol, which is an important synthetic intermediate of various drugs. Its enantiomers were derivatized with methyl (5-fluoro-2,4-dinitro-phenyl)-(S)-prolinate, which could be regarded as a structural variant of Marfey's reagent. The resultant diastereomers were further cyclized using bis(trichloromethyl)carbonate to form oxazolidin-2-one ring, leading to a distinct difference in their electronic circular dichroism (ECD) spectra. Time-dependent density functional theory (TDDFT) was then applied to simulate theoretical ECD spectra for unambiguous absolute configuration assignment of cyclized products without putative samples. Both pairs of diastereomers could be separated on a reverse-phase C18 column using a mobile phase of methanol or acetonitrile and water with UV detection at 336 nm. The tandem reactions could be integrated into one-pot synthesis with high yield. Thus, exact absolute optical purity of 2-amino-1-butanol could be directly determined by monitoring the peak ratios of these diastereomers. This method is well worth extending to the absolute optical purity assessment of other chiral amino alcohols.
Introduction
Amino alcohols are important structural elements that are prevalently stemming from natural products. With the continuous concern about exploring chiral molecules, enantiopure amino alcohols are drawing increasing attention in recent years.1 Their potentials were excavated to synthesize biologically active compounds or building blocks for catalysts and auxiliaries in asymmetric synthesis.2 Enantiomerically pure amino alcohols have also emerged as pivotal intermediates in medicinal chemistry (Fig. 1).3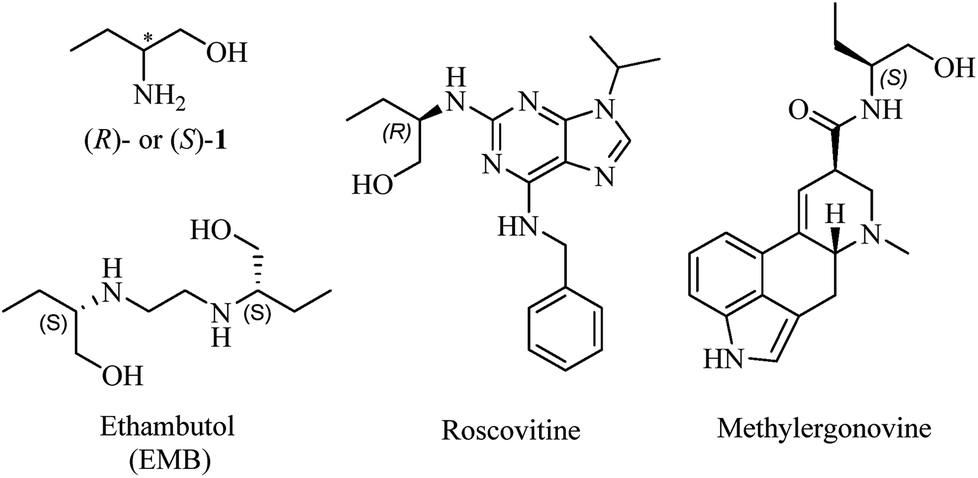 | ||
| Fig. 1 Chemical structures of 1 and drugs containing chiral 2-amino-1-butanol moiety. | ||
The most representative and immediate instance is the application of 2-amino-1-butanol (1, Fig. 1) to synthesize ethambutol (EMB), one of the front-line drugs against tuberculosis.4 (S,S)-EMB is the most effective agent against almost all the strains of Mycobacterium sps., causing an enhancement in activity of up to 200-fold than (R,R)-enantiomer and meso-isomer.5 Moreover, it is reported that optical impurity of (R,R)-EMB could lead to serious visual impairment, which is the main side-effect in the clinic application of EMB.6 The classical and widespread methods to synthesize EMB mainly involved reacting 1,2-dihaloethane with (S)-1.7 Accordingly, it is apparent that the enantiopurity of 1 dominates the optical purity of EMB. For the sake of gaining perfect antimycobacterial activity of EMB, the optical purity of (S,S)-configuration must be impeccable. Moreover, sustained efforts are carried out to explore derivatives of chiral 1 for more powerful antimycobacterial activity, with R isomer being more active than S isomer in some cases.8 Therefore, taking the above-mentioned reasons into account, it is imperative and significant to establish an efficient and concise method to determine the absolute optical purity of chiral 1.
Compound 1 is a highly polar molecule with low molecular weight and lack of sufficient UV chromophores. Conventional chromatographic analysis methods such as normal-phase high-performance liquid chromatography or gas-chromatography (GC) could only be utilized indirectly for its optical purity detection after derivatization using chiral stationary phase.9 There exists a few relevant reports of solutions to this thorny problem, such as use of 1,1′-bi-2-naphthol derivatives by virtue of NMR to determine their enantiomeric excesses (ee's), which showed up inevitable flaws in its accuracy and sensitivity.10 Tremendous efforts have also been made to seek for novel chirality sensing of amino alcohols using stereodynamic or fluorescence probes.11 Nevertheless, taking the advantages of accuracy and feasibility into consideration, RPLC could be a favourable choice for the rapid ee routine assessment in a laboratory.
It is widely shared that besides single-crystal X-ray diffraction, chiroptical spectroscopy including electronic circular dichroism (ECD) spectroscopy is an alternative way to establish absolute configurations of chiral molecules. In recent years, comparison of experimental ECD spectra with theoretical ECD curve obtained using quantum-chemical calculation and time-dependent density functional theory (TDDFT) has been widely applied in the absolute configuration assignments of chiral drugs and natural products.12
Marfey's reagent is regarded as the most successful derivatizing reagent for indirect resolution of chiral amino acids and amines on the standard reverse-phase column.13 It could also be used to determine the content of monoethanolamine in drug substance.14 The characteristics and applications of Marfey's reagent have been fully reviewed by Bhushan.15 However, to the best of our knowledge, there has been no report on ECD spectra of these diastereomeric derivatives and absolute configuration assignments without putative samples.
In this study, we describe a simple and efficient approach to determine absolute configuration and enantiomeric composition of 1 using RPLC without putative samples. This method is based on forming the corresponding epimeric derivatives upon reaction of 1 with methyl (5-fluoro-2,4-dinitrophenyl)-(S)-prolinate, which could be regarded as a structural variant of Marfey's reagent. Their optical purity and absolute configuration verification were achieved by RPLC and ECD spectra analysis adopting a ring-locking strategy, respectively. This methodology could competently determine the enantiopurity of chiral 1 as expected.
Results and discussion
Derivatization route
Synthetic route of diastereomers is depicted in Scheme 1. First, we used methyl (S)-prolinate as nucleophile reagent to quantitatively substitute one fluorine atom of 2. In this step, due to the strong electron-withdrawing effect of two nitro groups, nucleophilic aromatic substitution could readily take place to afford compound 3 with excellent yield and purity. The remaining fluorine atom of 3 was substituted by (S)/(R)-1 at a refluxing temperature in THF to form diastereomers 4a or 4b.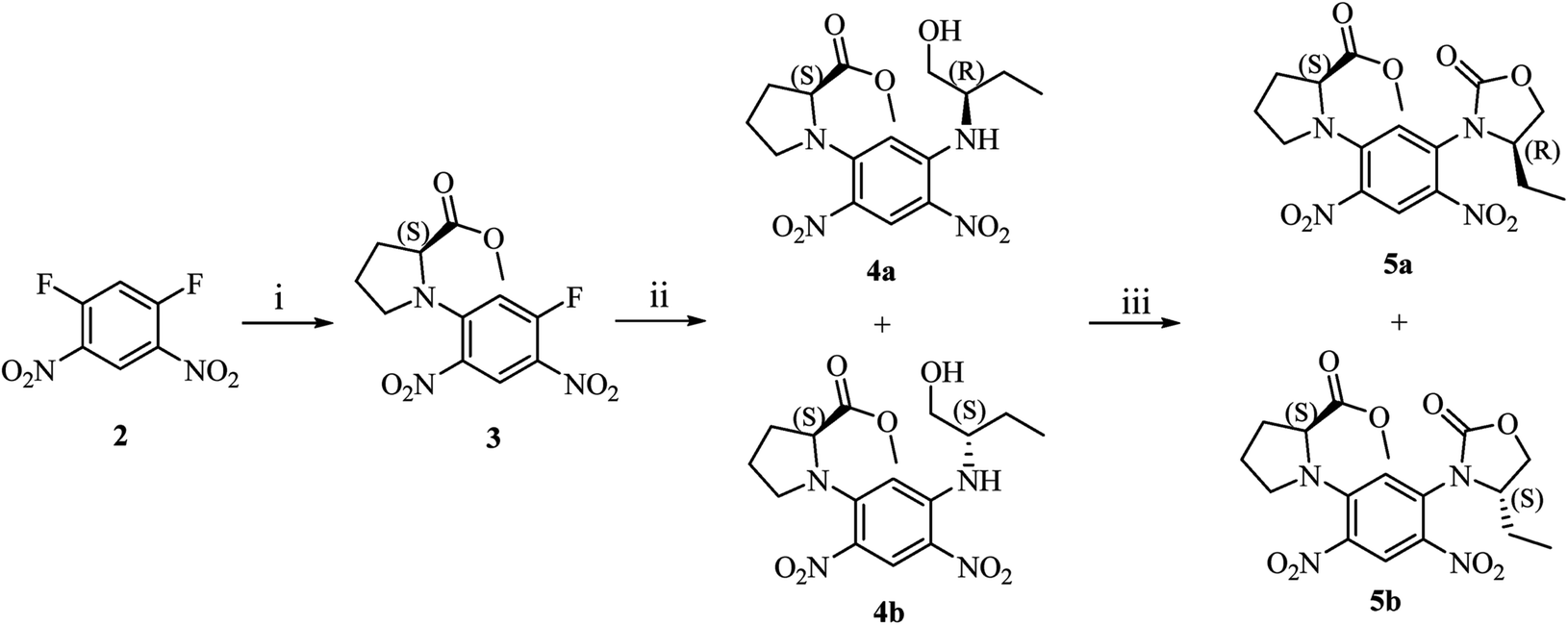 | ||
| Scheme 1 (i). Methyl (S)-prolinate hydrochloride, DIPEA, THF, r.t., 5 h; (ii). (S)/(R)-1, DIPEA, THF, refluxing, 2 h; (iii). bis(trichloromethyl) carbonate, DIPEA, dichloromethane, r.t., 1 h. | ||
With compounds 4a and 4b in hand, we tried to assign their absolute configurations by ECD spectroscopy. However, their ECD spectra were assumed to be fairly similar as shown in Fig. 2. It is due to the fact that the dominant chiral factor disturbing the aromatic chromophore was the chiral carbon atom on the pyrrolidine ring, rather than that on the flexible alkyl chain. Accordingly, a ring-locking strategy was proposed to tether amino and hydroxyl groups with a carbonyl group, which could lock the conformation of flexible alkyl chain near the chiral centre. This was achieved by adopting bis(trichloromethyl)carbonate as acylation reagent to construct five-membered heterocyclic compounds 5a and 5b.
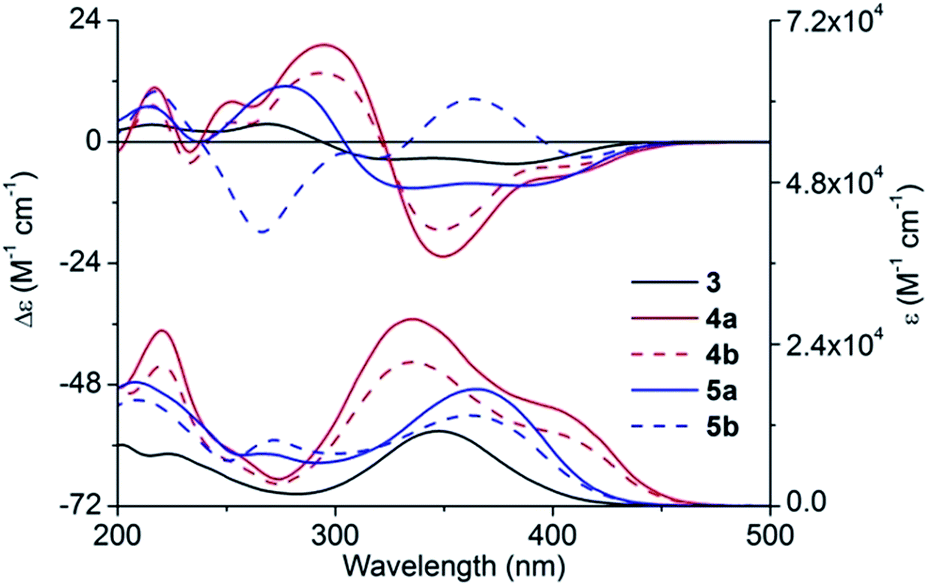 | ||
| Fig. 2 Experimental ECD and UV-vis absorption spectra of 3, 4a/4b, and 5a/5b in methanol. | ||
Experimental UV-vis absorption and ECD spectra
Experimental UV-vis absorption and ECD spectra of 3, 4a/4b and 5a/5b in methanol are demonstrated in Fig. 2. Substitution of a fluorine atom of 3 by (S)/(R)-1 led to the transformation of the strong absorption at 348.0 nm into two peaks, with one red shifting up to 50 nm (from 348 to 402 nm) and the other blue shifting to 335.5 nm. Moreover, the absorption intensity also increased significantly. However, the cyclization decreased the absorption intensity and a new peak was formed at about 270 nm, which corresponded to the n → π* electronic transition of 2-oxazolidone moiety.In ECD spectra, compound 3 showed two negative Cotton effects (CEs) in the wavelength range between 300 and 450 nm, and two positive CEs from 200 to 300 nm. As expected, 4a and 4b exhibited quite similar ECD curves, which were substantially different from that of 3. Two negative CEs of equal intensity above 300 nm transformed into one intense negative CE and a very weak negative CE of 4a/4b. Compound 5a showed a similar ECD pattern as 3 with part of CEs being red-shifted or blue-shifted about 10 nm. However, ECD spectrum of 5b possessed a positive CE at 363 nm and a negative CE at 266 nm, where all the other compounds showed CEs of opposite signs.
Structural elucidation
Quantum chemical calculation parameters might greatly influence calculation results; consequently, ECD calculation of 3 was first carried out with caution using various functionals and basis sets.16 In total, 28 conformers of (S)-3 were taken into account for the theoretical simulation (for main conformers, see Fig. S1, ESI†). All the theoretical ECD spectra of (S)-3 possessed a similar tendency as the experimental ECD curve. Amongst all, ECD prediction at the Cam-B3LYP/6-31 + G(d,p) level using the B3LYP/6-31G(d) geometries offered the optimal fit of CE positions and relative strengths after a red shift of 20 nm (Fig. 3).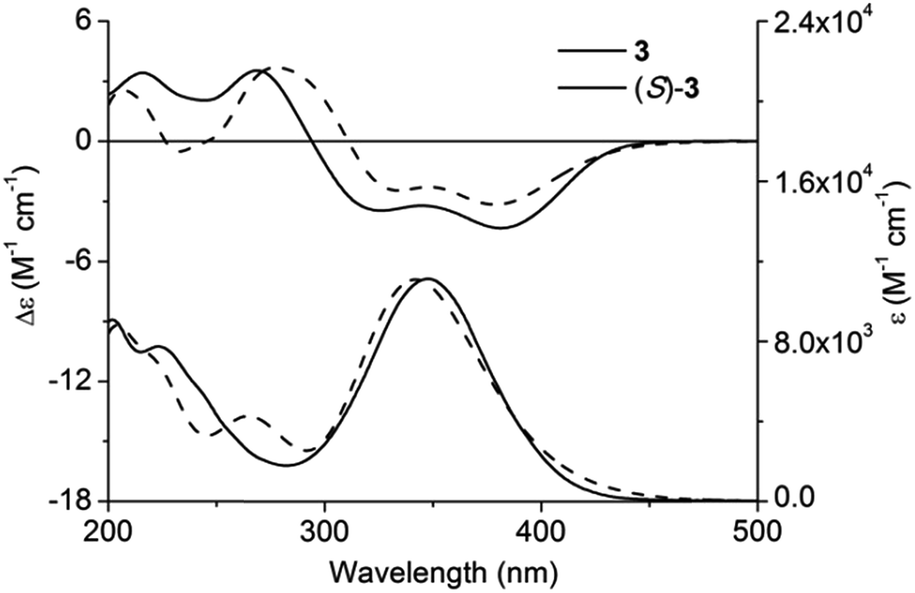 | ||
| Fig. 3 Experimental and calculated ECD and UV-vis absorption spectra of (S)-3 and 3 in methanol. | ||
Chiral centres of 4a and 4b were not involved during the cyclization step; accordingly, their absolute configurations could be inferred from 5a and 5b. Unlike ECD spectra of 4a and 4b, the remarkable difference between ECD spectra of 5a and 5b facilitated their absolute configuration assignment.
Adopting the above-mentioned calculation method, theoretical ECD spectra of (2S,5′R)-5 and (2S,5′S)-5 were obtained and compared with experimental ECD data of 5a and 5b (Fig. 4 and 5; for main conformers, see Fig. S2 and S3†). Thus, their absolute configurations were assigned by comparison of theoretical and experimental ECD spectra. Albeit, it could be anticipated that a better match could be achieved by selecting suitable functional and basis set conformations. It is clear that the absolute configuration of 5a is assigned as (2S,5′R), and that of 5b is (2S,5′S).
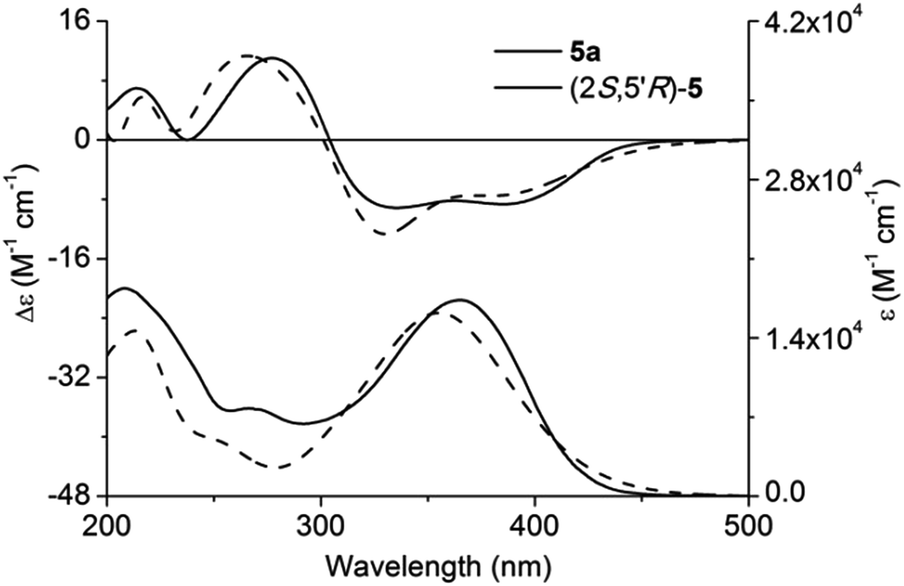 | ||
| Fig. 4 Experimental and calculated ECD and UV-vis absorption spectra of (2S,5′R)-5 and 5a in methanol. | ||
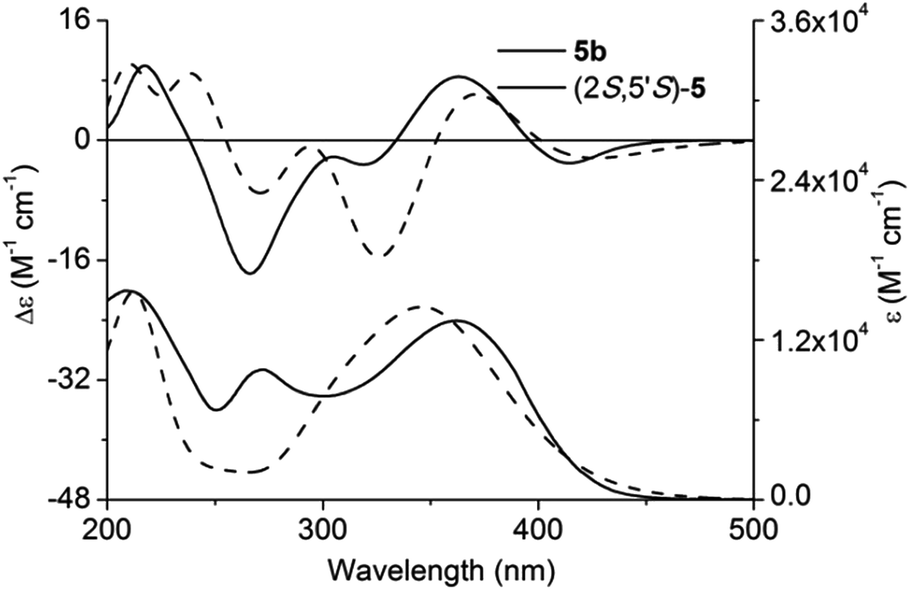 | ||
| Fig. 5 Experimental and calculated ECD and UV-vis absorption spectra of (2S,5′S)-5 and 5b in methanol. | ||
HPLC analysis of diastereomers
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) as a mobile phase allowed the most satisfactory separation of 4a and 4b with a maximum peak resolution with retention times of 15.27 min and 13.91 min, respectively (Fig. 6). Moreover, the equivalent peak areas of 4a and 4b demonstrated that the chirality of 1 exerted no effect on its chemical reactivity with 3, indicating that the optical purity of 1 was preserved after the conversion to 4a/4b. Moreover, the elution sequence changed as 5a (16.178 min) followed by 5b (18.165 min) after cyclization using a mobile phase of methanol–water (55:45). The optimal absorption signals in terms of signal-to-noise ratio and sensitivity were obtained at 336 nm.
:50) as a mobile phase allowed the most satisfactory separation of 4a and 4b with a maximum peak resolution with retention times of 15.27 min and 13.91 min, respectively (Fig. 6). Moreover, the equivalent peak areas of 4a and 4b demonstrated that the chirality of 1 exerted no effect on its chemical reactivity with 3, indicating that the optical purity of 1 was preserved after the conversion to 4a/4b. Moreover, the elution sequence changed as 5a (16.178 min) followed by 5b (18.165 min) after cyclization using a mobile phase of methanol–water (55:45). The optimal absorption signals in terms of signal-to-noise ratio and sensitivity were obtained at 336 nm.
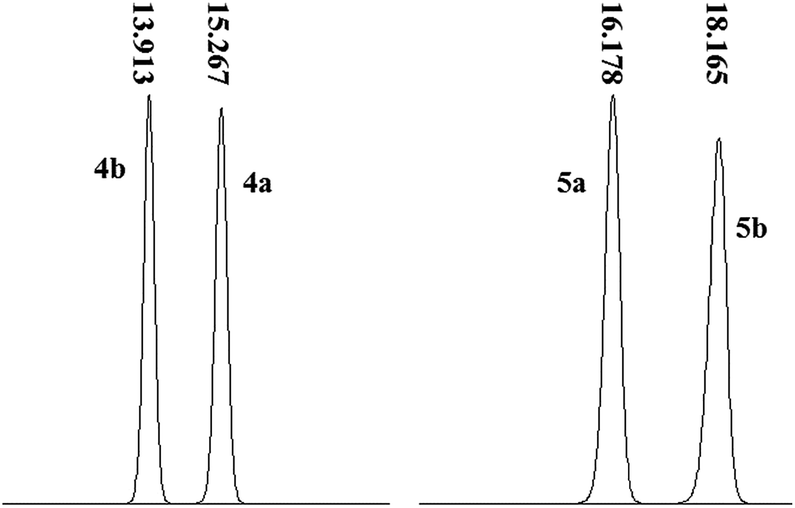 | ||
| Fig. 6 RPLC profiles of 4a/4b and 5a/5b. | ||
| Validation parameter | 4a | 4b | ||||
|---|---|---|---|---|---|---|
| LOD, LOQ | ||||||
| LOD (μg mL−1) | 0.001 | 0.001 | ||||
| LOQ (μg mL−1) | 0.003 | 0.003 | ||||
|
||||||
| Linearity | ||||||
| Calibration range (μg mL−1) | 1000–1.9 | 1000–1.9 | ||||
| Calibration points | 10 | 10 | ||||
| Regression equations | A = 0.0434C + 0.00323 | A = 0.0361C + 0.0012 | ||||
| Coefficient of determination (R2) | 0.9999 | 0.9996 | ||||
| Relative response factor (RRF) | 1.202 | 1.000 | ||||
|
||||||
| Accuracy | ||||||
| Recovered (μg mL−1) | 600 | 500 | 400 | 600 | 500 | 400 |
| % Recovery | 101.2 | 98.5 | 99.8 | 99.6 | 98.2 | 100.5 |
| % RSD (n = 3) | 0.78 | 0.45 | 0.88 | 0.78 | 0.76 | 0.45 |
The accuracy of the method was validated through recovery experiments by spiking known amounts of 4a and 4b at concentrations of 300, 400 and 500 μg mL−1. Each preparation was analysed in triplicate (n = 3) and percent recovery was calculated. The recovery was found to be between 98.5% and 101.2% with the RSD (n = 9) less than 0.88% (Table 1). The LOD and LOQ for 4a/4b were determined by the signal-to-noise ratio (S/N) method. The LOD and LOQ obtained for 4a/4b were 0.001 μg mL−1 and 0.003 μg mL−1, respectively, which is more sensitive than the GC method (LOD is 0.05 μg mL−1).17
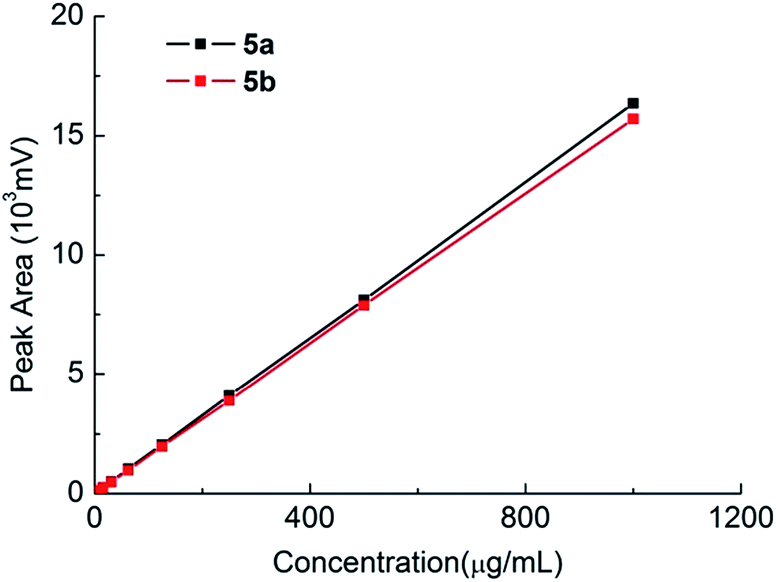 | ||
| Fig. 7 Standard curves of 5a and 5b. | ||
Application of this method
It is demonstrated that both 4a/4b and 5a/5b could be used for the enantiopurity assessment of chiral 1. However, for the user's convenience, 4a and 4b were easier to obtain and thus selected for optical purity analysis. The external standard method could be adopted to determine the accurate concentrations of 4a and 4b and the optical purity could then be calculated according to their ratio. In addition to the external standard method, a peak area normalization method with a correction factor is a feasible way to detect the exact absolute optical purity of chiral 1 without the use of standard samples. It means that the absolute optical purity of 1 could be quantitatively determined by the peak area ratios of two diastereomers, which clearly surpasses the optical rotation method in accuracy.Four commercially available samples were then assessed for their enantiomeric excess using both the external standard method and the peak area normalization method. The samples were first reacted with 3 to provide the reaction mixture, which was analysed after a simple sample treatment.
As listed in Table 2, both analytical methods provided extremely close ee values. Therefore, in a typical routine determination without requirement of system suitability evaluation, the peak area normalization method with a correction factor is sufficient to provide reliable results without preparing standard curves.
Experimental section
Reagents and apparatus
Commercially available reagents were used without further purification. Reactions were monitored by thin-layer chromatography (TLC) using silica gel precoated glass plates visualized with UV light. Flash chromatography columns were packed with 300–400 mesh silica gel. Gradient flash chromatography was conducted with a continuous gradient from petroleum ether (PE) to ethyl acetate (EA), listed as volume/volume ratios. 1H and 13C NMR data were recorded using the Varian Mercury (500 MHz) spectrometer with TMS as an internal standard. ESI-HRMS data were collected on the Thermo Exactive plus Orbitrap mass spectrometer. Melting points were measured on the Yanaco MP-J3 microscope melting point apparatus. RPLC analysis was carried out using the Shimadzu LC-20A HPLC instrument (Kyoto, Japan) equipped with a UV/VIS detector (SPD-20A) and an auto sampler (SIL-20A). The Shimadzu LC Solution software was used for data acquisition and system suitability calculations. Separation was performed on the Ultimate® XB-C18 column (5 μm, 4.6 × 250 mm). Optical rotations were measured on the SGW-5 automatic polarimeter (INESA Inc., Shanghai) in acetonitrile at D line (589.3 nm) at room temperature (r.t.). ECD spectra were recorded on the Jasco J-815 spectrometer (Tokyo, Japan) in methanol.Synthesis
m.p.: 72–73 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.61 (d, J = 8.0 Hz, 1H), 6.56 (d, J = 13.6 Hz, 1H), 4.47 (t, J = 6.8 Hz, 1H), 3.76 (s, 3H), 3.52–3.62 (m, 1H), 3.30–3.37 (m, 1H), 2.46–2.56 (m, 1H), 2.10–2.22 (m, 2H), 1.96–2.07 (m, 1H). 13C NMR (125 MHz, CDCl3) δ (ppm): 171.3, 159.1, 156.9, 146.2 (d, J = 12.1 Hz), 133.1, 126.8, 104.5 (d, J = 26.9 Hz), 62.8, 53.3, 52.5, 31.2, 24.7 (see Fig. S4†). ESI-MS calcd for C12H13N3O6F [M + H]+ 314.0783, found 314.0766 (see Fig. S5†). [α]25D = −644.4 (c = 0.5, CH3CN). UV (CH3OH) λmax (logε): 348.0 (4.05), 223.5 (3.89) nm; ECD (CH3OH) λmax (Δε): 381.5 (−4.34), 345.0 (−3.21), 325.0 (−3.46), 269.0 (+3.53), 244.5 (+2.04), 216.0 (+3.42) nm.
:1). Then the solvent was evaporated under reduced pressure, and the residue was washed with distilled water and dried in a vacuum drying oven to obtain 4a as a yellow solid (467 mg) in a yield of 97.6%, which was used without further purification.m.p.: 57–58 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.78 (s, 1H), 8.32 (d, J = 7.6 Hz, 1H), 5.92 (s, 1H), 4.40 (t, J = 6.8 Hz, 1H), 3.75 (s, 3H), 3.66–3.71 (m, 2H), 3.56–3.64 (m, 1H), 3.44–3.54 (m, 1H), 3.23–3.30 (m, 1H), 2.42–2.51 (m, 1H), 2.09–2.22 (m, 2H), 1.79–2.03 (m, 3H), 1.59–1.72 (m, 1H), 1.06 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ (ppm): 173.1, 147.7, 146.6, 129.5, 129.0, 123.7, 95.8, 64.2, 63.4, 57.0, 53.2, 52.4, 30.95, 25.1, 24.8, 10.9 (see Fig. S6†). ESI-MS calcd for C16H23N4O7F [M + H]+ 383.1561, found 383.1545 (see Fig. S7†). [α]25D = −918.2 (c = 0.2, CH3CN). UV (CH3OH) λmax (logε): 402.0 (4.16), 335.5 (4.44), 220.5 (4.42) nm; ECD (CH3OH) λmax (Δε): 410.0 (−6.35), 392.0 (−7.48), 349.5 (−22.66), 295.0 (+19.17), 261.5 (+6.95), 252.5 (+7.96), 233.5 (−1.99), 217.0 (+10.73) nm.
m.p.: 55–56 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.79 (s, 1H), 8.39 (d, J = 7.6 Hz, 1H), 5.97 (s, 1H), 4.42 (t, J = 6.8 Hz, 1H), 3.75–3.87 (m, 2H), 3.73 (s, 3H), 3.57–3.65 (m, 1H), 3.47–3.56 (m, 1H), 3.24–3.31 (m, 1H), 2.42–2.53 (m, 1H), 2.08–2.19 (m, 2H), 1.87–2.01 (m, 1H), 1.69–1.82 (m, 2H), 1.56–1.68 (m, 1H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 172.7, 147.9, 146.8, 129.5, 128.9, 123.6, 95.8, 64.3, 63.0, 56.8, 53.0, 52.6, 31.1, 25.1, 24.5, 10.6 (see Fig. S8†). ESI-MS calcd for C16H23N4O7F [M + H]+ 383.1561, found 383.1540 (see Fig. S9†). [α]25D = −668.8 (c = 0.4, CH3CN). UV (CH3OH) λmax (logε): 402.0 (4.03), 335.5 (4.33), 220.5 (4.32) nm; ECD (CH3OH) λmax (Δε): 410.0 (−4.67), 392.0 (−5.17), 348.5 (−17.38), 293.5 (+13.61), 259.5 (+3.52), 252.5 (+3.86), 233.0 (−4.18), 216.0 (+7.18) nm.
:3) afforded 5a as a yellow solid (159 mg) in a yield of 92.9%.m.p.: 153–154 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.65 (s, 1H), 6.65 (s, 1H), 4.64–4.73 (m, 1H), 4.52 (t, J = 6.8 Hz, 1H), 4.14–4.22 (m, 2H), 3.75 (s, 3H), 3.51–3.60 (m, 1H), 3.18–3.26 (m, 1H), 2.45–2.55 (m, 1H), 2.11–2.23 (m, 2H), 1.95–2.06 (m, 1H), 1.51–1.76 (m, 3H), 0.92 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 171.5, 156.9, 144.5, 135.12, 134.8, 133.7, 126.8, 118.8, 68.6, 62.8, 59.4, 53.1, 52.3, 30.8, 25.8, 24.9, 8.6 (see Fig. S10†). ESI-MS calcd for C17H21N4O8F [M + H]+ 409.1354, found 409.1340 (see Fig. S11†). [α]25D = −588.1 (c = 0.1, CH3CN). UV (CH3OH) λmax (logε): 365.0 (4.24), 267.0 (3.89), 208.5 (4.26) nm; ECD (CH3OH) λmax (Δε): 385.5 (−8.65), 363.0 (−8.20), 335.5 (−9.14), 277.0 (+11.01), 237.5 (0), 214.0 (+6.97) nm.
m.p.: 151–152 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.51 (s, 1H), 6.65 (s, 1H), 4.65 (t, J = 8.0 Hz, 1H), 4.48 (t, J = 6.8 Hz, 1H), 4.24–4.32 (m, 1H), 4.18–4.23 (m, 1H), 3.74 (s, 3H), 3.50–3.59 (m, 1H), 3.28–3.36 (m, 1H), 2.46–2.56 (m, 1H), 2.00–2.23 (m, 3H), 1.56–1.77 (m, 2H), 0.90 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 171.8, 155.8, 144.3, 134.7, 134.22, 133.4, 126.6, 115.0, 68.3, 62.9, 58.6, 53.2, 52.3, 31.0, 25.3, 24.9, 8.7 (see Fig. S12†). ESI-MS calcd for C17H21N4O8F [M + H]+ 409.1354, found 409.1355 (see Fig. S13†). [α]25D = −153.0 (c = 0.2, CH3CN). UV (CH3OH) λmax (logε): 362.0 (4.13), 271.5 (3.99), 208.5 (4.20) nm; ECD (CH3OH) λmax (Δε): 414.5 (−3.03), 363.0 (+8.48), 318.5 (−3.23), 305.0 (−2.19), 266.0 (−17.81), 217.5 (+9.95) nm.
RPLC analysis conditions
:50 (v/v) or 55:45 (v/v). Mobile phases were filtered through a 0.45 μm membrane filter (Millipore, Bedford, USA) and degassed before use. The flow rate was 1.0 mL min−1 with UV detection at 336 nm. Each sample was injected three times with an injection volume of 10 μL.Conclusions
Two pairs of diastereomers were designed and synthesized to assess the absolute enantiopurity of 1. Their absolute configurations were assigned unambiguously using the TDDFT calculation of ECD spectra via the ring-locking strategy. The linearity, RRF, LOD, LOQ, and recovery rates were obtained for 4a/4b and 5a/5b. The method could serve as an alternative approach to detect absolute optical purity of 1 by regular RPLC without using chiral stationary phase. Furthermore, it consequently affords a promising prospect for analysis of other chiral amino alcohols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the CAMS Innovation Fund for Medical Sciences (CIFMS, No. 2016-I2M-3-009) and the Fundamental Research Funds for the Central Institutes (No. 2016ZX350003).Notes and references
- (a) Y. J. Chen, Y. H. Chen, C. G. Feng and G. Q. Lin, Org. Lett., 2014, 16, 3400–3403 CrossRef CAS PubMed; (b) V. Jha, N. B. Kondekar and P. Kumar, Org. Lett., 2010, 12, 2762–2765 CrossRef CAS PubMed; (c) O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2012, 10, 4311–4326 RSC; (d) A. W. Buesking and J. A. Ellman, Chem. Sci., 2014, 5, 1983–1987 RSC.
- (a) C. P. Pradeep and S. K. Das, Coord. Chem. Rev., 2013, 257, 1699–1715 CrossRef CAS; (b) M. M. Wanderley, C. Wang, C. D. Wu and W. Lin, J. Am. Chem. Soc., 2012, 134, 9050–9053 CrossRef CAS PubMed; (c) N. Caldwell, C. Jamieson, I. Simpson and T. Tuttle, Org. Lett., 2013, 15, 2506–2509 CrossRef CAS PubMed.
- (a) M. P. Chelopo, S. A. Pawar, M. K. Sokhela, T. Govender, H. G. Kruger and G. E. Maguire, Eur. J. Med. Chem., 2013, 66, 407–414 CrossRef CAS PubMed; (b) M. Mujahid, R. G. Gonnade, P. Yogeeswari, D. Sriram and M. Muthukrishnan, Bioorg. Med. Chem. Lett., 2013, 23, 1416–1419 CrossRef CAS PubMed; (c) A. G. Rossi, D. A. Sawatzky, A. Walker, C. Ward, T. A. Sheldrak, N. A. Riley, A. Caldicott, M. Martinez-Losa, T. R. Walker, R. Duffin, M. Gray, E. Crescenzi, M. C. Martin, H. J. Brady, J. S. Savill, I. Dransfield and C. Haslett, Nat. Med., 2006, 12, 1056–1064 CrossRef CAS PubMed; (d) R. Hajjo, C. M. Grulke, A. Golbraikh, V. Setola, X. P. Huang, B. L. Roth and A. Tropsha, J. Med. Chem., 2010, 53, 7573–7586 CrossRef CAS PubMed.
- A. Zumla, P. Nahid and S. T. Cole, Nat. Rev. Drug Discovery, 2013, 12, 388–404 CrossRef CAS PubMed.
- (a) R. G. Wilkinson, R. G. Shepherd, J. P. Thomas and C. Baughn, J. Am. Chem. Soc., 1961, 83, 2212–2213 CrossRef CAS; (b) B. Blessington and A. Beiraghi, J. Chromatogr. A, 1990, 522, 195–203 CrossRef CAS.
- (a) J. Tomaszewski and M. M. Rumore, Drug Dev. Ind. Pharm., 1994, 20, 119–139 CrossRef CAS; (b) F. W. Fraunfelder, A. A. Sadun and T. Wood, Expert Opin. Drug Saf., 2006, 5, 615–618 CrossRef CAS PubMed.
- R. G. Wilkinson, M. B. Cantrall and R. G. Shepherd, J. Med. Chem., 1962, 5, 835–845 CrossRef CAS.
- (a) G. M. Dobrikov, V. Valcheva, M. Stoilova-Disheva, G. Momekov, P. Tzvetkova, A. Chimov and V. Dimitrov, Eur. J. Med. Chem., 2012, 48, 45–56 CrossRef CAS PubMed; (b) G. M. Dobrikov, V. Valcheva, Y. Nikolova, I. Ugrinova, E. Pasheva and V. Dimitrov, Eur. J. Med. Chem., 2013, 63, 468–473 CrossRef CAS PubMed; (c) Z. Petkova, V. Valcheva, G. Momekov, P. Petrov, V. Dimitrov, I. Doytchinova, G. Stavrakov and M. Stoyanova, Eur. J. Med. Chem., 2014, 81, 150–157 CrossRef CAS PubMed.
- (a) N. W. Fadnavis, M. Sharfuddin and S. K. Vadivel, Tetrahedron: Asymmetry, 1999, 10, 4495–4500 CrossRef CAS; (b) D. W. Armstrong, W. Li and C. D. Chang, Anal. Chem., 1990, 62, 914–923 CrossRef CAS PubMed.
- (a) F. Francalanci, P. Cesti, W. Cabri, D. Bianchi, T. Martinengo and M. Foa, J. Org. Chem., 1987, 52, 5079–5082 CrossRef CAS; (b) S. K. Mishra, S. R. Chaudhari and N. Suryaprakash, Org. Biomol. Chem., 2014, 12, 495–502 RSC.
- (a) E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1997, 119, 6345–6359 CrossRef CAS; (b) M. M. Wanderley, C. Wang, C. D. Wu and W. Lin, J. Am. Chem. Soc., 2012, 134, 9050–9053 CrossRef CAS PubMed; (c) K. W. Bentley, Y. G. Nam, J. M. Murphy and C. Wolf, J. Am. Chem. Soc., 2013, 135, 18052–18055 CrossRef CAS PubMed; (d) C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC; (e) P. Zhang and C. Wolf, Chem. Commun., 2013, 49, 7010–7012 RSC; (f) C. Wang, E. Wu, X. Wu, X. Xu, G. Zhang and L. Pu, J. Am. Chem. Soc., 2015, 137, 3747–3750 CrossRef CAS PubMed; (g) A. Zeus, R. Ding and C. Wolf, Org. Biomol. Chem., 2016, 14, 1934–1939 RSC.
- (a) N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC; (b) X. C. Li, D. Ferreira and Y. Ding, Curr. Org. Chem., 2010, 14, 1678–1697 CrossRef CAS PubMed; (c) G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC.
- (a) P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS; (b) K. Fujii, Y. Ikai, T. Mayum, H. Oka, M. Suzuki and K. I. Harada, Anal. Chem., 1997, 69, 3346–3352 CrossRef CAS; (c) K. Harada, K. Fujii, K. Hayashi, M. Suzuki, Y. Ikai and H. Oka, Tetrahedron Lett., 1996, 37, 3001–3004 CrossRef CAS; (d) Y. Hou, M. D. B. Tianero, J. C. Kwan, T. P. Wyche, C. R. Michel, G. A. Ellis, E. Vazquez-Rivera, D. R. Braun, W. E. Rose, E. W. Schmidt and T. S. Bugni, Org. Lett., 2012, 19, 5050–5053 CrossRef PubMed; (e) J. G. Adamson, T. Hoang, A. Crivici and G. A. Lajoie, Anal. Biochem., 1992, 202, 210–214 CrossRef CAS PubMed; (f) Y. Nozawa, N. Sakai, K. Arai, Y. Kawasaki and K. Harada, J. Microbiol. Methods, 2007, 70, 306–311 CrossRef CAS PubMed; (g) H. Yambe, S. Kitamura, M. Kamio, M. Yamad, S. Matsunaga, N. Fusetani and F. Yamazaki, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15370–15374 CrossRef CAS PubMed; (h) S. Vijayasarathy, P. Prasad, L. J. Fremlin, R. Ratnayake, A. A. Salim, Z. Khalil and R. J. Capon, J. Nat. Prod., 2016, 79, 421–427 CrossRef CAS PubMed.
- K. K. Ngim, J. Zynger and B. Downey, J. Chromatogr. Sci., 2007, 45, 126–130 CAS.
- (a) R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231–247 CrossRef CAS PubMed; (b) R. Bhushan and H. Brückner, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3148–3161 CrossRef CAS PubMed; (c) R. Bhushan and V. Kumar, Biomed. Chromatogr., 2008, 22, 906–911 CrossRef CAS PubMed.
- G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS PubMed.
- I. N. Stan’kov, A. A. Sergeeva and S. N. Tarasov, J. Anal. Chem., 2000, 55, 150–154 CrossRef.
- (a) L. Li and Y. K. Si, Chirality, 2012, 24, 987–993 CrossRef CAS PubMed; (b) L. Li, C. Li, Y. K. Si and D. L. Yin, Chin. Chem. Lett., 2013, 24, 500–502 CrossRef CAS; (c) L. Li, B. B. Yang and Y. K. Si, Chin. Chem. Lett., 2014, 25, 1586–1590 CrossRef CAS.
- Gaussian 09, Revision C. 01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, ESI-HRMS and computation data for 3, 4a/4b, 5a/5b. See DOI: 10.1039/c7ra09362d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |