
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Osteocytes regulate osteoblast differentiation and osteoclast activity through Interleukin-6 under mechanical loading†
Zhichao Hao‡
ab,
Yuanyuan Ma‡a,
Jun Wu‡ cd,
Xianxian Lie,
Helin Chenb,
Jiefei Shen*b and
Hang Wang*b
cd,
Xianxian Lie,
Helin Chenb,
Jiefei Shen*b and
Hang Wang*b
aGuanghua School of Stomatology, Hospital of Stomatology, Sun Yat-sen University, Guangdong Provincial Key Laboratory of Stomatology, Guangzhou 510055, China. E-mail: haozi0548@163.com
bState Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu 610041, China. E-mail: wanghang@scu.edu.cn; shenjiefei@scu.edu.cn
cKey Laboratory of Sensing Technology and Biomedical Instrument of Guangdong Province, School of Engineering, Sun Yat-sen University, Guangzhou, 510006, China
dKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, Sun Yat-sen University, Guangzhou, 510275, China
eSichuan Academy of Medical Sciences and Sichuan Provincial People's Hospital, Chengdu 610041, China
First published on 27th October 2017
Abstract
Osteocytes are the major mechanosensors that respond to mechanical strain and regulate bone formation and resorption. Previous studies have indicated that IL-6 is a mechano-sensitive cytokine. However, there are few data describing the effect of IL-6 on osteoblasts and osteoclasts under mechanical loading. The purpose of this study was to evaluate whether osteocytes affect osteoblast differentiation and osteoclast activity via IL-6 production. Murine primary calvarial osteoblasts and RAW264.7 macrophage cells were cultured in conditioned medium collected from murine long bone osteocytes Y4 (MLO-Y4) subjected to compressive cyclic force (CCF) with or without IL-6 and soluble IL-6 receptor (sIL-6R). The results showed that MLO-Y4 osteocytes increased ALP activity and osteoblast markers expression in murine calvarial osteoblasts and inhibited osteoclastogenesis and osteoclast differentiation in osteoclast precursors via soluble factors in the CCF-induced conditioned medium (CCF CM). Typically, the osteocyte-induced effects were significantly greater in the presence of exogenous IL-6/sIL-6R. However, when the CCF CM was treated with the IL-6 neutralizing antibody, the stimulating effect was significantly blocked. Activation of Janus protein tyrosine kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) and extracellular signal-regulated kinase (ERK) signal transduction pathways were involved in IL-6-induced osteoblasts and osteoclast differentiation and activities. In conclusion, osteocytes regulate osteogenic response and osteoclast activity under mechanical loading through IL-6 production via the STAT3 and ERK signaling pathways. These pathways not only induce distinct biological responses, but also coordinate with each other to have detrimental effects on bone homeostasis.
1 Introduction
Bone tissue is in a constant state of bone turnover under physiological loading. Mechanically induced adaptation of bone tissue is mainly mediated by the balance between bone formation and bone resorption.1,2 Bone tissue remodeling is ultimately regulated by the coordinated activities of bone cells: osteoblasts, of mesenchymal origin, that deposit bone matrix; osteoclasts, of hematopoietic origin, that resorb bone; osteocytes, descendants of osteoblasts, that sense mechanical load and their progenitors. Increasing evidence has confirmed that osteocytes, which make up 90–95% of all the bone cells in the adult skeleton, are the major mechanosensory cells that respond to mechanical strain and translate the force into biochemical signals.3,4Osteocytes are believed to regulate bone remodeling under mechanical loading by signaling to other effector bone cells via soluble autocrine and paracrine factors, and direct cell–cell contact. The role of osteocytes in directly controlling the differentiation and activity of either osteoclasts or osteoblasts has been determined in recent years. After mechanical loading, osteocytes may send anabolic signals that are released rapidly including nitric oxide (NO) and prostaglandins (PGE), and other small molecules such as ATP to induce osteoblast activation.5–7 As the major source of the cytokine receptor activator of nuclear factor kappa-B ligand (RANKL), osteocytes subjected to mechanical strain can control osteoclastogensis and thus bone resorption.8 Osteocytes markers, such as dentin matrix protein 1 (DMP1), matrix extracellular phosphoglycoprotein (MEPE), and sclerostin, which are also mediated by mechanical loading, may control bone metabolism and remodeling.9,10 Previous studies have argued that IL-6 is a new mechano-sensitive cytokine, which may play an important role in the biomechanical control of bone remodeling.11,12
Adequate IL-6 is critical to bone homeostatic balance by controlling bone formation and degradation. IL-6 has a dual effect on cell proliferation, osteoblast and osteoclast differentiation and cell apoptosis. IL-6 enhances cell proliferation in UMR-106 cells;13 whereas it does not affect the growth of human bone marrow stromal osteoprogenitor cells.14 This cytokine increases the expression of osteoblastic markers such as alkaline phosphatase (ALP), osteocalcin (OCN) and bone sialoprotein and induces bone nodule formation.15–18 Several studies have confirmed the inhibitory effect of IL-6 on RANKL-induced osteoclast formation and bone resorption.19,20 IL-6 can also prevent apoptosis of osteoblastic cells induced by serum removal or addition of tumor necrosis factor-α (TNF-α).21 However, IL-6 appears to show an inhibitory effect on osteoblstic marker expression and bone formation in vitro and it enhances apoptosis.22,23 IL-6 is a pre-osteoclastic factor inducing the production of RANKL by cells in bone tissue, which in turn enhances in osteoclast survival, differentiation and function.24,25 Moreover, IL-6−/− mice have delayed callus maturity and mineralization and retarded fracture healing compared to the wild type mice.26 However, chronic IL-6 over-expression enhances bone resorption and causes low bone mass.27,28
Upon binding either to its membrane IL-6 receptor (mIL-6R) or its sIL-6R, IL-6 triggers homodimerization of signal-transducing molecular gp130, followed by initiation of two major downstream signaling pathways, JAK/STAT, or Src-homology domain 2 containing protein-tyrosine phosphatase (SHP2)/mitogen-activated protein kinase (MAPK, mainly ERK).29 IL-6-type cytokines have been shown to influence osteoblast differentiation and osteoclast activity via complex and often contradictory mechanisms.30 In summary, results from studies in vitro and in vivo are still controversial with regard to whether IL-6 is an anabolic or catabolic factor for bone metabolism. The purpose of this study was to evaluate whether osteocytes, as the major mechanosensory cells in bone, regulate osteoblast differentiation and osteoclastogenesis through IL-6 and if so, to identify the molecule signaling mechanisms responsible for this process. The hypothesis of this study was that osteocytes affected osteoblast and osteoclast differentiation and function under mechanical loading through IL-6.
2 Material and methods
2.1 MLO-Y4 culture and mechanical stimulation
The MLO-Y4 cell line, generously gifted by Prof. Lynda F. Bonewald (Department of Oral Biology, University of Missouri at Kansas City, Kansas City, Missouri, USA), was cultured in α-modified essential medium (Hyclone, Logan, UT, USA) supplemented with 5% calf serum and 5% fetal bovine serum (Gibico, Grand Island, NY, USA), 100 U mL−1 penicillin and 100 mg mL−1 streptomycin (Sigma-Aldrich, St. Louis, MO, USA). Then osteocytes were subjected to uniaxial compressive cyclic force at 2 Hz with 2000 με for 10 min, 30 min, 1 h, 3 h or 6 h by a four-point bending system based on beam-deflection theory. Briefly, the equation for the maximum magnitude of the strain is ε = 3dx/a(3l − 4a) (a denotes the distance between the inner and outer contacts; d denotes the thickness of the plate, l denotes the span between the outer contacts, x is the vertical displacement of the inner contacts and ε = 3dx/a(3l − 4a) is the strain at the surface of the plate). The frequency of the strain equals to that of the beam deflection and the strain cycle is shown in a sinusoidal pattern.31 The holographic interferometry measurements confirmed that a uniformly deformed strain was distributed.32 Control cells were maintained in a static culture. Conditioned medium was collected at 48 h after 1 h mechanical loading or static culture according to previous studies.12,33,342.2 IL-6 and gene analysis of osteocytes subjected to mechanical loading
The concentration of IL-6 was determined using a commercially available ELISA kit (Senxiong, Shanghai, China) according to the manufacture's instructions. After incubation for 48 h starting from the stimulation, supernatants of MLO-Y4 cells were collected and centrifuged at 1000 rpm for 10 min to remove pellet and cellular debris. Supernatants were used as samples and stored at −80 °C. Optical absorbance was then measured in an ELISA reader (HTS7000+, PerkinElmer, Wlatham, MA, USA). All experiments above were performed in triplicate and each sample was tested in duplicate.Real-time reverse transcription polymerase chain reaction (real-time RT-PCR) was used for genetic analysis. Total RNA (1 μg) was extracted using TRIzol method (Invitrogen, Gaithersburg, MD, USA) according to manufacturer's protocol. Reverse transcription was performed using a PCR Master Mix Kit (Takara, Tokyo, Japan) for IL-6, RANKL, OPG and OCN. To standardize the annealing temperature, a number of cycles were run for each set of primers. Real-time PCR was performed with a SYBR® Premix Ex Taq™ (Takara, Japan) and the sense and antisense PCR primers used are listed in Table 1. GAPDH was used as a housekeeping gene to correct the tested gene expression. All the RT-PCR experiments were run in triplicate for different samples and each mRNA sample was tested in triplicate.
| Gene | Primer sequences (5′-3′) | |
|---|---|---|
| IL-6 | Forward | TCCAGAAGACCAGAGGAAAT |
| Reverse | TCCAGAAGACCAGAGGAAAT | |
| RANKL | Forward | TCCAGAAGACCAGAGGAAAT |
| Reverse | GGAAGGGTTGGACACCTGAATG | |
| OPG | Forward | TGACCTCTGTGAAAGCAGCGTG |
| Reverse | GTTCTCTCAATCTCTTCTGGGC | |
| OCN | Forward | GAGGGCAATAAGGTAGTGAACAGAC |
| Reverse | AATAGTGATACCGTAGATGCGTTTG | |
| GAPDH | Forward | ACTCAAGATTGTCAGCAAT |
| Reverse | CCATCCACAGTCTTCTGGGT | |
| ALP | Forward | GAATCAAATGTTCAGGGTGGT |
| Reverse | TGGCACGTTAAAGGTAATCAG | |
| RunX2 | Forward | CTCTTCTGGAGCCGTTTATGT |
| Reverse | GTTTCTTAGGGTCTTGGAGTGA | |
| Ctsk | Forward | GAAGAAGACTCACCAGAAGCA |
| Reverse | TCCAGGTTATGGGCAGAGATT | |
| MMP-9 | Forward | CCACCGAGCTATCCACTCAT |
| Reverse | GAGTCTGGGGTCTGGTTTCA | |
| Trap (Acp5) | Forward | CGTCTCTGCACAGATTGCAT |
| Reverse | AAGCGCAAACGGTAGTAAGG | |
2.3 Isolation and culture of osteoblasts
Primary calvarial osteoblasts were isolated from 2–3 days-old C57BL/6 mice by sequential collagenase digestion.35 Briefly, cells released from the first digestion were discarded and the cells released from the second digestion were cultured in α-MEM (Hyclone, USA) containing 10% fetal calf serum (Gibco, USA), 100 U mL−1 penicillin and 100 mg mL−1 streptomycin (Sigma-Aldrich, USA), 10 mM – glycerophosphate, and 50 μg mL−1 ascorbic acid (Sigma-Aldrich, USA). The medium and reagents were renewed every 3 days.Murine calvarial osteoblasts were seeded at a density of 2.0 × 105 cells per mL in 6-well plates and cultured for 1 day. The culture medium was then replaced with filter-sterilized conditioned medium from MLO-Y4 osteocytes, with or without CCF mixed with fresh culture medium in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. For the IL-6 and sIL-6R supplementation assays, osteoblasts were treated with normal cell culture media and conditioned media supplemented with IL-6 (25 ng mL−1, R&D system, Abington, UK) and sIL-6R (100 ng mL−1, R&D system, UK). For blocking experiments, neutralizing IL-6 antibody (5 μg mL−1, R&D System, UK) was added to CCF CM. Alkaline phosphatase (ALP) activity was measured at days 1, 3, 5 and 7 of culture, and osteoblastic gene expression was measured at day 5. For Alizarin Red staining, the cells were cultured for 21 days.
:1 volume ratio. For the IL-6 and sIL-6R supplementation assays, osteoblasts were treated with normal cell culture media and conditioned media supplemented with IL-6 (25 ng mL−1, R&D system, Abington, UK) and sIL-6R (100 ng mL−1, R&D system, UK). For blocking experiments, neutralizing IL-6 antibody (5 μg mL−1, R&D System, UK) was added to CCF CM. Alkaline phosphatase (ALP) activity was measured at days 1, 3, 5 and 7 of culture, and osteoblastic gene expression was measured at day 5. For Alizarin Red staining, the cells were cultured for 21 days.
2.4 ALP activity and osteogenic gene expression
ALP activity was assessed by a ρ-nitrophenyl-phosphate colorimetric assay (Sigma-Aldrich, USA) and the protein content was measured using the Bicinchoninic Acid Protein Assay Kit (Pierce, Rockford, IL, USA). RT-PCR was performed to analyze the expression of osteogenic genes ALP, OCN and Runx2 expression. Sequences of primers used for these RT-PCR experiments are also listed in Table 1.2.5 Alizarin red staining
After fixation with 10% formalin, murine calvarial osteoblasts were washed with distilled water, and stained with 2% Alizarin red S solution, pH 4.1 (Sigma-Aldrich, USA) for 2 min, followed by incubation with 100 mM cetylpyridinium chloride (CPC, Sigma-Aldrich, USA) for 1 h at room temperature to dissolve and release calcium-bound alizarin red. Absorbance of the released Alizarin red was then measured at 570 nm.2.6 Proliferation assay
Murine primary calvarial osteoblast cells were incubated in 96-well plates at a concentration of 2.0 × 105 cells per mL in a-MEM containing 10% FBS for 1 day. Cells were then treated with the five different media previously mentioned for 1, 3, 5, and 7 days. Cell proliferation was measured using the CCK8 Cell Proliferation Assay System (Dojindo Molecular Technologies, Inc., Gaithersburg, MD, USA), according to the manufacturer's instructions.2.7 Cell culture and osteoclast differentiation assays
Murine RAW264.7 macrophage cells (American Type Culture Collection, Promochem, Molsheim, France) were cultured in α-MEM (Hyclone, USA) containing 10% fetal calf serum (Gibco, USA). RAW264.7 cells were scraped and put back at 37 °C for 2 min. Nonadherent cells were then seeded in fresh medium at 5 × 103 cells in 48-well plates. After 2 h, recombinant mouse RANKL (PeproTech EC Ltd., London, UK), was added at the concentration of 50 ng mL−1. At day 2, the cells were incubated continuously in the five different culture systems used previously for murine calvarial osteoblasts supplemented with RANKL (50 ng mL−1). All the media used for osteoclast culture were supplemented with recombinant mouse RANKL (PeproTech EC Ltd., London, UK). The medium was renewed every 2 or 3 days. At day 7 after cell plating, cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) using a leukocyte acid phosphatase kit (Sigma-Aldrich, USA). The numbers of TRAP-positive multinucleated osteoclasts containing three or more nuclei were counted using a light microscope (Leica, Wetzlar, Germany).2.8 Molecular characterization of osteoclast differentiation using RT-PCR
TRAP, matrix metalloproteinase-9 (MMP-9) and Cathepsin K (Ctsk) expression were analysed by RT-PCR, as described earlier. The sequences of primers used for these RT-PCR experiments are also listed in Table 1. All the RT-PCR experiments were run in triplicate for different samples and each mRNA sample was tested in triplicate.2.9 Western blot
Murine calvarial osteoblasts and RAW264.7 macrophage cells were cultured in the presence or absence of the ERK inhibitor U0126 (10 μM, Cell Signaling Technology, Medford, MA, USA) or the STAT3 inhibitor S3I-201 (100 μM, Selleck Chemicals, Houston, TX, USA) for 1 h, and were then stimulated with or without IL-6 and sIL-6R for 15 min. Total protein was collected from murine osteoblasts and RAW264.7 cells using a total protein extraction kit (KeyGen Biotech, China) and centrifuged at 13000 rpm at 4 °C for 15 min. The supernatant was removed and stored at −80 °C for protein detection. After measuring protein concentration by the BCA method, all the samples containing 30 μg of protein were separated on 5% and 10% SDS–polyacrylamide gels, and electro-transferred transferred onto polyvinylidene difluoride membranes. Membranes were incubated for 1 h with 5% bovine serum albumin in PBS buffer to block non-specific protein binding. Subsequently, the membranes were incubated with primary antibodies against phosphorylated (p)-STAT3 or STAT3 and phosphorylated (p)-ERK1/2 or ERK1/2 (All Cell Signaling Technology, USA) at 4 °C overnight, washed with TBST and probed with a horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Finally, protein brands were detected using an enhanced chemiluminescence (ECL) Western Blot Detection Kit (Amersham Pharmacia Biotech, Little Chalfont, UK). The band intensity ratios of phosphorylated form and total protein were analysed using Quantity One software (Bio-Rad, Hercules, CA, USA). Results were reproduced in three independent experiments on different samples.
2.10 Statistical analysis
Statistical analyses were performed using one-way or two-way ANOVA with SPSS 20.0 software, followed by multiple comparisons testing using the Tukey test. P < 0.05 was considered statistically significant. Data are presented as mean ± SD.3 Results
3.1 CCF increases IL-6 and OCN expression and decreases the RANKL/OPG ratio in MLO-Y4 cells
Mechanical loading by CCF significantly increased IL-6 expression compared to the unloaded condition in MLO-Y4 cells (P < 0.05). IL-6 secretion detected in the supernatant increased after 10 min (798 pg mL−1) and 30 min (1080 pg mL−1) of mechanical loading, reached a maximum after 1 h (1673 pg mL−1, P < 0.001) and then progressively decreased at 3 h (1300 pg mL−1) and 6 h (1193 pg mL−1) groups (Fig. 1A). With the increasing CCF duration, IL-6 mRNA level also increased significantly in a time-dependent manner, reached a peak at 3 h (4.73 fold, P < 0.001) and then decreased, but remained higher than the control group after 6 h (1.57 fold, P < 0.05) of CCF loading (Fig. 1B).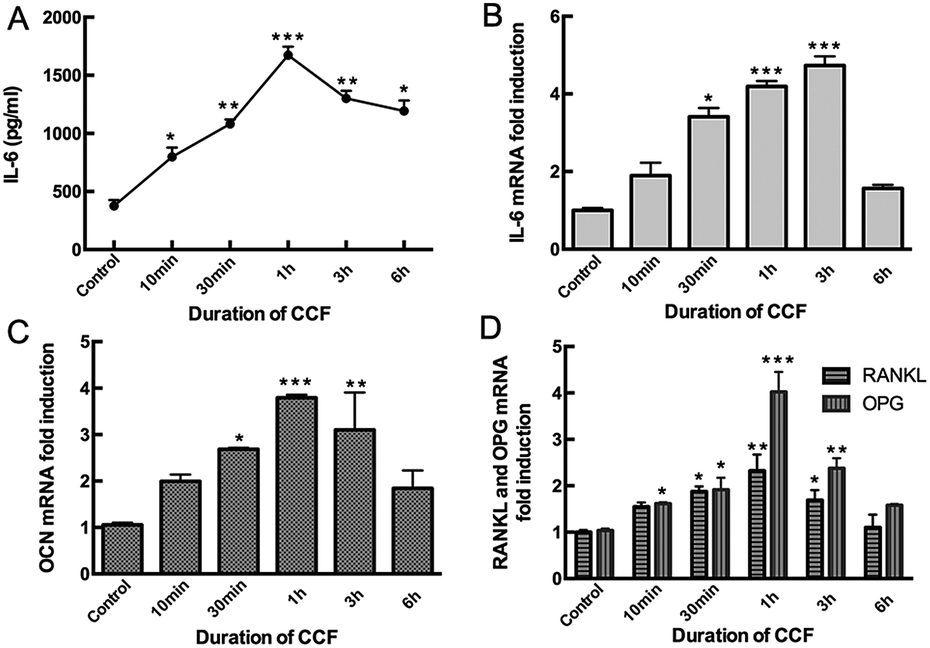 | ||
| Fig. 1 MLO-Y4 osteocytes respond to CCF. MLO-Y4 osteocytes were subjected to CCF at a frequency of 2 Hz with 2000 με for 10 min, 30 min, 1 h, 3 h, and 6 h. Both IL-6 secretion (A) and mRNA expression (B) were upregulated in a time-dependent manner; (C) CCF also enhanced OCN mRNA gene expression in the same tendency; (D) the RANKL/OPG ratio decreased significantly after 1 h of CCF strain and then gradually returned to baseline levels. Data are shown as mean ± SD from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001. | ||
Moreover, application of 2000 με CCF at a frequency of 2 Hz also increased OCN mRNA levels in osteocytes. This stimulating effect was statistically significant after 30 min (2.69 fold) of CCF loading (P < 0.05), reached a maximum after 1 h (3.79 fold, P = 0.001) and then decreased after 3 h (3.10 fold) and 6 h (1.84 fold, Fig. 1C) of strain. RANKL and OPG mRNA levels showed the same time-dependent tendency (Fig. 1D). However, after exposure to CCF for 1 h, the RANKL/OPG ratio was significantly deceased to approximately 57% of the unloaded condition. RANKL/OPG ratio then increased to 71% of the baseline level after 3 h of CCF and 69% of baseline level after 6 h of CCF. Based on these observations and our previous studies, we obtained conditioned media from static osteocytes (control group) and from CCF-stimulated osteocytes at 48 h after 1 h of mechanical loading for subsequent experiments.
3.2 Osteocytes significantly promote ALP activity and osteogenic differentiation through IL-6, but do not affect cell proliferation in osteoblasts
Cell proliferation was increased progressively at days 1, 3 and 5 of incubation (p < 0.001). While no significant differences were found between day 5 and day 7. The cells in the 96-well plate might reach the saturation density since day 5. No significant differences in cell proliferation were found among the different culture conditions (P > 0.05). The results indicate that CCF CM and IL-6/sIL-6R have no influence on osteoblastic cells proliferation (Fig. 2A).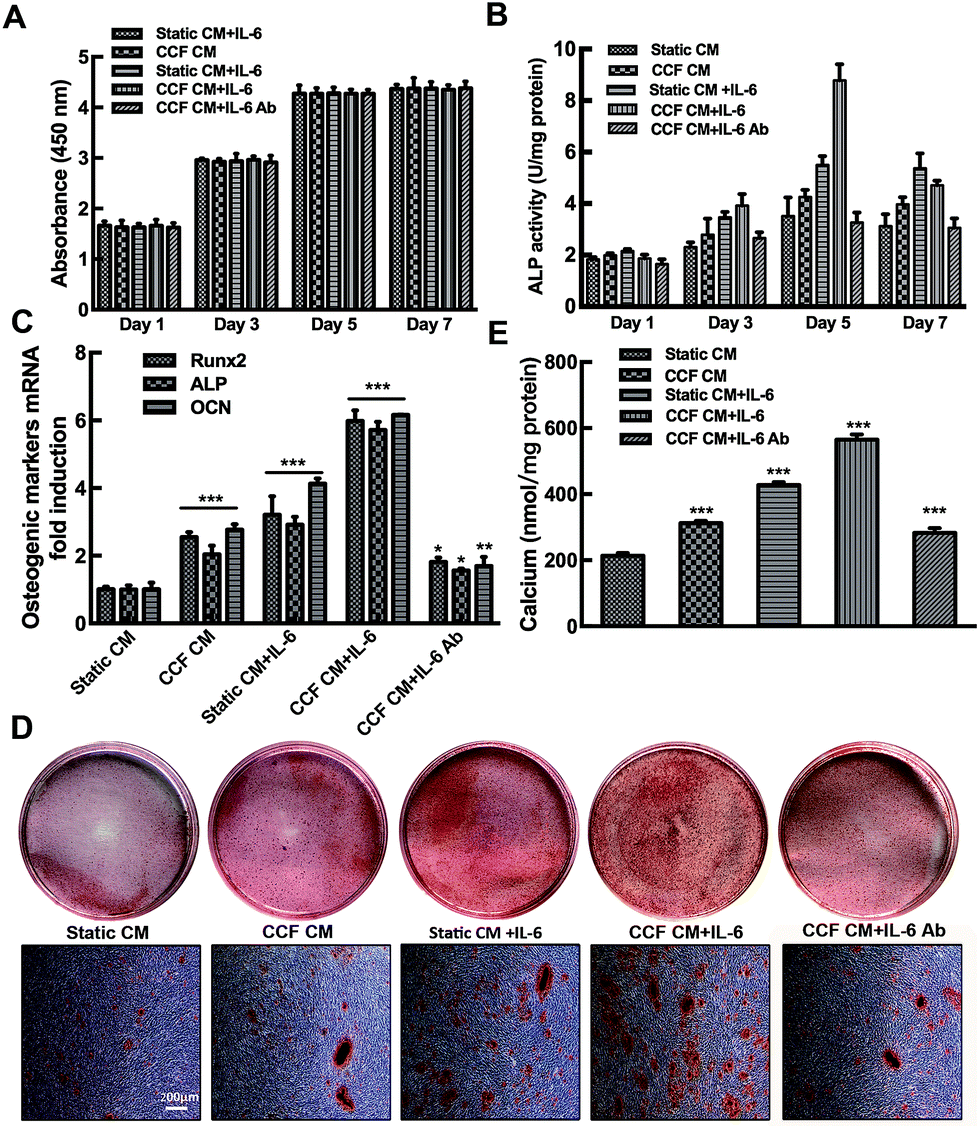 | ||
| Fig. 2 MLO-Y4 osteocytes enhance osteoblast differentiation and mineralization under mechanical strain through IL-6. (A) CCF CM and IL-6 had no significant effect on osteoblasts proliferation. (B) CCF CM and IL-6 increase ALP activity in murine calvarial osteoblasts. (C) Both CCF CM and IL-6 both increased Runx2, ALP, and OCN mRNA expression. (D) Murine calvarial osteoblasts showed enhanced mineralization using Alizarin red staining after CCF CM and IL-6 treatments compared to static conditioned medium (control group, static CM). (E) CPC assays were performed on mineralizing osteoblasts. The results indicated that CCF CM and IL-6 treatments significantly promoted mineralization of murine calvarial osteoblasts. IL-6 antibody treatments significantly inhibited the stimulating effect of CCF CM (B–E). Data are shown as mean ± SD from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001. | ||
CCF CM and IL-6/sIL-6R significantly enhanced osteoblast ALP activity of osteoblasts in a time-dependent manner (P < 0.001). The ALP activity reached a peak at day 5 and then decreased at day 7 (Fig. 2B). As shown in Fig. 2C, CCF CM and IL-6 significantly increased Runx2, ALP and OCN gene expression (P < 0.001). Moreover, quantification of Alizarin red staining showed that IL-6 and CCF CM promoted the mineralization of murine calvarial osteoblasts on day 21 (Fig. 2D). Quantification of the Alizarin red staining showed that the stimulating effect was significant compared to control group (P < 0.001, Fig. 2E). Conditioned medium from static osteocytes had no distinct effects on osteoblasts differentiation. Osteoblast markers mRNA expression and bone nodule formation increased significantly even when IL-6/sIL-6R was present in static conditioned medium. The neutralizing IL-6 antibody decreased but did not completely abrogate, the CCF CM stimulation (P < 0.05, Fig. 2B–E).
3.3 STAT3 activation is necessary to the IL-6-stimulated osteoblast differentiation
To further investigate the mechanism of osteocyte-stimulated osteoblast differentiation induced by IL-6, we evaluated the two main downstream signaling pathways. Phosphorylation levels of STAT3 and ERK1/2 after IL-6 and CCF CM treatment were detected by Western blotting, results were calculated as the ratio of phosphorylated form to the total protein. The results showed that ERK1/2 and STAT3 activation both were involved in osteocyte-stimulated osteoblast differentiation. However, U0126, a specific inhibitor of the ERK1/2 pathway, did not reverse the upregulation of ALP activity and osteoblast differentiation. This indicates that ERK1/2 is not the essential signaling pathway for osteocyte-stimulated osteoblast differentiation under mechanical loading (ESI†). As shown in Fig. 3A, the p-STAT3/STAT3 ratio increased significantly after CCF CM treatment, up to 2.1-fold compared to the control group. IL-6 treatment also enhanced the activation of STAT3 up to 4.55-fold. STAT3 activation reached a peak at 5.68-fold, when the murine calvarial osteoblasts were treated with both CCF CM and IL-6 treatments. The level of p-STAT3 was obviously inhibited by addition of IL-6 antibody to CCF CM and returned to the control level (1.53 fold, P = 0.51). Pretreatment with the specific STAT3 inhibitor, S3I-201, completely inhibited CCF CM and IL-6-induced phosphorylation of STAT3 (Fig. 3B).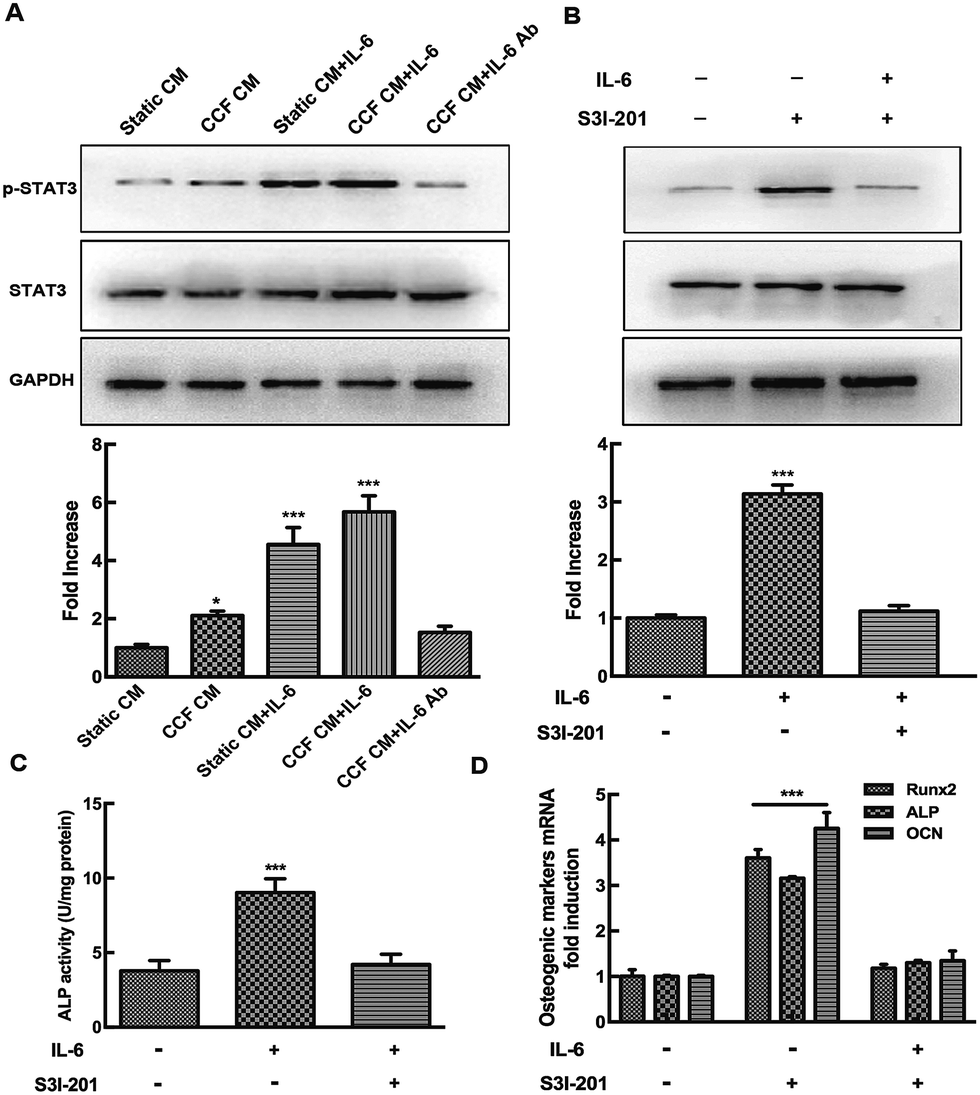 | ||
| Fig. 3 The STAT3 signaling pathway controls osteocyte-stimulated osteoblast differentiation induced by IL-6 under mechanical loading. Western blot analysis was performed to detect the phosphorylation level of STAT3 (p-STAT3). (A) The ratio of p-STAT3 to STAT3 increased significantly when the osteoblasts were treated with CCF CM or IL-6 treatment, and reached a peak when exposed to both the two treatments. STAT3 phosphorylation was inhibited by the addition of IL-6 antibody to CCF CM. (B) The specific STAT3 inhibitor, S3I-201, blocked the upregulation of p-STAT3 induced by IL-6. The positive effects of IL-6 on ALP activity and the expression of osteoblastic genes were reversed by inhibition of the STAT3 signaling pathway (C and D) data are shown as mean ± SD from three independent experiments. *P < 0.05; ***P < 0.001. | ||
The effects of specific STAT3 inhibitor (S3I-201) on ALP activity and the expression of osteoblastic genes (Runx2, ALP and osteocalcin) were assessed to further identify the association of STAT3 signaling pathways with the upregulation of osteoblast differentiation. The results showed that the positive effect of IL-6 on osteoblast differentiation was almost completely blocked by STAT3 inhibition (Fig. 3C and D), indicating that STAT3 plays an essential role during the osteocyte-stimulated osteoblast differentiation induced by IL-6 under mechanical loading.
3.4 Osteocytes have a suppressive effect on osteoclast differentiation through IL-6
Different culture systems with and without IL-6 and CCF CM were used to examine the osteocyte-stimulated osteoclastogenesis under mechanical loading (Fig. 4). In different culture media, RAW264.7 differentiated into TRAP-positive multinuclear cells also upon RANKL stimulation. In the CCF culture system, differentiation of RAW264.7 was significantly suppressed (69% of the control group), as shown by the number of TRAP-positive multinuclear cells. The single addition of IL-6 also strongly inhibited the osteoclast differentiation. Differentiated osteoclast numbers decreased to 19% of the control group with the addition of both CCF CM and IL-6. Moreover, the results indicate that IL-6 antibody treatment, even in the presence of CCF CM, strongly enhances osteoclastogenesis. | ||
| Fig. 4 Osteocytes inhibited osteoclastogenesis and osteoclastic markers expression through IL-6. (A) Effects of CCF CM and IL-6 on TRAP + multinucleated cell formation. (A) TRAP + cell number in osteocyte CCF CM moderately decreased to 69% of the control group. The presence of IL-6 reduced the number of TRAP + multinucleated cells to 32% of the control group. CCF CM and IL-6 treatment strongly reduced osteoclastogenesis. The inhibitory effect of CCF CM and IL-6 treatment was reversed by IL-6 antibody treatment. (B) CCF CM and IL-6 both decreased osteoclastic markers, Ctsk, TRAP, and MMP-9 expression. IL-6 neutralizing antibody blocked the CCF CM-induced inhibitory effect on osteoclastogenesis and osteoclastic markers expression. Data are shown as mean ± SD from three independent experiments. *P < 0.05; ***P < 0.001. | ||
Moreover, we further investigated the inhibitory effect on osteoclast differentiation by measuring markers gene expression. The osteoclastic gene markers, Ctsk, TRAP, and MMP-9 were suppressed significantly by CCF CM culture. The addition of IL-6 also decreased the mRNA expression of Ctsk, TRAP and MMP-9 to 49.7%, 31.9% and 32.9% of controls respectively. The mRNA expression decreased markedly when the RAW264.7 cells were treated with both CCF CM and IL-6 treatments. The CCF CM-induced inhibitory effect on osteoclast differentiation was strongly blocked by the addition of IL-6 antibody. However, the levels of gene expression remained lower than the control group (P < 0.05).
3.5 Concomitant activation of the STAT3 and ERK1/2 signaling pathways is involved in the inhibitory effect on osteoclast differentiation
The levels of p-STAT3 and p-ERK1/2 increased up to 2.4-fold and 2.1-fold of the control group, respectively, after RAW264.7 cells were exposed CCF CM (P < 0.001) and thereafter, increased when the osteoclasts were subjected to IL-6 stimulation, and were even higher after both CCF CM and IL-6 treatment (P < 0.001). The levels of p-STAT3 and p-ERK1/2 returned to the control level when IL-6 neutralizing antibody was added to CCF CM (P > 0.05, Fig. 5A). | ||
| Fig. 5 STAT3 and ERK1/2 are involved in negative effect induced by IL-6 on osteoclast differentiation. (A) RAW264.7 cells were pretreated with U0126 (10 μM) and/or S3I-201 (100 μM) for 1 h prior to 15 min of stimulation with IL-6. The high levels of p-STAT3 and p-ERK1/2 were blocked by the addition of IL-6 neutralizing antibody to CCF CM. (B) Cross-inhibition between IL-6-induced STAT3 and ERK signaling pathways. Combination of the inhibitors of the two signaling pathways totally inhibited IL-6-induced phosphorylation of the two kinases. Data are shown as mean ± SD from three independent experiments. *P < 0.05; ***P < 0.001. | ||
To examine the relationship between STAT3 and ERK1/2 in this process, we measured their phosphorylation levels after pretreatment with specific inhibitors. The STAT3 inhibitor S3I-201 abrogated STAT3 phosphorylation, but activated ERK effectively (Fig. 5B). Meanwhile, the MEK/ERK inhibitor, U0126, blocked the ERK signaling pathway, but enhanced STAT3 activation. IL-6-induced p-STAT3 and p-ERK1/2 signaling pathways may intersect to negatively regulate each other and co-regulate downstream functions. Combination of the two specific inhibitors totally inhibited IL-6-induced phosphorylation of the two kinases.
The negative effects of IL-6 on osteoclast differentiation and osteoclastic markers genes expression were partially reversed by inhibition of ERK or STAT3, while they were completely blocked by combining the specific inhibitors of the two signaling pathways (Fig. 6).
 | ||
| Fig. 6 STAT3 and ERK1/2 blockers reversed the negative effect of IL-6 on osteoclast differentiation. RAW264.7 cells were pretreated with U0126 (10 μM) and/or S3I-201 (100 μM) for 1 h prior to IL-6 (25 ng mL−1) stimulation, and then osteoclastogenesis and osteoclastic markers were analyzed. The IL-6-induced inhibitory effect was totally blocked by pretreatment with specific inhibitors of the two kinases. Data are shown as mean ± SD from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001. | ||
4 Discussion
The results of this study showed that osteocytes, as the major mechanotransducers in bone tissue, enhanced osteogenic response in murine calvarial osteoblasts and inhibited osteoclast formation from macrophage precursors by soluble factors in the CCF CM. Typically, IL-6 was introduced directly into the CCF CM to enhance the osteocyte-stimulated effect. Most interestingly, we found that when murine calvarial osteoblasts and osteoclast precursors were treated with IL-6 neutralizing antibody and CCF CM simultaneously, the stimulating effect was blocked significantly but still higher than the control group exposed to Static conditioned medium. These data confirm that osteocytes can directly control the differentiation and activity of both osteoblasts and osteoclasts under mechanical loading via soluble factors. Furthermore, the regulation was exerted by production of the mechano-sensitive cytokine, IL-6.During vigorous activities, maximal strain measured for the human and animal locomotion ranges from 2000 to 3500 με (1000 με = 0.1% change in length from the original length) at a low frequency of 1–3 Hz.36,37 In this study, MLO-Y4 osteocytes were exposed to CCF with 2000 με at a frequency of 2 Hz. Thus the strains applied on MLO-Y4 cells fall within physiological range in our experimental conditions. It is known that physical mechanical loading enhances anabolic response in bone mass through a positive shift in the balance between bone formation and bone resorption. Recent evidences have confirmed the idea that osteocytes are indeed an essential mediator of osteoblasts and osteoclasts, especially under mechanical loading. The results that CCF promoted IL-6 production in osteocytes were in good agreement with those of our previous studies and other reports.11,12,33,38
IL-6 type cytokines are important regulators of osteoblast activities. However, the role of IL-6 in osteoblast regulation is still controversial and not fully clarified. In this study, osteoblast proliferation did not show any significant differences when the cells were cultured in the presence of secreted factors from CCF-induced osteocytes or/and IL-6 factor. Kaneshiro S. et al. also reported that IL-6 did not affect osteoblast proliferation.23 However, another study found that IL-6 in combination with soluble sIL-6R markedly decreased the proliferation of the human osteoblastic cell line MG-63.39 Inversely, the proliferation marker Ki67 was increased by IL-6-enhanced soluble factors secreted by osteocytes under mechanical loading.38 Duration of osteogenic differentiation of osteoblasts or their progenitors ranged from 2 weeks to 4 weeks.40–46 When the cells were treated with osteogenic factors, remarkable mineralization may occur between 2 to 3 weeks. However, for time-dependent differentiation or inhibitory treatments, osteodifferentiation should be further evaluated for a long period at least up to 4 weeks. In this study, marked mineralization was detected after murine osteoblasts were incubated for 3 weeks. We showed that either CCF CM or IL-6/sIL-6 enhanced ALP activity, expression of key osteoblast differentiation markers (Runx2, ALP, and OCN) and bone mineralization in murine calvarial osteoblasts. The osteogenic response was even greater when the osteoblasts were synergistically co-cultured with IL-6 in the CCF-induced conditioned medium. The neutralizing antibody to IL-6 inhibited CCF CM-induced osteoblast differentiation, strongly suggesting that IL-6 mediated osteogenic response in osteoblasts cultured in CCF CM, which is consistent with the finding of previous studies.15,39 In addition, IL-6/sIL-6R stimulates the differentiation of mesenchymal stem cells (MSCs) toward the osteoblastic lineage and ALP activity.17,47 In the contrast, IL-6 treatments on osteoblast precursors or osteoblasts have shown either no effect or a small effect.13,48 Other reports even indicate an inhibitory effect of IL-6-type cytokines on bone formation and marker expression in vitro.23,49 The possible explanation is that the effects of IL-6 cytokines on the osteogenic response on osteoblasts or osteoblasts precursors were evaluated in the absence of IL-6R. Osteoblasts, MSCs and other bone stromal cells produce IL-6 cytokines; thus osteoblasts/stromal cells are exposed to an IL-6-rich environment. As osteoblasts and osteoprogenitors express low levels of mIL-6R,17,50 sIL-6R is necessary for IL-6 to exert its effects on osteoblast differentiation. The absence of IL-6R in these cells prevents the osteogenic effect of IL-6. On the other hand, the effects of IL-6 on osteoblasts depend on the stage of differentiation. IL-6-type cytokines may act as a differentiation promoter on osteoblast precursors at the early stage, indicated by high mRNA levels of Runx2 (a master gene for the differentiation of progenitor cells into osteoblasts) and ALP.47,51,52 However, the effects on more mature cells and OCN expression at the late stage of osteoblast differentiation are still controversial.23,53 The terminal differentiation of osteoblasts is to osteocyte, which is characterized by reduced proliferation, osteoblastic marker expression, bone nodule formation and enhanced apoptosis.22,38
We also conducted an experiment to determine whether osteocytes may function as mechanotransducers by controlling osteoclast differentiation via soluble factors. The results showed that RAW264.7 macrophages cultured in CCF CM with or without IL-6 showed significantly decreased osteoclast formation and expression of osteoclastic markers relative to those cultured in the conditioned medium from the static control. The addition of IL-6 alone into the static CM also had inhibitory effects on osteoclast differentiation. The neutralizing IL-6 antibody, even in the presence of CCF CM, strongly abolished the osteoclastogenesis and mRNA expression of osteoclastic markers. The results confirmed that osteocytes inhibited local osteoclast differentiation via IL-6 under mechanical loading. Our finding was consistent with the results of several previous studies, which have indicated that IL-6 reduced the RANKL-induced osteoclastogenesis and expression of osteoclast markers in osteoclast precursors. IL-6 even diverts RAW264.7 cells to the macrophage lineage to suppress RANKL-induced osteoclast formation.19,20 In the contrast, other studies have implicated IL-6 in pathological situations related to bone loss, such as multiple myeloma,54 Paget's disease,55 periodontal disease,56 and rheumatoid arthritis.57 The pre-osteoclastic activity of IL-6 and its soluble receptor is enhanced by an indirect mechanism due to the production of RANKL by cells in bone tissue, which in turn results in osteoclast formation and differentiation.24,25 Besides, Kudo et al. have argued that IL-6 may directly promote osteoclastogenesis through a RANKL-independent mechanism.58
In this study, we proved that the neutralizing antibody to IL-6 only partially blocks osteocyte-stimulated effects on osteoblast and osteoclast activities via soluble factors under mechanical loading. It is possible that other factors such as DMP1, ATP and IGF-1 may be involved in the regulation of osteocytes-induced bone remodeling in a synergistic manner. In addition, we observed that mechanical loading increased OCN expression and decreased RANKL/OPG ratio in osteocytes, which may also enhance the anabolic effects of IL-6 under mechanical loading.
Gp130 is found in almost all cells and organs, whereas mIL-6R expression in osteoblasts and osteoclast macrophages was weak.19,39,50 Thus, the transsignaling by IL-6/sIL-6R appears to be critical to the IL-6 pleiotropic functions of IL-6. It is known that IL-6 exerts its action through the JAK/STAT3 and SHP2/ERK signal transduction pathways, although their main implications in osteoblasts and osteoclasts are still controversial.17,20,29,59–61 Using the specific inhibitors S3I-201 and U0126 to block the STAT3 and ERK1/2 signaling pathways, we confirmed that concomitant activation of the two signal was involved in IL-6-induced osteoclastogenesis and osteoclast differentiation in the presence of exogenous sIL-6R. IL-6-induced STAT3 and ERK signaling pathways negatively regulated each other in cultured osteoclastic macrophages RAW264.7. When one signal was abolished, another signal was enhanced by reciprocal regulation due to the crosstalk between IL-6 activated signaling pathways. However, the specific inhibitor U0126 did not abolished the stimulating effects of IL-6 in murine calvarial osteoblasts. Only the specific inhibition of STAT3 by S3I-201 totally abolished IL-6-induced osteoblast differentiation, suggesting that the STAT3 signal in osteoblasts plays a key role in osteoblast differentiation. Other studies also argue IL-6 promotes osteoblast differentiation through the gp130-mediated STAT3 pathway in vitro.15,17 Itoh et al. using STAT3 knock out transgenic mice have revealed important in vivo roles of IL-6 in the bone formation.62 On the other hand, activation of STAT3 signal in osteoblasts or stromal cells by IL-6 stimulation is the key event leading to RANKL production for induction of osteoclast formation. Previous studies have indicated the inhibitory effect of MAPK in osteoclastogenesis.57,59,63 The role of STAT3 in osteoclasts is still controversial. Duplomb et al. have confirmed that IL-6 inhibits RANKL-induced osteoclastogenis through STAT3 activation.19 STAT3 is a pro-osteoclastic molecule that acts by enhancing RANKL expression; in the absence of this pathway, expression of RANKL is reduced.64 Moreover, Sims et al. used gp130 knock in transgenic mice to attenuate either the STAT1/3 or MAPK signaling pathways and showed that IL-6-induced STAT1/3 signal in osteoblasts enhance RANKL-dependent osteoclastogenesis, whereas osteoclast formation is inhibited by gp130 family cytokines, acting through MAPK signal within the osteoclast precursors.65 In physiological conditions, the primary function of IL-6 cytokines act via gp130 signal in osteoblast lineage is to stimulate osteoblast differentiation, rather than promote bone resorption. However, under pathological conditions when IL-6 and mIL-6R or sIL-6R are abundant, the gp130 signal pathway in osteoblasts might lead to osteoclast differentiation.23,38,53,61
5 Conclusions
Osteocytes not only represent the final descendent of the osteoblasts, but they are also major mechanosensory cells that regulate bone remodeling by controlling bone formation and bone resorption. These functions are exerted by the production of IL-6 under mechanical loading, which in turn enhances the osteogenic response and inhibits osteoclast differentiation through activation of the STAT3 and ERK signaling pathways. In conclusion, the present study reveals a key role for STAT3 during the IL-6-induced osteogenic response, and demonstrates the balance between STAT3 and MAPK ERK signals in bone resorption. Activation of the two signaling pathways by IL-6 induces distinct biological responses, which interact with each other to transduce an integrated signal for bone metabolism.Conflicts of interest
The authors have no conflicts of interest to report.Acknowledgements
We thank Dr L. F. Bonewald from the Department of Oral Biology, University of Missouri at Kansas City, Kansas City, Missouri, USA, for the gift of the MLO-Y4 cell line. This work was financially supported by the National Natural Science Foundation of China (No. 81300856) and College Students Innovative Project of Sun Yat-sen University (No. 201701129).References
- J. H. Chen, C. Liu, L. You and C. A. Simmons, J. Biomech., 2010, 43, 108–118 CrossRef PubMed.
- S. Allison, P. Baldock and H. Herzog, Peptides, 2007, 28, 320–325 CrossRef CAS PubMed.
- L. F. Bonewald, J. Bone Miner. Res., 2011, 26, 229–238 CrossRef CAS PubMed.
- B. S. Noble, Arch. Biochem. Biophys., 2008, 473, 106–111 CrossRef CAS PubMed.
- A. F. Taylor, M. M. Saunders, D. L. Shingle, J. M. Cimbala, Z. Zhou and H. J. Donahue, Aust. J. Pharm., 2007, 292, C545–C552 CAS.
- Y. Kitase, L. Barragan, H. Qing, S. Kondoh, J. X. Jiang, M. L. Johnson and L. F. Bonewald, J. Bone Miner. Res., 2010, 25, 2657–2668 CrossRef PubMed.
- J. Rubin, T. C. Murphy, L. Zhu, E. Roy, M. S. Nanes and X. Fan, J. Biol. Chem., 2003, 278, 34018–34025 CrossRef CAS PubMed.
- Y. Y. Kong, H. Yoshida, I. Sarosi, H. L. Tan, E. Timms, C. Capparelli, S. Morony, A. J. Oliveira-dos-Santos, G. Van, A. Itie, W. Khoo, A. Wakeham, C. R. Dunstan, D. L. Lacey, T. W. Mak, W. J. Boyle and J. M. Penninger, Nature, 1999, 397, 315–323 CrossRef CAS PubMed.
- D. G. Winkler, M. K. Sutherland, J. C. Geoghegan, C. Yu, T. Hayes, J. E. Skonier, D. Shpektor, M. Jonas, B. R. Kovacevich and K. Staehling-Hampton, EMBO J., 2003, 22, 6267–6276 CrossRef CAS PubMed.
- S. Harris, J. Gluhak-Heinrich, M. Harris, W. Yang, L. Bonewald, D. Riha, P. Rowe, A. Robling, C. Turner and J. Feng, J. Musculoskeletal Neuronal Interact., 2007, 7, 313 CAS.
- C. Sanchez, O. Gabay, C. Salvat, Y. Henrotin and F. Berenbaum, Osteoarthritis Cartilage, 2009, 17, 473–481 CrossRef CAS PubMed.
- W. Chen, Y. Ma, H. Ye, Y. He, X. Li, J. Li, Z. Zhu and H. Wang, Biochem. Biophys. Res. Commun., 2010, 401, 339–343 CrossRef CAS PubMed.
- M. A. Fang and T. J. Hahn, J. Bone Miner. Res., 1991, 6, 133–139 CrossRef CAS PubMed.
- C. H. Kim, S. L. Cheng and G. S. Kim, Endocr. Res., 1997, 23, 181–190 CrossRef CAS PubMed.
- T. Bellido, V. Z. Borba, P. Roberson and S. C. Manolagas, Endocrinology, 1997, 138, 3666–3676 CrossRef CAS PubMed.
- D. Heymann and A.-V. Rousselle, Cytokine, 2000, 12, 1455–1468 CrossRef CAS PubMed.
- A. Erices, P. Conget, C. Rojas and J. J. Minguell, Exp. Cell Res., 2002, 280, 24–32 CrossRef CAS PubMed.
- L. C. Yeh, M. C. Zavala and J. C. Lee, J. Cell. Physiol., 2002, 190, 322–331 CrossRef CAS PubMed.
- L. Duplomb, M. Baud'Huin, C. Charrier, M. Berreur, V. Trichet, F. Blanchard and D. Heymann, Endocrinology, 2008, 149, 3688–3697 CrossRef CAS PubMed.
- F. Yoshitake, S. Itoh, H. Narita, K. Ishihara and S. Ebisu, J. Biol. Chem., 2008, 283, 11535–11540 CrossRef CAS PubMed.
- R. L. Jilka, R. S. Weinstein, T. Bellido, A. M. Parfitt and S. C. Manolagas, J. Bone Miner. Res., 1998, 13, 793–802 CrossRef CAS PubMed.
- C. Chipoy, M. Berreur, S. Couillaud, G. Pradal, F. Vallette, C. Colombeix, F. Redini, D. Heymann and F. Blanchard, J. Bone Miner. Res., 2004, 19, 1850–1861 CrossRef CAS PubMed.
- S. Kaneshiro, K. Ebina, K. Shi, C. Higuchi, M. Hirao, M. Okamoto, K. Koizumi, T. Morimoto, H. Yoshikawa and J. Hashimoto, J. Bone Miner. Metab., 2013, 32, 378–392 CrossRef PubMed.
- K. T. Steeve, P. Marc, T. Sandrine, H. Dominique and F. Yannick, Cytokine Growth Factor Rev., 2004, 15, 49–60 CrossRef CAS.
- P. Palmqvist, E. Persson, H. H. Conaway and U. H. Lerner, J. Immunol., 2002, 169, 3353–3362 CrossRef CAS.
- X. Yang, B. F. Ricciardi, A. Hernandez-Soria, Y. Shi, N. Pleshko Camacho and M. P. G. Bostrom, Bone, 2007, 41, 928–936 CrossRef CAS PubMed.
- F. De Benedetti, N. Rucci, A. Del Fattore, B. Peruzzi, R. Paro, M. Longo, M. Vivarelli, F. Muratori, S. Berni and P. Ballanti, Arthritis Res., 2006, 54, 3551–3563 CrossRef CAS PubMed.
- A. Rufo, A. Del Fattore, M. Capulli, F. Carvello, L. De Pasquale, S. Ferrari, D. Pierroz, L. Morandi, M. De Simone and N. Rucci, J. Bone Miner. Res., 2011, 26, 1891–1903 CrossRef CAS PubMed.
- P. Heinrich, I. Behrmann, S. Haan, H. Hermanns, G. Muller-Newen and F. Schaper, Biochem. J., 2003, 374, 1–20 CrossRef CAS PubMed.
- F. Blanchard, L. Duplomb, M. Baud'huin and B. Brounais, Cytokine Growth Factor Rev., 2009, 20, 19–28 CrossRef CAS PubMed.
- E. R. C. Draper and A. E. Goodship, J. Biomech., 2003, 36, 1497–1502 CrossRef PubMed.
- M. Bottlang, M. Simnacher, H. Schmitt, R. A. Brand and L. Claes, Biomed. Eng., 1997, 42, 305–309 CrossRef CAS PubMed.
- J. Yin, Z. Hao, Y. Ma, S. Liao, X. Li, J. Fu, Y. Wu, J. Shen, P. Zhang, X. Li and H. Wang, Cell Biol. Int., 2014, 38, 591–598 CrossRef CAS PubMed.
- J. Liu, T. Liu, Y. Zheng, Z. Zhao, Y. Liu, H. Cheng, S. Luo and Y. Chen, Biochem. Biophys. Res. Commun., 2006, 348, 1167–1173 CrossRef CAS PubMed.
- P. Ducy, M. Starbuck, M. Priemel, J. Shen, G. Pinero, V. Geoffroy, M. Amling and G. Karsenty, Genes Dev., 1999, 13, 1025–1036 CrossRef CAS PubMed.
- C. T. Rubin and L. E. Lanyon, J. Theor. Biol., 1984, 107, 321–327 CrossRef CAS PubMed.
- S. P. Fritton, K. J. McLeod and C. T. Rubin, J. Biomech., 2000, 33, 317–325 CrossRef CAS PubMed.
- A. Bakker, R. Kulkarni, J. Klein-Nulend and W. Lems, J. Dent. Res., 2014, 93, 394–399 CrossRef CAS PubMed.
- R. Nishimura, K. Moriyama, K. Yasukawa, G. R. Mundy and T. Yoneda, J. Bone Miner. Res., 1998, 13, 777–785 CrossRef CAS PubMed.
- T. R. Nayak, H. Andersen, V. S. Makam, C. Khaw, S. Bae, X. Xu, P. L. Ee, J. H. Ahn, B. H. Hong, G. Pastorin and B. Ozyilmaz, ACS Nano, 2011, 5, 4670–4678 CrossRef CAS PubMed.
- E. Birmingham, G. Niebur, P. McHugh, G. Shaw, F. Barry and L. McNamara, Eur. Cells Mater., 2012, 23, 13–27 CrossRef CAS PubMed.
- E. Balint, P. Szabo, C. Marshall and S. Sprague, Bone, 2001, 28, 21–28 CrossRef CAS PubMed.
- C. Shui and A. Scutt, J. Bone Miner. Res., 2001, 16, 731–741 CrossRef CAS PubMed.
- J. de Boer, R. Siddappa, C. Gaspar, A. van Apeldoorn, R. Fodde and C. van Blitterswijk, Bone, 2004, 34, 818–826 CrossRef CAS PubMed.
- G. D'Ippolito, S. Diabira, G. A. Howard, B. A. Roos and P. C. Schiller, Bone, 2006, 39, 513–522 CrossRef PubMed.
- S. Sundelacruz, M. Levin and D. L. Kaplan, PLoS One, 2008, 3, e3737 Search PubMed.
- Y. Taguchi, M. Yamamoto, T. Yamate, S. C. Lin, H. Mocharla, P. De Togni, N. Nakayama, B. F. Boyce, E. Abe and S. C. Manolagas, Proc. Assoc. Am. Physicians, 1998, 110, 559–574 CAS.
- A. J. Littlewood, L. A. Aarden, D. B. Evans, R. G. G. Russell and M. Gowen, J. Bone Miner. Res., 1991, 6, 141–148 CrossRef CAS PubMed.
- Y. P. Li and P. Stashenko, J. Immunol., 1992, 148, 788–794 CAS.
- T. Bellido, N. Stahl, T. J. Farruggella, V. Borba, G. D. Yancopoulos and S. C. Manolagas, J. Clin. Invest., 1996, 97, 431–437 CrossRef CAS PubMed.
- K. Iwasaki, M. Komaki, K. Mimori, E. Leon, Y. Izumi and I. Ishikawa, J. Dent. Res., 2008, 87, 937–942 CrossRef CAS PubMed.
- S. Fukuyo, K. Yamaoka, K. Sonomoto, K. Oshita, Y. Okada, K. Saito, Y. Yoshida, T. Kanazawa, Y. Minami and Y. Tanaka, Rheumatology, 2014, 53, 1282–1290 CrossRef CAS PubMed.
- Y. Li, C.-M. Bäckesjö, L.-A. Haldosén and U. Lindgren, Cytokine, 2008, 43, 165–173 CrossRef CAS PubMed.
- B. Klein, J. Wijdenes, X.-G. Zhang, M. Jourdan, J.-M. Boiron, J. Brochier, J. Liautard, M. Merlin, C. Clement and B. Morel-Fournier, Blood, 1991, 78, 1198–1204 CAS.
- G. D. Roodman, N. Kurihara, Y. Ohsaki, A. Kukita, D. Hosking, A. Demulder, J. F. Smith and F. R. Singer, J. Clin. Invest., 1992, 89, 46–52 CrossRef CAS PubMed.
- B. G. Loos, J. Craandijk, F. J. Hoek, P. M. W.-V. Dillen and U. V. D. Velden, J. Periodontol., 2000, 71, 1528–1534 CrossRef CAS PubMed.
- P. K. Wong, I. K. Campbell, P. J. Egan, M. Ernst and I. P. Wicks, Arthritis Res., 2003, 48, 1177–1189 CrossRef CAS PubMed.
- O. Kudo, A. Sabokbar, A. Pocock, I. Itonaga, Y. Fujikawa and N. A. Athanasou, Bone, 2003, 32, 1–7 CrossRef CAS PubMed.
- N. Franchimont, S. Wertz and M. Malaise, Bone, 2005, 37, 601–606 CrossRef CAS PubMed.
- X. H. Liu, A. Kirschenbaum, S. Yao and A. C. Levine, in Vitamins & Hormones, ed. L. Gerald, Academic Press, 2006, vol. 74, pp. 341–355 Search PubMed.
- R. W. Johnson, H. J. Brennan, C. Vrahnas, I. J. Poulton, N. E. McGregor, T. Standal, E. C. Walker, T.-T. Koh, H. Nguyen, N. C. Walsh, M. R. Forwood, T. J. Martin and N. A. Sims, J. Bone Miner. Res., 2014, 29, 1492–1505 CrossRef CAS PubMed.
- S. Itoh, N. Udagawa, N. Takahashi, F. Yoshitake, H. Narita, S. Ebisu and K. Ishihara, Bone, 2006, 39, 505–512 CrossRef CAS PubMed.
- H. Hotokezaka, E. Sakai, K. Kanaoka, K. Saito, K.-I. Matsuo, H. Kitaura, N. Yoshida and K. Nakayama, J. Biol. Chem., 2002, 277, 47366–47372 CrossRef CAS PubMed.
- T. Mori, T. Miyamoto, H. Yoshida, M. Asakawa, M. Kawasumi, T. Kobayashi, H. Morioka, K. Chiba, Y. Toyama and A. Yoshimura, Int. Immunol., 2011, 23, 701–712 CrossRef CAS PubMed.
- N. A. Sims, B. J. Jenkins, J. M. Quinn, A. Nakamura, M. Glatt, M. T. Gillespie, M. Ernst and T. J. Martin, J. Clin. Invest., 2004, 113, 379–389 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09308j |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |