
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic hollow mesoporous hydroxyapatite microsphere with controlled morphology, entrapment efficiency and degradability for cancer therapy
Wei Qiao†
 ac,
Xinmiao Lan†b,
James K. H. Tsoia,
Zhuofan Chend,
Richard Y. X. Su*b,
Kelvin W. K. Yeung*c and
Jukka P. Matinlinna*a
ac,
Xinmiao Lan†b,
James K. H. Tsoia,
Zhuofan Chend,
Richard Y. X. Su*b,
Kelvin W. K. Yeung*c and
Jukka P. Matinlinna*a
aDental Materials Science, Applied Oral Sciences, Faculty of Dentistry, The University of Hong Kong, Rm.1F30, 1/F, Prince Philip Dental Hospital, 34 Hospital Road, Sai Ying Pun, Hong Kong S.A.R., P. R. China. E-mail: jpmat@hku.hk; Fax: +852 25489464; Tel: +852 28590380
bDepartment of Oral and Maxillofacial Surgery, Faculty of Dentistry, The University of Hong Kong, Rm.2B88, 2/F, Prince Philip Dental Hospital, 34 Hospital Road, Sai Ying Pun, Hong Kong S.A.R., P. R. China. E-mail: richsu@hku.hk; Fax: +852 28575570; Tel: +852 28590267
cDepartment of Orthopaedics and Traumatology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Rm.515, 5/F, Professorial Block, Queen Mary Hospital, 102 Pokfulam Road, Hong Kong S.A.R., P. R. China. E-mail: wkkyeung@hku.hk; Fax: +852 28174392; Tel: +852 22554654
dDepartment of Oral Implantology, Hospital of Stomatology, Guanghua School of Stomatology, Institute of Stomatological Research, Sun Yat-sen University, Guangzhou, P. R. China
First published on 19th September 2017
Abstract
Hollow hydroxyapatite (HAp) microspheres bearing hierarchical structures have been one of the most popular carriers for drug delivery. However, it's been challenging to find an approach that can simultaneously control the morphology, degradability and drug loading/releasing properties of the microsphere. In this study, hollow mesoporous microspheres composed of nano-sized HAp crystals were hydrothermally synthesized from hollow CaCO3 precursors prepared via sodium dodecyl sulfate (SDS)-assisted precipitation. The particle size, hollow structure and wall thickness of the fabricated hollow mesoporous carbonated HAp microsphere (CHAM) could be facilely regulated by tailoring the concentration of SDS in the reaction. The achieved carbonate-containing CHAMs were demonstrated to be suitable for drug delivery through either surface attachment or intracellular engulfment without causing cytotoxicity. In addition, their distinguished biodegradability and sustained drug release properties in a pH-dependent manner might allow them to be used in cancer treatment by targeting the acidic microenvironment in a solid tumor. Moreover, the CHAM prepared using SDS at its critical micelle concentration (CMC) was suggested to be optimal for anti-cancer therapy due to its highest entrapment efficiency of cis-diammineplatinum(II) dichloride (CDDP) and the strongest killing effect on human squamous cell carcinoma in vitro.
1. Introduction
Hydroxyapatite (HAp) microspheres are among the most extensively studied carriers for the delivery of drugs, proteins and genes. As the major inorganic component of human hard tissues (e.g. bones and teeth), the biocompatibility and biodegradability of HAp microsphere are superior to many other hybrid microspheres.1–3 Moreover, the facile modification of HAp microspheres with relatively low cost and high efficiency allows them to be very versatile for different medical applications. In recent years, a number of studies have been focusing on optimizing the drug release profile of HAp based delivery systems through the control of particle size at the nanoscale,4,5 the addition of hierarchical structures6–9 or the modification of organic components.10,11 Hollow HAp microsphere bearing mesoporous structure is one of the most popular design among the drug delivery system, due to its high specific surface area, low density, high drug loading efficiency, and controlled releasing kinetic.12–15 It was reported that hollow mesoporous microsphere built by hydroxyapatite nanofibers displayed higher drug loading capacity and a controlled pH-responsive drug release profile, which is able to kill cancer cells with less damage to normal cells.16 Indeed, the distinguished properties of hollow HAp microsphere have allow it to be used for the delivery of antibiotics,6,17–19 anticancer drugs13 or cytokines20,21 in various applications.There have been a number of methods reported for the fabrication of hollow HAp microsphere, including the template assisted hydrothermal reaction,15,16,22 spray freezing and drying,23,24 ice-template spray freezing,19,25 microwave assisted transformation,26 etc. In order to achieve the hollow space in the HAp microspheres, a core template that can be sacrificed following the formation of HAp shell, or a hollow shaped precursor that can be directly transformed into HAp is needed. Although a variety of reagents (e.g., poly(aspartic acid),27 hexadecyl trimethylammonium bromide (CTAB),22 Deoxyribonucleic acid (DNA)15 block copolymer,28 and sodium dodecyl sulfate (SDS)6,26) have been known be able to produce hollow structures in the HAp microsphere, reports on the regulation of the hollow size and wall thickness are still lacking. The size of microsphere decides its loading capacity and target location, while the wall thickness and degradability of the microsphere are closely related to the release profile of the loaded drugs. Therefore, it's both vital and challenging to develop HAp microsphere with controlled morphology, degradability and drug loading/releasing properties based on their specific usage.
Sodium dodecyl sulfate, SDS is a synthetic organic compound with the formula CH3(CH2)11SO4Na. Its amphiphilic properties, as well as low cytotoxicity or carcinogenicity, allow it to be widely used in the production of detergents, cosmetic, medicine and food.29 It was used, together with polyvinylpyrrolidone (PVP), to achieve hollow CaCO3 microspheres via a precipitation reaction at a relatively low concentration.30 However, in some recent studies, the concentration of SDS used in the preparation of hollow HAp microspheres could be up to 0.1 M (28.8 mg mL−1),6,12,26 which is much higher than its critical concentration to cause cytotoxicity.31 Even multiple washing and rinsing steps have been claimed in the SDS assisted synthesis, considering the sustained release profile of the hollow microspheres, there still remains some concerns about the side effects that might be caused by the residual SDS. Moreover, even though SDS is among one of the most frequently used anionic surfactant for the synthesis of hollow structure in HAp microspheres, investigations on the effects of its concentration on the morphology of microspheres are extremely limited. It has been known for long that SDS micelles can be formed at its critical micelle concentration (CMC)32 (i.e. 8.2 mM in pure water at 25 °C (ref. 33)), and grow with the increase in the concentration of surfactant.34 This implies that the size and morphology of the hollow HAp microspheres or their precursors might be controlled by the concentration of SDS used in the synthesis.
Therefore, in the present study, different concentrations of SDS were used in fast precipitation for the preparation of hollow CaCO3 precursors with a different particle size, hollow structure and wall thickness. The precursors were then directly transformed into hollow mesoporous microspheres composed of nano-sized carbonated hydroxyapatite crystals through hydrothermal reaction. The morphology, chemical composition, phrase purity, size distribution, porosity and degradability of the samples were systematically characterized for fully elucidating the effects of SDS concentration on the microspheres. In addition, the microspheres were loaded with cis-diammineplatinum(II) dichloride (CDDP) for evaluating drug loading efficiency and releasing properties in different pH environment. The biocompatibility of the microspheres and the cytotoxicity of the CDDP loaded microspheres were tested in vitro using a human squamous cell carcinoma cell line. The extracellular attachment of the microspheres was observed under scanning electron microscopy, while the intracellular engulfment of the rhodamine B isothiocyanate (RITC) conjugated microspheres was visualized using confocal microscopy. The aim of this study was to provide a straight-forward approach to achieve hollow mesoporous carbonated HAp microsphere (CHAM) with controlled size, loading capability and degradability for sustained drug delivery, and to test their potential applications as drug carriers targeting cancer cells in vitro.
2. Experimental section
2.1. Preparation of HAp microspheres
Hollow mesoporous CaCO3 precursors were prepared by fast precipitation using sodium dodecyl sulfate (SDS, Sigma Aldrich, USA) as the surfactant. In brief, 250 mL 0.2 M CaCl2 (AnalaR grade, BDH, Poole, UK) was mixed with an equal volume of SDS at different concentrations (i.e. 1 mg mL−1, 2.33 mg mL−1, 4.66 mg mL−1, and 10 mg mL−1) under vigorous stirring. Then same volume of 0.2 M Na2CO3·10H2O (AnalaR grade, BDH) was added to the solution and stirred for 1.5 h. The achieved CaCO3 templates were obtained by filtering off the liquid with a vacuum pump. Then, the samples were rinsed eight times with distilled water and two times with ethanol before being dried in an oven at 120 °C overnight.The dried CaCO3 precursors were mixed with 0.2 M Na2HPO4 (AnalaR grade, BDH), and drop-wisely added with 0.2 M ammonia (AnalaR grade, BDH) until the pH value reached 11. Then the solution was transferred into a Teflon-lined stainless steel autoclave for a hydrothermal reaction at 180 °C for 24 h. The resulting solids were collected by centrifugation, thoroughly rinsed with distilled water and ethanol, and then dried at 120 °C for 24 h. According to the concentration of SDS used in the precursor preparation, the as-prepared samples were noted as A-CHAM (1 mg mL−1 SDS), B-CHAM (2.33 mg mL−1 SDS), C-CHAM (4.66 mg mL−1 SDS) and D-CHAM (10 mg mL−1 SDS), respectively. All of the samples were separately stored in a vacuum desiccator over silica gel before further testing. The schematic diagram for sample preparation were illustrated in Fig. 1.
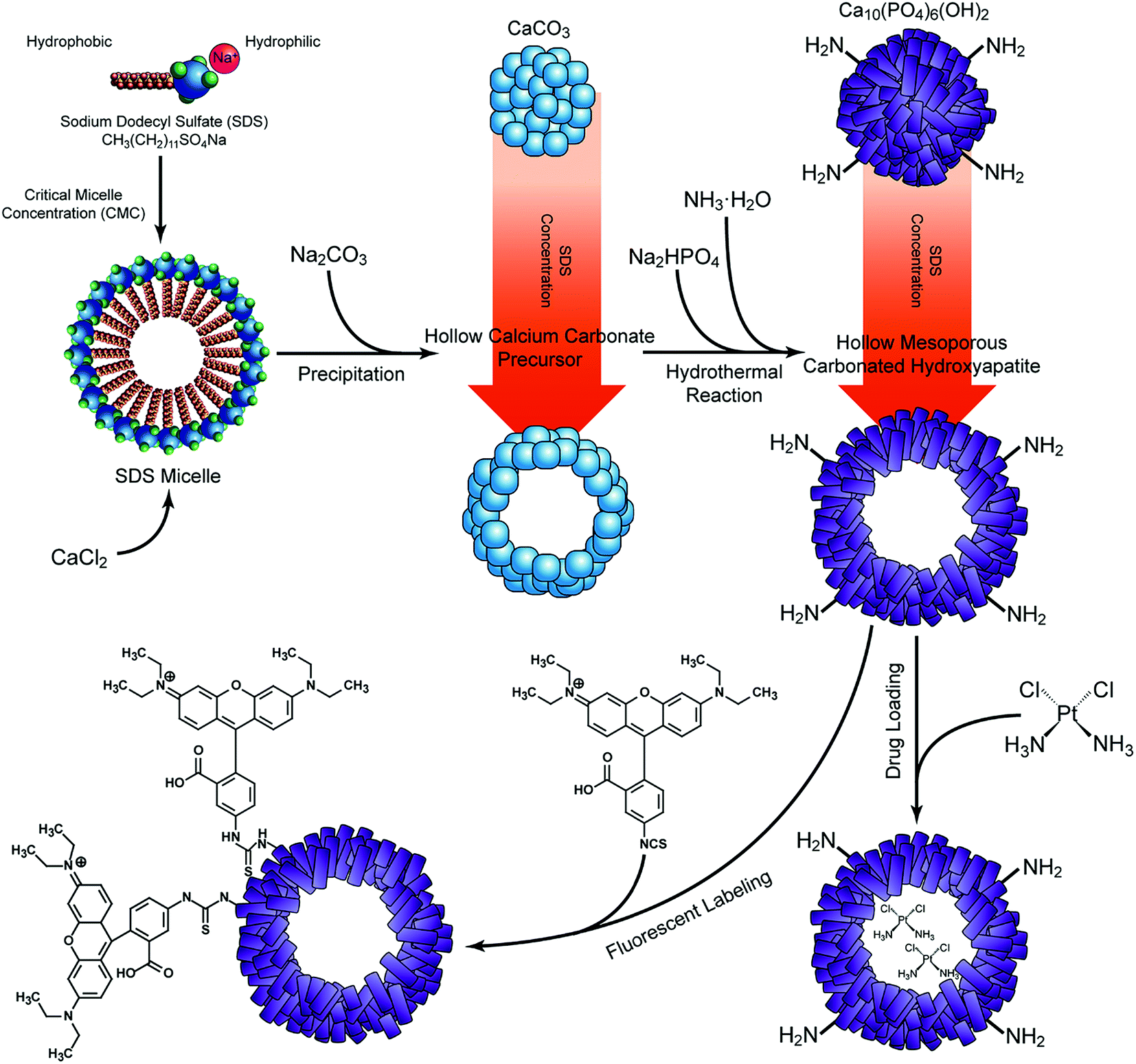 | ||
| Fig. 1 Schematic diagram showing the synthesis of CHAMs, as well as the drug loading and fluorescent labeling process. | ||
2.2. Sample characterization
The morphology of the four groups of CHAMs were observed under transmission electron microscopy (TEM, CM100, Philips Electron Optics, The Netherlands) and scanning electron microscopy (SEM, S-4800, Hitachi, Tokyo, Japan). For TEM observation, images were taken in the bright field mode at an accelerating voltage of 100 kV, and the selected area electron diffraction (SAED) patterns were captured for each sample. The SEM images of the CHAMs and the crystals were captured at 5 kV, with the elemental composition and distribution examined by energy-dispersive X-ray spectroscopy (EDX) and SEM-EDX mapping. The functional groups of CHAMs were identified using Fourier-transform infrared spectroscopy (FTIR, Vector 33, Bruker Optics, Ettlingen, Germany). IR spectra were collected in the transmittance mode with a scanning range of 4000–400 cm−1 at a 0.2 cm−1 resolution. Phrase analysis of CHAMs was done by using X-ray diffractometer (XRD, Empyrean, Panalytical, Eindhoven, The Netherlands). A scanning speed of 10° (2θ)/min and a step size of 0.01° were adopted over a 2θ range of 20–60°. A stoichiometric HAp pattern (JCPDS card #09-0432) were used as references for peak fitting in Jade software (v. 6.1, Materials Data, Livermore, California, USA). The particle size and the zeta potential of CHAMs were determined by dispersing the samples in 1 mM KCl solution, and measured by a dynamic light scattering particle analyzer (Nanotrac Wave II, Microtract, FL, USA). The Brunauer–Emmett–Teller (BET) specific surface area and the Barrett–Joyner–Halenda (BJH) pore-size distribution were recorded using a Quantachrome QuadraSorb SI instrument (Quantachrome, USA).2.3. Degradation
For in vitro degradation tests, 20 mg of A-CHAM, B-CHAM, C-CHAM and D-CHAM were, respectively, incubated in 10 mL phosphate buffered saline (PBS) solution at the pH of 3, 5 and 7, and the medium were maintained in a 37 °C shaker at 300 rpm for one month. 2 mL of the supernatant was collected from the mixture by centrifugation at designated time intervals, and the withdrawn solution was replaced by the same volume of fresh PBS at specific pH value. The degradation of CHAMs was determined by measuring the released calcium ions in the harvested supernatant with inductively coupled plasma atomic emission spectroscopy (ICP-OES, Thermo, Waltham, MA, USA). All the tests were repeated and carried out at least three times, and the data are shown in mean values with standard deviations.2.4. Drug entrapment efficiencies and releasing profiles
1.5 mL cis-diammineplatinum(II) dichloride (CDDP, Sigma-Aldrich, USA) solution at a concentration of 0.4 mg mL−1 was added to 20 mg of A-CHAM, B-CHAM, C-CHAM and D-CHAM, respectively. A tube filled with 0.4 mg mL−1 CDDP without any sample was set as a control. After gentle vortexing, the suspension was maintained in a shaker at 37 °C at 300 rpm in darkness. After 24 h, the CDDP loaded CHAMs were obtained by centrifugation, rinsed with distilled water twice, and freeze dried overnight. The unloaded CDDP in the supernatant was determined using ICP-OES, by measuring the concentration of platinum. The CDDP entrapment efficiency was calculated according to the formula: CDDP entrapment efficiency (%) = (Ptcontrol − Ptsupernatant)/Ptcontrol × 100%, while the loaded CDDP amount was calculated with the expression: CDDP loading content (mg per 20 mg CHAM) = (Ptcontrol − Ptsupernatant) × 1.5.In vitro drug releasing tests were done by adding PBS solution at the pH of 5 and 7 to the CDDP loaded CHAMs at a solid/liquid ratio of 2 mg mL−1, respectively. The mixture was maintained in a shaker at 37 °C at 300 rpm in darkness, 2 mL of the medium was periodically extracted for the measurement of platinum using ICP-OES, and the residual mixture was supplemented by 2 mL fresh PBS at the specific pH value. All the tests were executed at least three times.
2.5. Cytotoxicity assay
FaDu cells, a well-established human head and neck squamous cell carcinoma cell line was received from American Type Culture Collection (ATCC, VA, USA), and maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco, NY, USA) supplemented with 10% (vol/vol) fetal bovine serum (Gibco, USA) and 100 U mL−1 penicillin–streptomycin (Gibco, USA) at 37 °C in humidified atmosphere with 5% CO2. For studying the cytotoxicity of the hollow microspheres, FaDu cells were seeded in 96-well plate at a density of 5 × 103 cells per well. After 24 h incubation, the culture medium was replaced by fresh DMEM supplemented with A-CHAM, B-CHAM, C-CHAM or D-CHAM at a series of concentrations (i.e. 0.5 mg mL−1, 0.2 mg mL−1, 0.1 mg mL−1, and 0.01 mg mL−1). The cell viability was tested after 48 h co-culture using MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. In brief, the culture medium was replaced by 100 μL MTT solution and further incubated for 4 h. Next, 100 μL SDS–HCl was added and mixed thoroughly using the micropipette. The plate was maintained at 37 °C in humidified chamber for 5 h to ensure complete dissolution of formazan crystals. The absorbance was read at the wavelength of 570 nm using a microplate spectrophotometer (SpectraMax 340, Molecular Devices, CA, USA). The cytotoxicity of the CDDP loaded CHAMs were done by treating the FaDu cells with DMEM supplemented with 0.1 mg mL−1 CDDP loaded A-CHAM, B-CHAM, C-CHAM or D-CHAM. The cell survival rate was calculated by comparing the cell viability of the studied group to that of control group. Each sample was performed in triplicates.The cellular attachment and engulfment of microspheres were visualized by SEM (LEO 1530, Oberkochen, Germany) and a confocal laser scanning microscope (CLSM, LSM700, Zeiss, Jena, Germany). For SEM observation, FaDu cells incubated with CDDP loaded and unloaded CHAMs for 48 h were thoroughly rinsed with PBS for three times, and then fixed with 2.5% glutaraldehyde at 4 °C overnight. The samples were dehydrated with gradient alcohols and a vacuum freeze drier (ES-2030, Hitachi, Kyoto, Japan). Then, the samples were coated with the gold sputter (E1010, Hitachi ion sputter, Tokyo, Japan) for 15 s before SEM observation at 5 kV. For CLSM observation, CHAMs were prestained by dispersing the microspheres in 0.05 mg mL−1 rhodamine B isothiocyanate (RITC, Sigma-Aldrich, USA) in PBS with vigorous stirring for 4 h. The RITC-labeled CHAMs were harvested by centrifugation, and washed with distilled water and ethanol, before being used for the incubation with FaDu cells. After 48 h, the cells were washed with PBS three times, fixed by 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Then, the cytoskeleton and nuclei of the FaDu cells were respectively stained with FITC-phallotoxins (Sigma-Aldrich, USA) and Hoechst 33342 (ThermoFisher Scientific, USA).
2.6. Statistical analysis
For drug entrapment efficiency and cell survival rate, data are shown in mean values with standard deviations. One-way analysis of variance (ANOVA) was performed with SPSS (v.13.0, IBM SPSS, USA), and ‘P < 0.05’ was defined as the level of significant difference.3. Results and discussion
3.1. Sample preparation and characterization
The SEM and the attached TEM images (Fig. 2a–d) showed the changes in the morphology of the CaCO3 precursors: with the addition of SDS, the size of the microspheres increased from around 2 μm in A-CHAM group, to more than 5 μm in D-CHAM group. Additionally, the hollow space in the spheres was also enlarged with the increase in SDS concentration. Specifically, in A-CHAM group, most of the microspheres remained solid without obvious hollow inside. Whilst in D-CHAM group, due to the expansion of the hollow space, the sphere wall became so thin to become disconnected at specific site. Fig. 2e–h shows the morphology of the fabricated CHAMs through hydrothermal reaction. It can be seen that the hollow space inside the microspheres can be more clearly seen in group C and D, which is similar to the situations observed in CaCO3 precursors. However, with the growth in the size of hollow, the sphere walls become thinner in D-CHAM (i.e. ∼0.8 μm), making the sphere more fragile. Consequently, unlike the other groups, more broken and collapsed microspheres, or even HAp debris can be found in D-CHAMs. This is consistent with the previous finding showing that, when the concentration of SDS exceed 20 mM, more dispersed CaCO3 particles, instead of homogeneous hollow microspheres were present.30 | ||
| Fig. 2 SEM images with inserted TEM images for calcium carbonate precursors of A-CHAM (a), B-CHAM (b), C-CHAM (c) and D-CHAM (d); SEM images with inserted TEM images showing the morphology of A-CHAM (e), B-CHAM (f), C-CHAM (g) and D-CHAM (h) (scale bar = 1 μm). | ||
The high magnification TEM images and the corresponding SAED patterns shown in Fig. 3a–d demonstrate the CHAMs to be composed of nano-sized HAp crystals, dramatically different from the sheet-like crystals in their precursors. The morphology of HAp crystals in microspheres was reported to have significant influence on the adsorption of drugs and proteins. In the current study, the magnified images reveal there exists some changes in the crystals shape among the four groups of CHAMs: there are more needle-like shaped crystals in A-CHAM and B-CHAM, while more rod or polygon shaped crystals were observed in C-CHAM and D-CHAM. This indicated that the concentration of SDS can also affects the nanostructure of the fabricated HAp microspheres as other crystal modifiers (e.g., inositol hexakisphosphate35 and cyclohexane-1,2,3,4,5,6-hexacarboxylic acid36) did in the synthesis of hollow HAp microspheres. Moreover, it is evident in Fig. 3e and d that the well-crystalized and oriented HAp crystals in nanoscale arrange to form a great number of micro-pore structures, ∼100 nm in diameter on the surface of the microspheres, which should have crucial roles in increasing the adsorption area and accessibility of the hollow space in the microspheres.
 | ||
| Fig. 3 Magnified TEM (scale bar = 100 nm) images with corresponding SAED patterns (scale bar = 5 1/nm) showing the nano-sized HAp crystals of A-CHAM (a), B-CHAM (b), C-CHAM (c) and D-CHAM (d). Magnified SEM (scale bar = 100 nm) images showing the morphology of the HAp crystals and mesoporous structures of A-CHAM (e), B-CHAM (f), C-CHAM (g) and D-CHAM (h). | ||
The phase purity of the CHAMs was confirmed by XRD and demonstrated in Fig. 4a. The XRD patterns of the four groups of CHAMs were found to be consistent with the stoichiometric HAp reference (JCPDS card #09-0432), and peaks indicating other phases were present. However, the relatively sharper peaks in low SDS used groups suggested that with the addition of SDS in sample preparation, the crystallinity of CHAMs was intriguingly decreased. Moreover, as demonstrated in the expanded patterns at 31.5–33.5° (2θ) in Fig. 4b, unlike the other peaks, the (2 1 1), (1 1 2) and (3 0 0) peaks gradually shifted to a higher diffraction angles. In addition, the FTIR spectra of CHAMs were investigated and are shown in Fig. 4c. Characteristic bands corresponding to phosphate at 1091 cm−1 (ν3), 1030 cm−1 (ν1), 601 cm−1 (ν4) and 564 cm−1 (ν4), as well as hydroxyl at 3576 cm−1 and 634 cm−1 were evident. In addition, bands indicating the presence of carbonate were observed at 1461–1410 cm−1 (ν3) and 873 cm−1 (ν2), suggesting the CHAMs prepared in this study to be B-type carbonated HAp.37
 | ||
| Fig. 4 XRD patterns (a), expanded X-ray reflections (b) and FTIR spectra (c) of four groups of CHAMs; representative EDX spectrum and EDX-mapping images showing the elemental composition and distribution of the microspheres (d). | ||
The composition of the CHAMs was analyzed by EDX, with the representative EDX spectra and the corresponding mapping figures shown in Fig. 4d. The detailed elemental composition of the four groups of CHAMs is listed in Table 1. It can be seen that the major elements within the microspheres were calcium, phosphorus, oxygen and carbon, while only a very little amount of sodium (less than 2%) was detected. The distribution of calcium, phosphate, oxygen and sodium in the hollow CHAMs were further demonstrated to be evenly inside and outside of the spheres (Fig. 4d), which, along with the XRD and FTIR results, indicates the CaCO3 precursors to be fully transformed into carbonated HAp via hydrothermal reaction. Nevertheless, the increase in SDS usage barely resulted in a significant difference in the element composition among the four groups of CHAMs. The Ca/P ratio of the four groups of CHAMs was also similarly at around 1.4 level, without any noticeable difference. The appreciable existence of carbonate substitution in HAp determined by FTIR was confirmed by EDX. Given this, it is expected to play an important role in the pH-selective drug release property of the CHAMs, because of its pH-dependent dissolution behavior. It was found the carbonate containing HAp released around 42% less doxorubicin hydrochloride (DOX) at the physiological pH, while it maintained the same release profile in acidic environment as its counterpart without carbonate incorporation.38 That said, the presence of sodium in CHAM, though little, could have dramatic effects on the physicochemical and biological properties of the said materials. Firstly, the incorporation of sodium decreases the crystallinity of HAp,39 resulting in increased drug entrapment efficiency and more sustained drug release. Secondly, the sodium content was reported to contribute to the biocompatibility and osteoconductivity of HAp,40 which benefit the potential use of CHAMs as a bone grafting materials. Recently, trace ions like strontium has been reported to affect the size, morphology, and surface area of the microspheres, which crucially promote their performance.27,41 Therefore, it is rational to propose the discrepancy among four groups of CHAMs might also be caused by the sodium content in SDS.
| Sample | Atomic percentage (wt%) | ||||
|---|---|---|---|---|---|
| Ca | P | O | C | Na | |
| A-CHAM | 10.3 ± 0.8 | 7.7 ± 0.6 | 60.2 ± 2.1 | 20.4 ± 1.6 | 1.4 ± 0.3 |
| B-CHAM | 10.6 ± 1.8 | 7.1 ± 1.2 | 56.7 ± 5.8 | 19.9 ± 1.3 | 1.4 ± 0.2 |
| C-CHAM | 10.6 ± 0.9 | 7.4 ± 0.3 | 58.9 ± 0.2 | 21.3 ± 1.2 | 1.8 ± 0.05 |
| D-CHAM | 11.3 ± 0.9 | 7.8 ± 0.5 | 60.0 ± 4.7 | 19.5 ± 3.8 | 1.5 ± 0.3 |
The effects of SDS on the size of the microspheres were further confirmed by particle analyzer. Fig. 5a–d demonstrate the particle size distribution of the four groups of CHAMs. In A-CHAM, more than 90% of the microspheres are under 3 μm in diameter, while in B-CHAM group, more particles around 3 μm in diameter or even larger were detected. When the concentration of SDS reached the CMC in C group, the particle size increased to around 3–5 μm in diameter. However, in D-CHAM, where SDS concentration exceeded CMC, the narrow distribution of microsphere seen in the other groups disappeared. Instead, two sub-groups with the diameter of 0.6 and 5 μm were observed, respectively. This can be explained by the coexistence of intact microspheres and the fragments due to the breakage of the microspheres in D group, as observed in SEM. These results were similar to the earlier investigation showing that, when with PVP, high level of SDS could actually contribute to dispersed particles, instead of evenly distributed hollow CaCO3 microspheres.30 Collectively, it can be concluded the concentration of SDS extensively used in the earlier publication6,12,26 (i.e., 0.1 M) is actually not the optimal choice for the preparation of hollow HAp microspheres.
 | ||
| Fig. 5 Particle size distribution of A-CHAM (a), B-CHAM (b), C-CHAM (c) and D-CHAM (d). Desorption isotherm and inserted pore size distribution of CHAMs measured by N2 adsorption (e). Cumulative calcium release showing the in vitro degradation of CHAMs in PBS at pH 3, 5 and 7 (f). | ||
3.2. Biodegradability
The N2 adsorption–desorption isotherm of the four groups of CHAMs, as demonstrated in Fig. 5e, all exhibit the type H3/4 hysteresis loop, indicating the samples to be mesoporous structures composed with nanocrystals with slit-shaped and panel-shaped pores.42 The pore size distribution of CHAMs measured from the adsorption branches of the isotherms using the Barrett–Joyner–Halenda (BJH) method (Fig. 5e insets) suggests the pore structures in the microspheres range from 3 nm to more than 100 nm, which is consistent with the magnified SEM figures shown previously. However, the addition of SDS seems to contribute to more pores in 20–100 nm range, which might be related to more brittle sphere wall in high SDS used group. Moreover, the Brunauer–Emmett–Teller (BET) surface areas of the microspheres increased from 17.58 m2 g−1 in A-CHAM to 25.12 m2 g−1 in D-CHAM.The degradation of as-prepared CHAMs was evaluated by measuring the released calcium ions into the PBS at different pH values. The results shown in Fig. 5f indicate that the degradation speeds of CHAMs are greatly influenced by the environmental pH value. In the initial 6 h, the CHAMs degraded much more quickly in acidic condition than in neutral condition. This can be caused by the fast dissolution of the less crystalized content, such as the incorporated carbonate. After the first day, the degradation rate in both acid and neutral condition slowed down, and the gradual dissolution of calcium was evident for up to 30 days. Moreover, we also noticed that the degradation behavior varies among the four groups of CHAMs. For instance, at pH 3 and pH 7, D-CHAM degraded faster than the other groups at first stage. This can be explained by the presence of small fragments from the burst microsphere, which dissolves more quickly than the intact microspheres. In addition, it can be seen that the degradation rate of B-CHAM remains to be the highest in the period of 30 day observation. We propose the slower degradation of A-CHAM can be explained by the increase in its crystallinity, while for C-CHAM, the reason can be the increased particle size and the subsequent decrease in the surface area.
3.3. Drug loading and releasing
The increase in the size of microsphere, the hollow space, and the surface area were found to have direct influence on the drug entrapment efficiency and releasing profile. Given this, CDDP, a widely-used chemotherapy drug showing a high level and broad spectrum of antitumor activity,43 was chosen as a model drug. It was previously reported that the adsorption of CDDP happens after its hydrolysis in solution, resulting in the absence of chlorine.44,45 Thus, the concentration of Pt in the solution was measured for investigating the drug entrapment efficiency and releasing profile of CDDP. The CDDP entrapment efficiency shown in Fig. 6a reveals that C-CHAM can carry significant more (P < 0.05) amount of CDDP than any other groups. This may be due to the decreased crystallinity of C-CHAM when compared with A-CHAM and B-CHAM, as more crystalized apatite was known to be compromised for loading sufficient amounts of CDDP.46 Moreover, the increased sphere size and hollow space might also contribute to the increase in loading capacity. Nevertheless, D-CHAM, which seems to bear an even larger size and hollow space, failed to favor drug loading as expected. It is possible that the collapsed or broken microspheres in D group can barely carry the drug as effective as the well-structured microsphere in C group did. | ||
| Fig. 6 CDDP entrapment efficiency of the four groups of CHAMs at 24 h (a); CDDP release profile of CHAMs measured by cumulative platinum release in PBS at pH 5 (b) and pH 7 (c). Cell viability of FaDu cells cultured with different concentrations of unloaded CHAMs (d). Cell cytotoxicity of CDDP loaded CHAMs to FaDu cells at 24 h and 72 h (e); asterisks denote a significant difference (according to ANOVA): *p < 0.05. | ||
Since the HAp crystal bounded CDDP was previously reported to be very stable, and its release in PBS at pH 7.4 can last for a long period of time.44 In the current study, the four groups of CDDP loaded CHAMs were respectively immersed in PBS at different pH values, and a total of 30 days were allowed for a clear understanding on the kinetics of drug release. As shown in Fig. 6b and c, the release of CDDP was determined by the cumulative Pt amount in the solution. For all the tested samples, the drug release profile clearly presents a two-stage manner: the quick release in the first 12 h followed by a period of slow and steady release. We propose that the initial bust release of Pt could be caused by the surface bound CDDP, while the later sustained release could originate from the inner encapsulated CDDP. This also explains why the slow release stage is more prominent in C-CHAM and D-CHAM, where hollow space within the microsphere is assured. The lasting release of CDDP after the first 24 h, as well as the delayed cytotoxicity of the CHAMs on day three (Fig. 6e) suggest the initial burst release of drug from microspheres was avoided. In addition, by comparing the releasing curve in Fig. 6b and c, it can be concluded that CDDP loaded on CHAMs released much faster in acidic condition, which is commonly found in the tumor microenvironment.47 The pH-dependent release profile could be explained by the distinct degradation of CHAMs in different pH environment, because the carbonate content in CHAMs are very sensitive to the change in the local pH value. Moreover, it is evident that the cumulative amount of released Pt from the four groups of CHAMs are proportional to the entrapment efficiency of the samples: C-CHAM > D-CHAM ≥ B-CHAM > A-CHAM. Collectively, C-CHAM might be considered as the optimal microsphere for the carrying and controlled release of CDDP.
3.4. Cytotoxicity and cellular response
The cytotoxicity of the unloaded and CDDP loaded CHAMs was done using an MTT assay on the FaDu cell line, a human head and neck squamous cell carcinoma cell line that sensitively responds to CDDP. Fig. 6d revealed that no cytotoxicity could be detected when the concentration of unloaded CHAMs was under 0.1 mg mL−1, while when the solid/liquid ratio exceed 0.2 mg mL−1, the cytotoxicity became detectable, especially in D-CHAM, where the highest concentration of SDS was used. The cell viability of FaDu cells co-cultured with 0.5 mg mL−1 microspheres dropped down to ∼75% in A-CHAM group and under 50% in D-CHAM group. Since with the increase in SDS usage, the residual SDS could become more difficult to eliminate, thus, our data provides a good reason to minimize the addition of SDS in sample preparation. In addition, the cell survival rate of FaDu cells treated with CDDP loaded CHAMs for 24 h or 72 h were shown in Fig. 6e, indicating C-CHAM to be the most effective group to kill the FaDu cells on both day one and three. This effect was more prominent on day three with the cell survival rate in group C to drop to only ∼20%, nearly half of that in group A and B, which is consistent to the drug entrapment efficiency results indicating C-CHAM to be the most effective carrier for CDDP. However, there seems to be no significant difference between C-CHAM and D-CHAM in terms of their cytotoxicity to the FaDu cell on either time point.The cellular response of FaDu cells to CHAMs was observed using SEM (Fig. 7a–d) and CLSM (Fig. 7e and f) respectively. Both the CDDP loaded and unloaded CHAMs could spontaneously attached to the seeded FaDu cells, but act on the cells in different ways. After the treatment of CDDP loaded CHAMs, the FaDu cells lost their cobblestone-like morphology and appeared in apoptosis-like morphology (Fig. 7a) characterized by cell shrinkage, cell detachment, as well as condensation and rounding of the cytoplasm.48 The unloaded CHAMs didn't lead to a prominent change in the morphology of the FaDu cells (Fig. 7c), but, instead, the invagination of cell membrane around the microsphere was evident (Fig. 7d), implying the particles were being pulled into the cell. To further prove the cell engulfment of the CHAMs, the microspheres were conjugated with RITC via the amine group on their surface. As shown in Fig. 7e, the attachment of RITC labeled CHAMs (red) to the FaDu cells stained with the nucleus (blue) and the cytoskeleton (green) could be clearly visualized under CLSM. The orthogonal views confocal images in Fig. 7f clearly demonstrated that one microsphere located intracellularly.
 | ||
| Fig. 7 SEM images (scale bar = 10 μm) showing the cell morphology of FaDu cells treated with CDDP loaded CHAMs (a), and unloaded CHAMs (c); the cell surface areas with the attached CHAMs in orange boxes were further shown in magnification (scale bar = 1 μm): CDDP loaded CHAM (b), unloaded CHAM (d); confocal microscopic image (scale bar = 10 μm) showing the RITC labeled microspheres (red) attached to the FaDu cells stained with nucleus (blue) and cytoskeleton (green) (e); cross-sectional view and lateral views showing a microsphere engulfed by a FaDu cell (f). | ||
Cancer cells are expected to carry negative surface charge due to their abnormal secretion of lactate across plasma membranes, which is rarely seen in normal cells.49 On the other hand, although HAp is normally neutral in surface charge, a small positive charge can be acquired through unequal adsorption of oppositely charged ions, which is quite common in physiological condition.50 In the current study, all four groups of CHAMs were determined to carry a positive zeta potential in 1 mM KCl solution. This positive surface charge makes HAp based microspheres a potential candidate for the delivery of cancer targeting drugs. Moreover, the biocompatibility and biodegradability of the CHAMs, as shown above, allow them to be recognized and engulfed by cells, suggesting there remains various applications to be discovered. For example, in some recent studies, hollow HAp microspheres were grafted with aliphatic poly(urethane-amine) to obtain thermal responsive drug release,51 or incorporated with Fe3O4 to gain magnetic properties.6 They could also be encapsulated in sodium alginate52 or collagen matrix53 to increase their clinical performance and workability.
4. Conclusion
Hollow CaCO3 precursors were prepared via fast precipitation using SDS at a much lower concentration than reported previously, and the precursors can be directly transformed into hollow mesoporous microspheres composed of nano-sized carbonated hydroxyapatite crystals through hydrothermal reaction. The hollow mesoporous CHAMs exhibit excellent biodegradability and sustained drug release properties in a pH-dependent manner, due to the distinct dissolution behavior of the sphere wall under acidic and neutral environment. The positively charged CHAMs can be loaded with CDDP to induce apoptosis of cancer cells through either direct attachment or engulfment. In addition, it was demonstrated that the particle size, hollow structure and wall thickness of the microspheres can be well controlled by simply adjusting the concentration of SDS in the reaction. The optimal morphology of CHAM was demonstrated to be achieved when using SDS at its CMC, indicated by it the highest entrapment efficiency of CDDP and the strongest killing effects on human squamous cell carcinoma cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was done in partial fulfillment of the requirements for the degree of PhD of the first authors at the Faculty of Dentistry, the University of Hong Kong. The authors would like to thank Dr Vivian Lui from the Chinese University of Hong Kong for kindly providing the FaDu cell line, and Huabin Fang from the State Key Lab of Inorganic–Organic Composites, Beijing University of Chemical Technology, for help with N2 adsorption test. We are also grateful for the technical support from Dr Karen H. M. Wong and Mr Paul K. D. Lee in the University of Hong Kong.References
- S. Saber-Samandari, N. Nezafati and S. Saber-Samandari, Crit. Rev. Ther. Drug Carrier Syst., 2016, 33, 41–75 CrossRef PubMed.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed.
- R. Z. LeGeros, Chem. Rev., 2008, 108, 4742–4753 CrossRef PubMed.
- Y. H. Yang, C. H. Liu, Y. H. Liang, F. H. Lin and K. C. W. Wu, J. Mater. Chem. B, 2013, 1, 2447–2450 RSC.
- K. Lin, C. Wu and J. Chang, Acta Biomater., 2014, 10, 4071–4102 CrossRef CAS PubMed.
- K. L. Lin, L. Chen, P. Y. Liu, Z. Y. Zou, M. L. Zhang, Y. H. Shen, Y. Q. Qiao, X. Y. Liu and J. Chang, CrystEngComm, 2013, 15, 2999–3008 RSC.
- Y.-J. Guo, Y.-Y. Wang, T. Chen, Y.-T. Wei, L.-F. Chu and Y.-P. Guo, Mater. Sci. Eng., C, 2013, 33, 3166–3172 CrossRef CAS PubMed.
- Y. S. Wang, M. S. Hassan, P. Gunawan, R. Lau, X. Wang and R. Xu, J. Colloid Interface Sci., 2009, 339, 69–77 CrossRef CAS PubMed.
- L. J. Hao, H. Yang, S. L. Du, N. R. Zhao and Y. J. Wang, Mater. Lett., 2014, 131, 252–254 CrossRef CAS.
- Y. H. Liang, C. H. Liu, S. H. Liao, Y. Y. Lin, H. W. Tang, S. Y. Liu, I. R. Lai and K. C. W. Wu, ACS Appl. Mater. Interfaces, 2012, 4, 6719–6726 Search PubMed.
- H. Xiong, S. Du, J. Ni, J. Zhou and J. Yao, Biomaterials, 2016, 94, 70–83 CrossRef CAS PubMed.
- K. Lin, X. Liu, J. Chang and Y. Zhu, Nanoscale, 2011, 3, 3052–3055 RSC.
- J. Wei, J. Shi, Q. Wu, L. Yang and S. K. Cao, J. Mater. Chem. B, 2015, 3, 8162–8169 RSC.
- M. Vukomanovic, S. D. Skapin, B. Jancar, T. Maksin, N. Ignjatovic, V. Uskokovic and D. Uskokovic, Colloids Surf., B, 2011, 82, 404–413 CrossRef CAS PubMed.
- C. Qi, Y. J. Zhu, B. Q. Lu, X. Y. Zhao, J. Zhao and F. Chen, J. Mater. Chem., 2012, 22, 22642–22650 RSC.
- W. Lai, C. Chen, X. Ren, I. S. Lee, G. Jiang and X. Kong, Mater. Sci. Eng., C, 2016, 62, 166–172 CrossRef CAS PubMed.
- Y. P. Guo, Y. B. Yao, Y. J. Guo and C. Q. Ning, Microporous Mesoporous Mater., 2012, 155, 245–251 CrossRef CAS.
- Y. J. Guo, Y. Y. Wang, T. Chen, Y. T. Wei, L. F. Chu and Y. P. Guo, Mater. Sci. Eng., C, 2013, 33, 3166–3172 CrossRef CAS PubMed.
- M. Yu, K. C. Zhou, Z. Y. Li and D. Zhang, Mater. Sci. Eng., C, 2014, 45, 306–312 CrossRef CAS PubMed.
- L. Xiong, J. Zeng, A. Yao, Q. Tu, J. Li, L. Yan and Z. Tang, Int. J. Nanomed., 2015, 10, 517–526 CrossRef PubMed.
- A. H. Yao, X. D. Li, L. Xiong, J. H. Zeng, J. Xu and D. P. Wang, J. Mater. Sci.: Mater. Med., 2015, 26(1), 25 CrossRef PubMed.
- C. M. Zhang, J. Yang, Z. W. Quan, P. P. Yang, C. X. Li, Z. Y. Hou and J. Lin, Cryst. Growth Des., 2009, 9, 2725–2733 CAS.
- T. Niwa, H. Shimabara, M. Kondo and K. Danjo, Int. J. Pharm., 2009, 382, 88–97 CrossRef CAS PubMed.
- T. Niwa, H. Shimabara and K. Danjo, Chem. Pharm. Bull., 2010, 58, 195–200 CrossRef CAS PubMed.
- M. Yu, K. C. Zhou, F. Q. Zhang and D. Zhang, Ceram. Int., 2014, 40, 12617–12621 CrossRef CAS.
- H. G. Chen and S. L. Leng, Ceram. Int., 2015, 41, 2209–2213 CrossRef CAS.
- S. Y. Park, K. I. Kim, S. P. Park, J. H. Lee and H. S. Jung, Cryst. Growth Des., 2016, 16, 4318–4326 CAS.
- B. P. Bastakoti, M. Inuoe, S. Yusa, S. H. Liao, K. C. Wu, K. Nakashima and Y. Yamauchi, Chem. Commun., 2012, 48, 6532–6534 RSC.
- C. S. L. Sulfate, J. Am. Coll. Toxicol., 1983, 2, 127–181 Search PubMed.
- L. N. Zhao and J. K. Wang, Colloids Surf., A, 2012, 393, 139–143 CrossRef CAS.
- A. S. Inacio, K. A. Mesquita, M. Baptista, J. Ramalho-Santos, W. L. C. Vaz and O. V. Vieira, PLoS One, 2011, 6(5), e19850 CAS.
- S. Hayashi and S. Ikeda, J. Phys. Chem., 1980, 84, 744–751 CrossRef CAS.
- P. Mukerjee and K. J. Mysels, Critical micelle concentrations of aqueous surfactant systems, DTIC Document, 1971 Search PubMed.
- F. H. Quina, P. M. Nassar, J. B. S. Bonilha and B. L. Bales, J. Phys. Chem., 1995, 99, 17028–17031 CrossRef CAS.
- D. Q. Xiao, Z. Tan, Y. K. Fu, K. Duan, X. T. Zheng, X. Lu and J. Weng, Ceram. Int., 2014, 40, 10183–10188 CrossRef CAS.
- D. Q. Xiao, X. Zhou, H. Y. Li, Y. K. Fu, K. Duan, X. Lu, X. T. Zheng and J. Weng, J. Eur. Ceram. Soc., 2015, 35, 1971–1978 CrossRef CAS.
- J. C. Elliott, Structure and chemistry of the apatites and other calcium orthophosphates, Elsevier, Amsterdam The Netherlands New York, 1994 Search PubMed.
- I. Rodriguez-Ruiz, J. M. Delgado-Lopez, M. A. Duran-Olivencia, M. Iafisco, A. Tampieri, D. Colangelo, M. Prat and J. Gomez-Morales, Langmuir, 2013, 29, 8213–8221 CrossRef CAS PubMed.
- S. Kannan, J. M. G. Ventura, A. F. Lemos, A. Barba and J. M. F. Ferreira, Ceram. Int., 2008, 34, 7–13 CrossRef CAS.
- J. S. Cho, S. H. Um, D. S. Yoo, Y. C. Chung, S. H. Chung, J. C. Lee and S. H. Rhee, J. Biomed. Mater. Res., Part B, 2014, 102, 1046–1062 CrossRef PubMed.
- F. Jiang, D. P. Wang, S. Ye and X. Zhao, J. Mater. Sci.: Mater. Med., 2014, 25, 391–400 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- A.-M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351 CrossRef CAS PubMed.
- A. Barroug and M. J. Glimcher, J. Orthop. Res., 2002, 20, 274–280 CrossRef CAS PubMed.
- B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L. Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007, 17, 2180–2188 CrossRef CAS.
- A. Barroug, L. T. Kuhn, L. C. Gerstenfeld and M. J. Glimcher, J. Orthop. Res., 2004, 22, 703–708 CrossRef CAS PubMed.
- V. Estrella, T. A. Chen, M. Lloyd, J. Wojtkowiak, H. H. Cornnell, A. Ibrahim-Hashim, K. Bailey, Y. Balagurunathan, J. M. Rothberg, B. F. Sloane, J. Johnson, R. A. Gatenby and R. J. Gillies, Cancer Res., 2013, 73, 1524–1535 CrossRef CAS PubMed.
- I. Bohm, Autoimmunity, 2003, 36, 183–189 CrossRef PubMed.
- B. Chen, W. Le, Y. Wang, Z. Li, D. Wang, L. Ren, L. Lin, S. Cui, J. J. Hu, Y. Hu, P. Yang, R. C. Ewing, D. Shi and Z. Cui, Theranostics, 2016, 6, 1887–1898 CrossRef CAS PubMed.
- H. Fu, M. N. Rahaman, D. E. Day and R. F. Brown, J. Mater. Sci.: Mater. Med., 2011, 22, 579–591 CrossRef CAS PubMed.
- Q. Wu, J. Shi, J. Wei, L. Yang and S. K. Cao, RSC Adv., 2015, 5, 70101–70108 RSC.
- Q. Zhong, W. Li, X. Su, G. Li, Y. Zhou, S. C. Kundu, J. Yao and Y. Cai, Colloids Surf., B, 2016, 143, 56–63 CrossRef CAS PubMed.
- R. Cholas, S. K. Padmanabhan, F. Gervaso, G. Udayan, G. Monaco, A. Sannino and A. Licciulli, Mater. Sci. Eng., C, 2016, 63, 499–505 CrossRef CAS PubMed.
Footnote |
| † The first two authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2017 |