
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClosed-loop solvometallurgical process for recovery of lead from iron-rich secondary lead smelter residues†
Federica Forte a,
Liesbeth Horckmansb,
Kris Broosb,
Eunyoung Kimb,
Frantisek Kukurugyab and
Koen Binnemans*a
a,
Liesbeth Horckmansb,
Kris Broosb,
Eunyoung Kimb,
Frantisek Kukurugyab and
Koen Binnemans*a
aKU Leuven, Department of Chemistry, Celestijnenlaan 200F, bus 2404, B-3001 Heverlee, Belgium. E-mail: Koen.Binnemans@kuleuven.be
bVITO NV, Boeretang 200, B-2400 Mol, Belgium
First published on 26th October 2017
Abstract
A solvometallurgical process based on the use of concentrated acetic acid as lixiviant is proposed as an alternative for conventional hydrometallurgical processes to recover lead from iron-rich industrial residues generated by recycling of spent lead-acid batteries in a secondary lead smelter. Under the optimal conditions, a high selectivity for lead was obtained: more than 90% of the lead content could be dissolved, while only a small amount of iron (<6%) was codissolved. Lead was quantitatively recovered from the acetic acid leachate by addition of a stoichiometric amount of sulphuric acid. Acetic acid was recycled by distillation and reused in the leaching step, so that a closed-loop process was obtained. The process was optimised for iron-rich residue (matte), but also a proof-of-principle is given for lead recovery from another lead-containing residue (slag). The main advantages of this solvometallurgical process are the low power consumption (room-temperature process), the low consumption of chemicals (only sulphuric acid is consumed), full recycling of the acetic acid and the limitation of waste water formation.
Introduction
Metal recovery from secondary sources has environmental and economic advantages because it allows delaying the depletion of primary ore deposits and requires much less energy than metal production from primary sources. Metallurgical slags produced by pyrometallurgical processes are potential secondary raw materials since they still contain considerable concentrations of metals. Ferrous slag, which includes iron slag, steel slag, alloy steel slag and ferroalloy slag is mainly treated through magnetic and gravity separation methods to recover chromium and nickel.1 Non-ferrous slag, which includes copper, zinc and lead slag, often has a high iron content, which is comparable to that of low-grade iron ores, and it can be used as a secondary iron source in a rotary kiln process.2 Metals other than iron can be recovered from non-ferrous slag. An extensive body of literature is available on the recovery of copper, zinc and cobalt from copper slag, which is usually treated by flotation,3 roasting4 and leaching.5Each year 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of lead slag are being produced by recycling of lead by secondary lead smelters, as well as 280000 tonnes of sludge by neutralisation of the sulphuric acid contained in lead-acid batteries.6 The particle size distribution of lead slag is very heterogeneous and there is a great variability in its chemical composition, which is strongly affected by the efficiency of the employed pyrometallurgical process.7 Some literature studies focus on the reuse of lead slag in construction materials,8,9 but only few studies have considered the recovery of metals from lead slag. Smaniotto et al. investigated the recovery of lead from lead slag of a battery recycling plant by using the chelating agent EDTA as a lixiviant (=leaching agent).7 However, EDTA is quite expensive and difficult to remove from aqueous waste streams. Shirinbayan et al. studied the kinetics of leaching lead from smelting furnace slag with hydrochloric acid.10 Ferracin et al. investigated the recovery of lead from sludges of exhausted lead-acid batteries by leaching with HBF4 followed by electrowinning of the dissolved lead.11 Major disadvantages of hydrometallurgical leaching processes are the generation of large volumes of waste water and the often limited selectivity, particularly if strong inorganic acids (H2SO4, HCl, HNO3) are used as leaching agent.
000 tonnes of lead slag are being produced by recycling of lead by secondary lead smelters, as well as 280000 tonnes of sludge by neutralisation of the sulphuric acid contained in lead-acid batteries.6 The particle size distribution of lead slag is very heterogeneous and there is a great variability in its chemical composition, which is strongly affected by the efficiency of the employed pyrometallurgical process.7 Some literature studies focus on the reuse of lead slag in construction materials,8,9 but only few studies have considered the recovery of metals from lead slag. Smaniotto et al. investigated the recovery of lead from lead slag of a battery recycling plant by using the chelating agent EDTA as a lixiviant (=leaching agent).7 However, EDTA is quite expensive and difficult to remove from aqueous waste streams. Shirinbayan et al. studied the kinetics of leaching lead from smelting furnace slag with hydrochloric acid.10 Ferracin et al. investigated the recovery of lead from sludges of exhausted lead-acid batteries by leaching with HBF4 followed by electrowinning of the dissolved lead.11 Major disadvantages of hydrometallurgical leaching processes are the generation of large volumes of waste water and the often limited selectivity, particularly if strong inorganic acids (H2SO4, HCl, HNO3) are used as leaching agent.
In the work of Kim et al. selective leaching of lead and other minor elements from secondary lead smelting residues was observed with several nitric acid-based systems.12 The authors found that in the presence of 0.15 M Fe(III) as oxidising agent, 90% of the lead was leaching, whereas iron codissolution was less than 25%. Selective leaching of copper, nickel and zinc was achieved by roasting at 600 °C followed by water leaching and pressurised leaching in 0.5 M HNO3. A selective leaching of lead, copper, nickel and zinc from the same residue was also observed by citrate leaching.13 Lead leaching in 1 M sodium citrate solution was highest (93%) in the presence of 0.5 M H2O2. In these conditions, iron was almost not leached (<0.6%). An oxidative roasting step prior to leaching improved the leachability of the target metals, with maximum leaching efficiency reached at 600 °C for lead (93%), copper (80%) and zinc (60%) and at 650 °C for nickel (53%).
To overcome the issue of lack of selectivity of inorganic chemistry, organic acids can be used as a lixiviant. For instance, acetic acid is known for its capability to dissolve lead compounds.14 Leaching of lead from galena (PbS) in acetic acid solutions with hydrogen peroxide as oxidizing agent was studied by Aydoğan et al.15 Nagib and Inoue investigated the recovery of lead and zinc from municipal solid waste incineration fly ash by leaching with several leaching agents, including acetic acid.16 Barrett et al. described a process based on a 3 M acetic acid solution to remove zinc, lead, copper and cadmium from carbon steel electric arc furnace dust.17 However, all these processes are still hydrometallurgical processes and generate aqueous waste streams. The formation of aqueous waste streams can be avoided by replacing water with an organic solvent and by working in a closed-loop process in which the organic solvent is recycled. Processes based on the use of solvents other than water are called “solvometallurgical processes”.18 Solvometallurgical processes can have a high selectivity. For instance, it was shown that the red phosphor Y2O3:Eu3+ could selectively be dissolved from a complex fluorescent lamp phosphor waste by using a carboxyl-functionalised ionic liquid.19 Abbott and coworkers demonstrated that sulfides and similar minerals could be oxidatively dissolved in deep-eutectic solvents based on choline chloride and ethylene glycol.20,21
In this paper, we describe a process to recover lead from residues from a secondary lead smelter, in which concentrated acetic acid is used as leaching agent to largely reduce the generation of waste water. A small amount of sulphuric acid is added in the separation step to precipitate lead as lead sulphate from the leachate. Acetic acid can easily be recycled through distillation and reused in the leaching step, thus ensuring a closed-loop process where only limited amounts of reagents are required. The recovered lead can be reused as a secondary raw material in a lead smelter, whereas the lead-depleted iron-rich residue can find application as a secondary iron source or as a building material. According to the 2011 GSK's solvent selection guide and its updates, very few issues concerning human health or environmental hazards are related to the use of acetic acid as a solvent, so that this process can be considered as a green chemical process.22–24
Experimental
Chemicals
Acetic acid (100%) was purchased from VWR Chemicals (Leuven, Belgium); H2SO4 (96%) and Triton X-100 from Acros Organics (Geel, Belgium). Water was always of ultrapure quality, deionised to a resistivity of 18.2 MΩ cm with a Sartorius Arium Pro ultrapure water system. The silicone solution in isopropanol was purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany) and the standard solutions (1000 μg mL−1 Ga, Dy and La) from Merck (Overijse, Belgium). All chemicals were used as received without any further purification.Analytical method
Sample characterisation was performed by Wavelength Dispersive X-ray Fluorescence (WD-XRF, Axios PANalytical). Measurements were performed on powders, pressed into pellets. Mineralogical analyses were performed by X-ray diffraction (X'Pert PRO, PANalytical; Cu tube, fine focus, wavelength λ(Kα1) = 1.5406 Å, 2θ = 2–120°, step size of 0.04°). The resulting diffractograms were analysed by the software HighScore Plus 4.0. The concentration of metals in solution was determined by Total Reflection X-ray Fluorescence spectroscopy (TXRF) with a Bruker S2 Picofox TXRF spectrometer equipped with a molybdenum source.25 Plastic microtubes were filled with a certain amount of the sample and diluted until 1 mL with ultrapure water/Triton solution (5% w/w).26 An appropriate internal standard was added, with the X-ray fluorescence energy as close as possible to the element to be determined in order to reduce the effects caused by secondary X-rays absorption. After shaking the samples on a vibrating plate (IKA MS 3 basic), a small droplet (5 μL) was put on a quartz carrier, previously treated with a silicone–isopropanol solution to avoid spreading of the droplet. The quartz carriers were then dried for 30 min at 60 °C prior to analysis. Each sample was measured for 500 s. The composition of the solid residue obtained after leaching was determined by EDXRF (Energy Dispersive X-ray Fluorescence). The purity of the obtained lead sulphate was determined via Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES, Perkin Elmer, Optima 3000 DV). Prior to analysis the sample was processed by microwave digestion with HCl, HNO3 and HBF4.Leaching tests
The leaching tests were performed in glass vials located on a mechanical shaker (TMS-200 Turbo Thermoshaker). The solid samples were contacted with concentrated acetic acid and several operative parameters were varied in order to determine the optimal operative conditions, namely the contact time (t = 1–4 h), the liquid-to-solid ratio (L/S = 5 mL g−1, 10 mL g−1, 20 mL g−1) and the temperature (T = 25 °C, 50 °C, 75 °C). The agitation was kept constant at 2500 rpm in all the experiments. The leachate was separated from the solid residue through centrifugation (5300 rpm, 30 min) and analysed by TXRF to determine the leaching efficiency EL (%), which was calculated according to eqn (1):
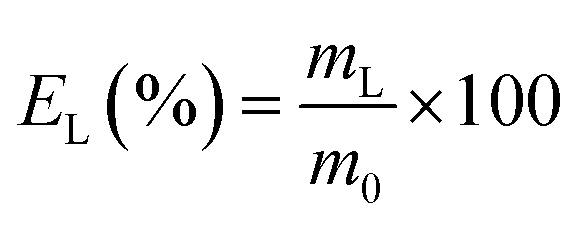 | (1) |
Water-leaching tests with ultrapure water (t = 24 h, L/S = 10 mL g−1) were performed on a leaching residue to verify the leachability of harmful elements and thus to assess a possible application of the residual solid matrix in for instance building materials.
Precipitation tests
Recovery of lead from the leachate was investigated through precipitation with sulphuric acid 1/10 v/v in water, i.e. 1.8 M. Addition of sulphuric acid leads to the formation of insoluble PbSO4. Attention was paid to determine the optimal conditions (in terms of sulphuric acid consumption) to minimise co-precipitation of iron.The solid residue was separated from the leachate through centrifugation (5300 rpm, 30 min) and the supernatant was analysed to determine the precipitation efficiency EP (%), which was calculated according to eqn (2):
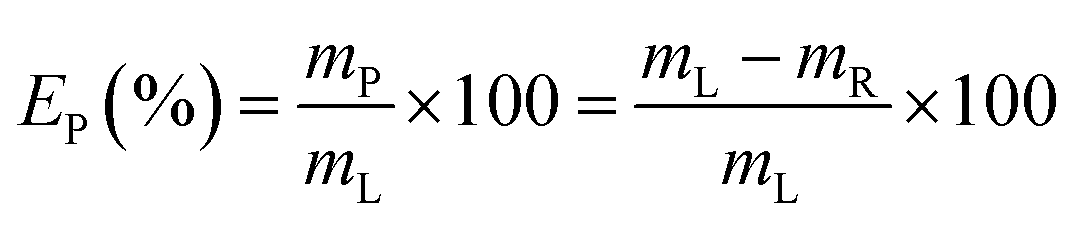 | (2) |
Acetic acid recovery
Recovery of acetic acid was investigated through distillation. The leachate obtained after a precipitation test with sulphuric acid was evaporated under reduced pressure at T = 50 °C by a rotary evaporator (Büchi R-210). The recovered solvent was used in a second leaching step to prove the feasibility of the proposed process.Results and discussion
Samples were collected in a secondary lead smelter plant which treats spent lead-acid batteries and other lead-containing residues to recycle lead. At the bottom of the furnace three layers are formed based on density: (1) top: slag, mainly consisting of oxides; (2) middle: matte, mainly constituted by sulphides; (3) bottom: lead bullion, i.e. impure metallic lead, which is further refined. The slag and the matte are tapped into “slag pots”. After cooling, the slag pots are turned over (Fig. 1) and the different layers are separated. The slag is broken and sieved; 2/3 of the material (i.e. particles of a diameter of more than 15 cm) is returned to the lead furnace, while 1/3 of the slag, together with the matte, is disposed of. | ||
| Fig. 1 Inverted slag pots, showing the slag at the bottom (light grey) and the matte (dark grey) at the top. | ||
Samples of slag and matte were pretreated through a milling step in order to obtain powders with particle size smaller than 125 μm. The chemical composition of the two materials was determined by WD-XRF analysis (Tables 1 and 2). The matte had a lead content of 8.2 wt% and the slag had a lead content of 4.0 wt%. The iron content of the matte (51.4 wt%) was considerably higher than that of the slag (36.6 wt%). The XRD pattern (ESI, S1†) of the iron stone sample shows that iron is present mainly as sulphide (FeS, troilite) and oxide (Fe3O4, magnetite; FeO, wüstite). XRD analyses were also performed on the matte after magnetic separation; it was found that lead is present in the ferromagnetic fraction as metallic lead (Pb) inside the FeS phase (ESI, S2†). In the slag, iron is present mainly as Fe2SiO4 (fayalite) and sulphide (FeS), whereas lead is present as sulphide (PbS, galena) and metallic lead (Pb) (ESI, S3†).
| Fe | S | Pb | Cu | Ni | Si | Cl | Cr | Mn | Zn | Sn |
|---|---|---|---|---|---|---|---|---|---|---|
| 51.4 | 20.5 | 8.2 | 1.0 | 0.2 | 2.6 | 0.1 | 0.2 | 0.3 | 0.5 | 0.2 |
| Fe | S | Pb | Cu | Si | Cl | Cr | Mn | Zn | Sn |
|---|---|---|---|---|---|---|---|---|---|
| 36.6 | 6.5 | 4.0 | 0.4 | 7.6 | 0.1 | 0.4 | 0.5 | 0.5 | 0.2 |
The leaching process with acetic acid was optimised for the matte, because this residue is the most interesting one from an economical point of view: the matte has a high lead content and the iron content of the residue obtained after removal of lead is so high that this material can be used as a secondary raw material for iron production. Moreover, lead is present in the matte in the form of elemental lead (metallic lead) which is readily attacked by acetic acid in the presence of oxygen by producing very soluble lead(II)acetate.27 The process parameters for recovery of lead from the matte must be optimised in order to obtain maximum recovery of lead, while having minimum codissolution of iron.
In Fig. 2 the leaching kinetics of lead and iron from the matte sample are reported. The lead leaching kinetics with acetic acid are quite fast: only 2 h were required to dissolve more than 90% of the lead. By increasing the contact time up to 4 h, the improvement in the dissolution efficiency for lead was negligible. The selected leaching agent shows a good selectivity of lead over iron. Iron is leached only to a small extent (<6%).
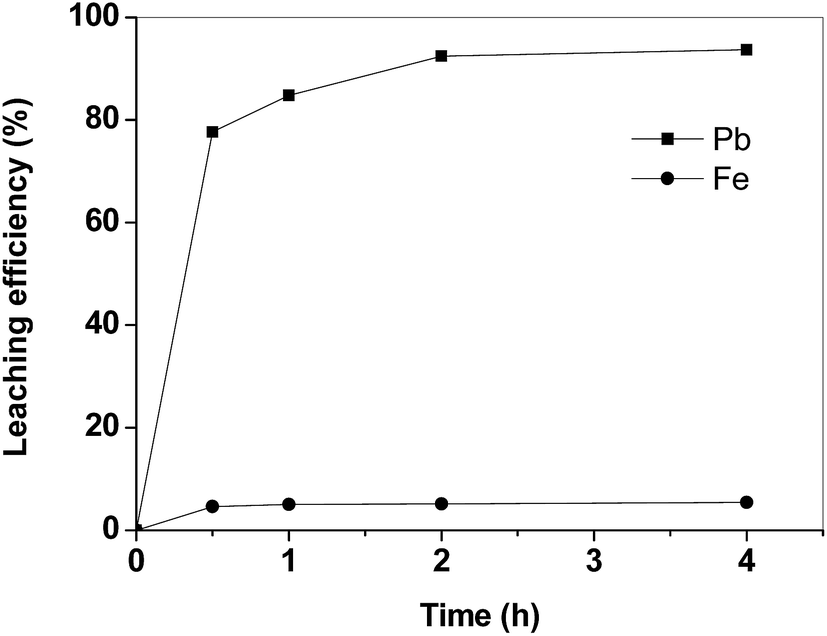 | ||
| Fig. 2 Leaching kinetics for lead and iron from the matte (T = 25 °C, L/S = 10 mL g−1). | ||
The efficiency of lead leaching decreased as a function of temperature, whereas also more iron was dissolved at higher temperatures (Fig. 3). This negative effect of the increasing temperature on the dissolution of lead is similar to what was found by Aydoğan et al. for longer contact times during the oxidative dissolution of galena (PbS) in acetic acid/hydrogen peroxide mixtures.15 The authors suggested that the produced lead sulphate immediately precipitated at the surface of the un-reacted galena, inhibiting further attack of hydrogen peroxide. Similar results were also obtained by Kim et al.,13 who observed that lead and copper leaching with sodium citrate solutions decreased when the temperature increased from 25 to 70 °C, whilst iron leaching efficiency increased. These results show that the leaching process with acetic acid is preferably performed at room temperature. By increasing the liquid-to-solid ratio (L/S) the leaching efficiency of lead increased (Fig. 4). In particular, by using a liquid-to-solid ratio of 20 mL g−1, about 95% lead could be dissolved together with only 6% of iron. A higher selectivity for lead over iron could be obtained by selecting a lower liquid-to-solid ratio (L/S = 5 mL g−1), but this resulted in a significantly lower lead leaching efficiency (<80%). In this study 20 mL g−1 was selected as the optimal value from a technical point of view (maximum lead leaching efficiency). However, a liquid-to-solid ratio of 10 mL g−1 can be considered feasible as well in term of economy of the process.
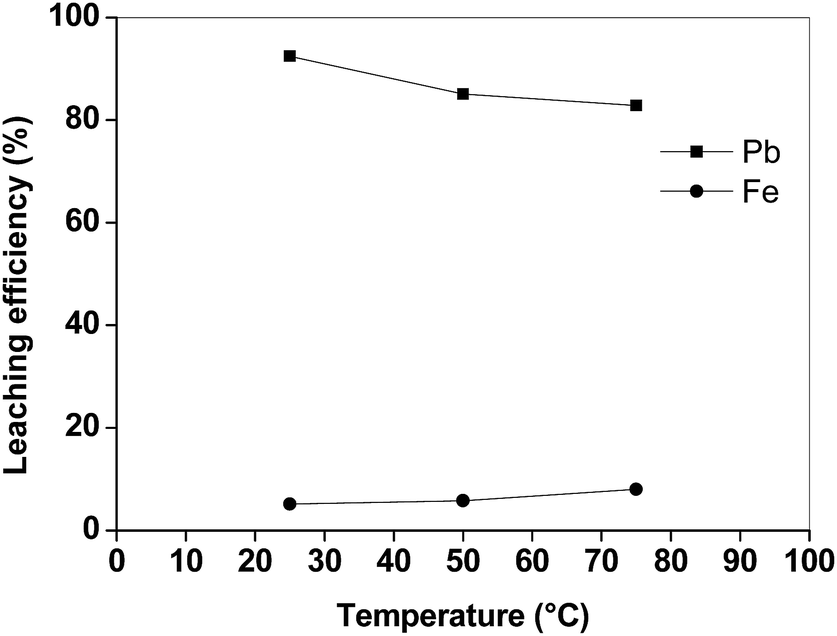 | ||
| Fig. 3 Lead and iron leaching efficiency from the matte as a function of the temperature (t = 2 h, L/S = 10 mL g−1). | ||
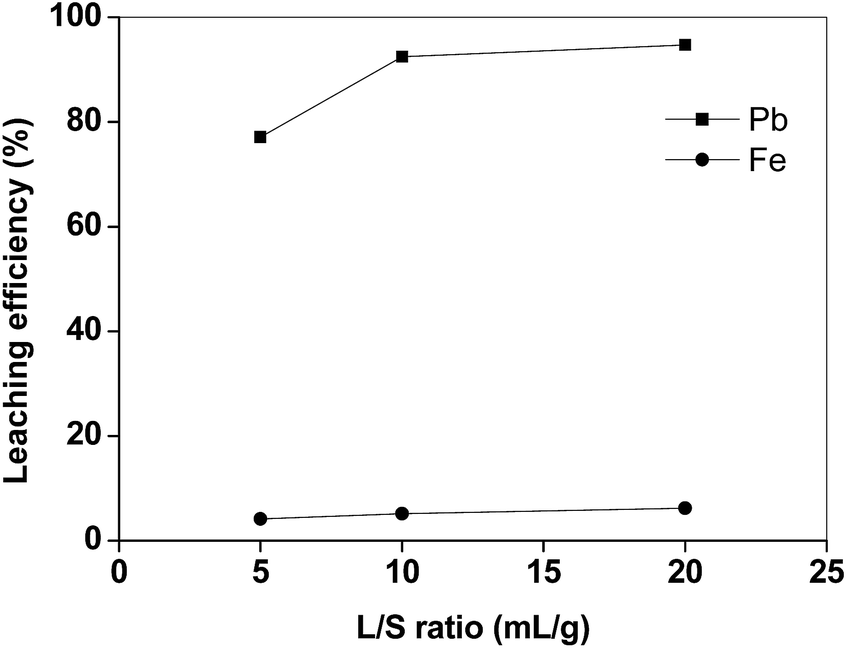 | ||
| Fig. 4 Lead and iron leaching efficiency from the matte as a function of the liquid-to-solid ratio (T = 25 °C, t = 2 h). | ||
The optimised set of parameters determined on the basis of the experiments described above is: time (t) = 2 h; temperature (T) = 25 °C; liquid-to-solid ratio (L/S) = 20 mL g−1. This set of parameters was used to determine the leaching efficiency and the concentration of the major and minor elements in the leachate (Table 3). The main elements in the leachate were found to be lead and iron, whilst nickel and zinc were present only at low concentrations. The concentrations of other elements (Cr, Mn, Sn,…) was below 1 ppm. The solid residue obtained after leaching the matte sample with the optimal operative conditions was separated from the leachate through vacuum filtration, washed with ultrapure water and dried in a vacuum oven at 50 °C until constant weight in order to determine the mass loss which was afterwards used to assess the mass balance. The mass loss was found to be about 15%.
| Pb | Fe | Ni | Zn | |
|---|---|---|---|---|
| Leaching efficiency (%) | 95 | 6 | 43 | 2 |
| Metal concentration (ppm) | 3706 | 1523 | 31 | 4 |
| RSD (%) | 2.5 | 3.7 | 3.5 | 5.3 |
The lead could be recovered from the leachate by addition of sulphuric acid. The addition of sulphuric acid resulted in the precipitation of PbSO4, while the protons of the sulphuric acid regenerate the acetic acid. The precipitation tests were performed on a leachate obtained after leaching the matte in the optimal conditions with acetic acid (T = 25 °C, t = 2 h, L/S = 20 mL g−1). The precipitation efficiency of PbSO4 was investigated as a function of the molar ratio (MR) of H2SO4 with respect to lead (Fig. 5). If a stoichiometric amount of H2SO4 is employed (MR = 1, which corresponds to around 1 microliter of H2SO4 1/10 v/v per mL of leachate), lead is quantitatively precipitated with partial iron co-precipitation (14%). The dosage of H2SO4 significantly affects the selectivity over iron: if a small excess of H2SO4 is used (MR = 1.3), co-precipitation of iron exceeds 20%. Lead(II)sulphate is obtained as white-colored solid product, as shown in Fig. 6. The residue was washed with ultrapure water and dried in a vacuum oven at 50 °C until constant mass. It was found that the amount of PbSO4 which can be obtained from the suggested process is around 115 mg per gram of matte.
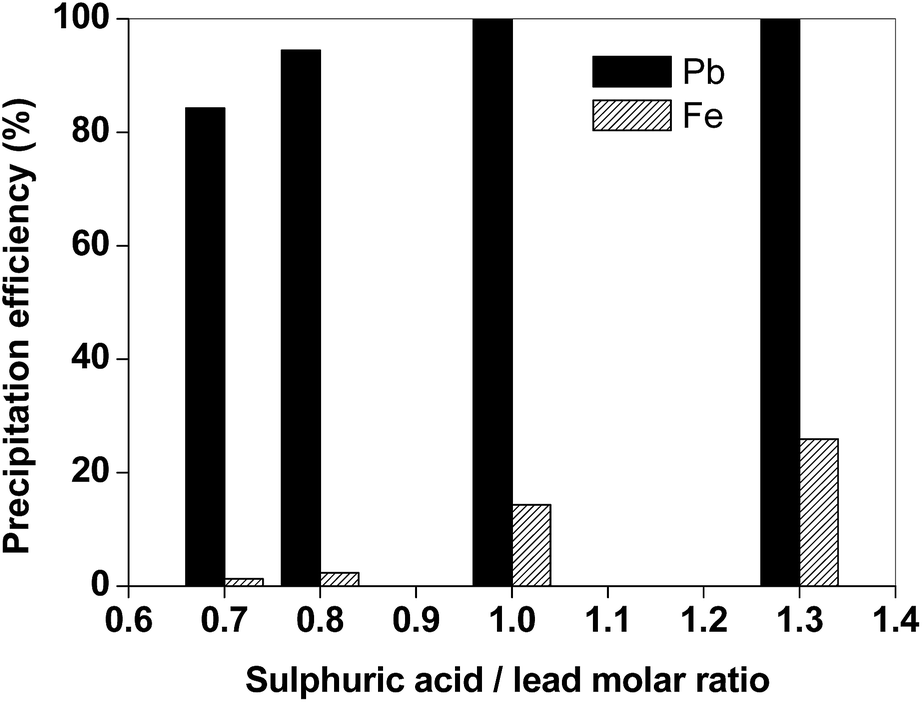 | ||
| Fig. 5 Efficiency for precipitation of lead and iron from the matte leachate (T = 25 °C, H2SO4 1/10 v/v). | ||
 | ||
| Fig. 6 PbSO4 precipitate obtained by addition of H2SO4 to the acetic acid leachate of the matte. | ||
The recovery process was performed on a larger scale to obtain a sufficient amount of solid residues for analysis purposes. A sample of 20 g of matte was contacted with 400 mL of acetic acid at 25 °C for 2 h in a beaker, agitated by a magnetic stirring bar. The leachate was separated from the solid residue by centrifugation and then treated by H2SO4 1/10 v/v (MR = 1, T = 25 °C) to precipitate lead as PbSO4. The recovered PbSO4 as well as the leaching residue were washed with ultrapure water and dried in a vacuum oven at 50 °C. Both samples were analysed by EDXRF (leaching residue) or ICP-AES (PbSO4). The chemical composition of the residue is reported in Table 4. The analysis confirmed the effectiveness of acetic acid as leaching agent, since the lead concentration in the residue was only 0.6%. Water-leaching tests were performed on this residue to verify the non-leachability of toxic elements (Table 5). It was found that most of the concentration values comply with the acceptance criteria in landfills for stable non-reactive hazardous waste and non-hazardous waste (Table 6).28 The only exception is nickel, which has a concentration (15 ppm) higher than the regulatory limit (10 ppm). Lead sulphate analyses by ICP-AES showed that the recovered product has a purity higher than 98%. The PbSO4 can be used as secondary raw material for lead production.
| Element | Concentration (wt%) | Element | Concentration (wt%) |
|---|---|---|---|
| Fe | 56.8 | Mn | 0.3 |
| Si | 2.7 | Zn | 0.3 |
| Cu | 1.2 | Ni | 0.2 |
| Pb | 0.6 | Cr | 0.2 |
| Element | Concentration (μg L−1) | Element | Concentration (μg L−1) |
|---|---|---|---|
| a Physical–chemical characteristics: pH = 4.7, T = 28 °C, σ (electrical conductivity) = 0.4 mS cm−1. | |||
| Cl | 4700 | Cu | 201 |
| SO4 | 163000 |
Pb | 3800 |
| As | 8.6 | Ni | 14900 |
| Cd | 14.4 | Zn | 2900 |
| Cr | <2.0 | ||
In order to prove the feasibility of a closed-loop recovery process, the recovery of acetic acid was investigated as well. An aliquot of 20 mL of the leachate obtained by leaching of matte with acetic acid and precipitation of lead as PbSO4 using the optimised process parameters was evaporated under reduced pressure. The recovered acetic acid was employed for a new leaching step which, as expected, gave the same leaching efficiency for both lead and iron. Based on the experimental results, a recovery process flow-sheet is here proposed, which includes three main steps (Fig. 7): (1) leaching with concentrated acetic acid (T = 25 °C, t = 2 h, L/S = 20 mL g−1); (2) precipitation of PbSO4 with H2SO4 1/10 v/v (T = 25 °C, MR = 1); (3) recovery of acid by distillation.
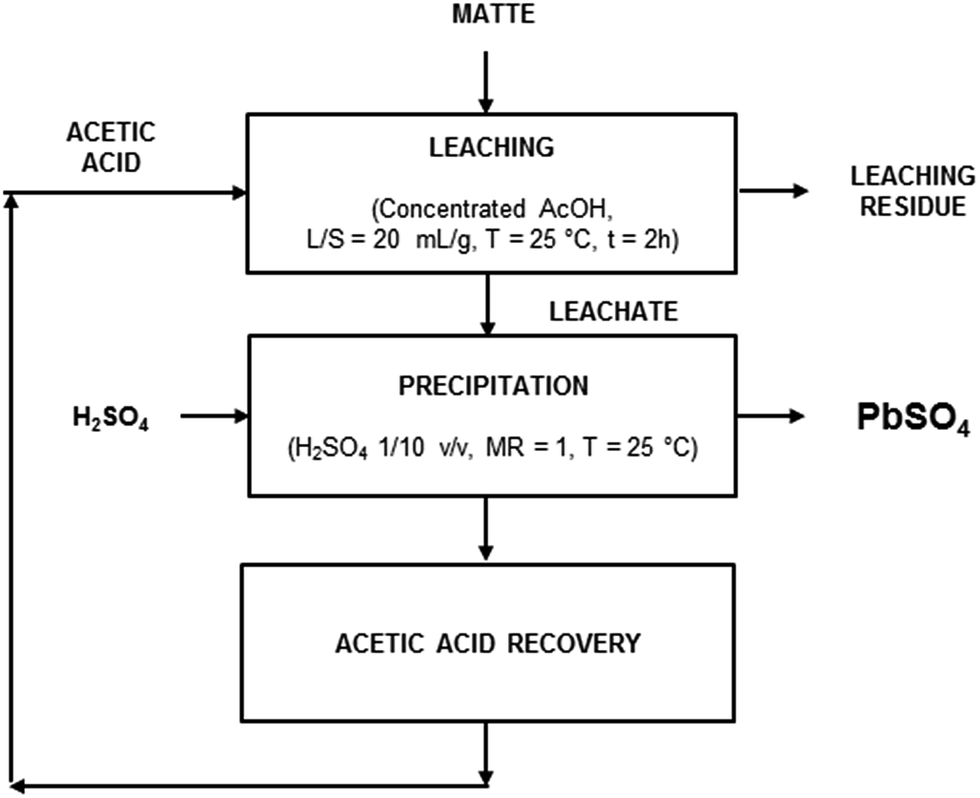 | ||
| Fig. 7 Flow sheet for the closed-loop process to recover lead from matte by leaching with acetic acid (AcOH). | ||
In Table 7 the mass balance of the whole process is reported. It was assessed by using 1 kg of matte as functional unit. The lead concentration in the matte is drastically reduced (from 8.2% in the initial sample to less than 1% in the leaching residue), whilst the iron concentration is slightly higher (∼57%) compared to the initial one (51.4%), showing that there is an enrichment in iron content. This is an advantage for the use of the residue as a secondary iron source. The proposed solvometallurgical process requires only a small number of reagents, namely acetic acid and sulphuric acid. Only sulphuric acid is consumed. Acetic acid can be recovered by distillation and re-used in the leaching step, thus reducing the volume of waste streams to be disposed of. During the distillation step, a residue was produced, but this was not studied further in this work. This can be attributed to the iron fraction dissolved during the leaching step and which is not precipitated in the separation step with sulphuric acid (around 27 g of iron per kg of matte). The leaching step is performed at room temperature, so the energy consumption is low. As mentioned above, the purity of the lead(II)sulphate is sufficient for use as a direct feed into a secondary lead smelter.
| In | Matte | 1 kg |
| Acetic acid | 20 L | |
| H2SO4 1/10 v/v | 20 mL | |
| Out | Residue leaching | 850 g ([Fe] ∼ 57%; [Pb] = 0.5–0.6%) |
| PbSO4 | 115 g |
The optimal conditions determined for the treatment of the matte sample (concentrated acetic acid, t = 2 h, T = 25 °C, L/S = 20 mL g−1) were applied for the leaching of the slag sample. The results are shown in Table 8. A lower leaching efficiency for lead was obtained. This can be explained by the differences in mineralogical composition between the two samples: in the matte, lead is mainly present as metallic lead, which is soluble in weak organic acids,14 whereas in the slag sample, lead is also present as sulphide (PbS), which requires oxidising conditions for leaching.16
| Pb | Fe | Sn | Zn | |
|---|---|---|---|---|
| Leaching efficiency (%) | 72 | 3 | 3 | 1 |
| Metal concentration (ppm) | 1370 | 548 | 2 | 2 |
As in the case of the matte sample, the main elements in the leachate of the slag were found to be lead and iron. The separation of lead from iron was thus the objective of the next step. In Fig. 8, the efficiency for precipitation of lead and iron is reported as a function of the molar ratio. The experimental data showed that precipitation with H2SO4 is selective towards lead: if a stoichiometric amount of H2SO4 is added, quantitative precipitation of lead was observed with only limited iron co-precipitation (10%).
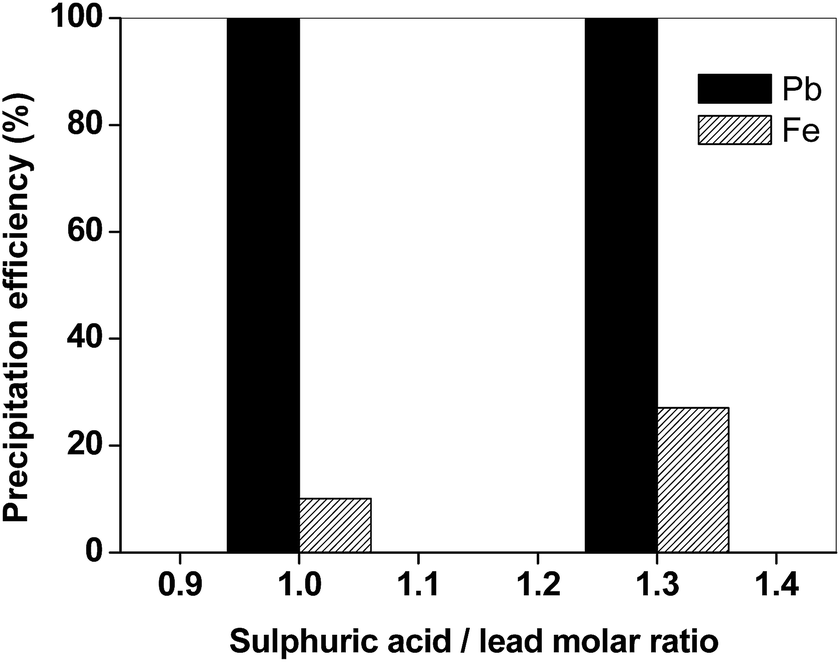 | ||
| Fig. 8 Efficiency for precipitation of lead and iron from the slag leachate (T = 25 °C, H2SO4 1/10 v/v). | ||
Conclusions
Lead can be efficiently recovered from iron-rich residues from a secondary lead smelter process for recycling of lead from lead-acid batteries (matte and slag) by a solvometallurgical process. The process is based on dissolution of lead by concentrated acetic acid, followed by precipitation of the dissolved lead as PbSO4 by addition of H2SO4. More than 90% of the lead present in the matte sample could be dissolved at room temperature, in a relatively short time (2 hours). The process is selective for lead, with codissolution of only small amounts of iron. Relatively pure PbSO4 could be obtained, with partial co-precipitation of iron, and this PbSO4 can be used as feed for a secondary lead smelter. Acetic acid could be recovered by distillation and reused in a second leaching step, showing the feasibility of a closed-loop process. The process was optimised for the recovery of lead from matte, in which lead is present in the metallic state. The solvometallurgical process was also applied to the slag, in which lead is present as PbS and metallic lead. Lead could be recovered from the slag by acetic acid leaching, but only the lead that was present in metallic state. Main advantages of this solvometallurgical process are the low power consumption (room-temperature process), the low consumption of chemicals (only sulphuric acid), full recycling of the acetic acid and the limitation of aqueous waste stream generation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the IWT Flanders for financial support (MIP ICON MaxiVia project, 2015–2017). This project was carried out with the support of the Flemish Region – Environmental and Energy Technology Innovation Platform (MIP).References
- H. Shen and E. Forssberg, Waste Manag., 2003, 23, 933–949 CrossRef CAS PubMed.
- M. Reuter, Y. Xiao and U. Boin, in VII International Conference on Molten Slags Fluxes and Salts, The South African Institute of Mining and Metallurgy, Johannesburg, 2004, vol. 20, pp. 349–356 Search PubMed.
- B. Das, B. Kanta Mishra, S. Angadi, S. Kumar Pradhan, S. Prakash and J. Mohanty, Waste Manag. Res., 2010, 28, 561–567 CrossRef CAS PubMed.
- E. Rudnik, L. Burzyńska and W. Gumowska, Miner. Eng., 2009, 22, 88–95 CrossRef CAS.
- Z. Yang, M. Rui-Lin, N. Wang-Dong and W. Hui, Hydrometallurgy, 2010, 103, 25–29 CrossRef.
- A. Lassin, P. Piantone, A. Burnol, F. Bodénan, L. Chateau, C. Lerouge, C. Crouzet, D. Guyonnet and L. Bailly, J. Hazard. Mater., 2007, 139, 430–437 CrossRef CAS PubMed.
- A. Smaniotto, A. Antunes, I. do N. Filho, L. D. Venquiaruto, D. de Oliveira, A. Mossi, M. Di Luccio, H. Treichel and R. Dallago, J. Hazard. Mater., 2009, 172, 1677–1680 CrossRef CAS PubMed.
- G. De Angelis, F. Medici, M. R. Montereali and L. Pietrelli, Waste Manag., 2002, 22, 925–930 CrossRef CAS PubMed.
- M. Penpolcharoen, Cem. Concr. Res., 2005, 35, 1050–1055 CrossRef CAS.
- M. Shirinbayan, H. Razavizadeh and M. R. Aboutalebi, Russ. J. Non-Ferr. Met., 2014, 55, 538–541 CrossRef.
- L. C. Ferracin, A. E. Chácon-Sanhueza, R. A. Davoglio, L. O. Rocha, D. J. Caffeu, A. R. Fontanetti, R. C. Rocha-Filho, S. R. Biaggio and N. Bocchi, Hydrometallurgy, 2002, 65, 137–144 CrossRef CAS.
- E. Kim, L. Horckmans, J. Spooren, K. C. Vrancken, M. Quaghebeur and K. Broos, Hydrometallurgy, 2017, 169, 372–381 CrossRef CAS.
- E. Kim, L. Horckmans, J. Spooren, K. Broos, K. C. Vrancken and M. Quaghebeur, Hydrometallurgy, 2017, 169, 290–296 CrossRef CAS.
- C. A. Sutherland, E. F. Milner, R. C. Kerby, H. Teindl, A. Melin and H. M. Bolt, in Ullmann's Encycl. Industrial Chem., Wiley-VCH, Weinheim, 2005, vol. 20, pp. 551–597 Search PubMed.
- S. Aydoğan, A. Aras, G. Uçar and M. Erdemoǧlu, Hydrometallurgy, 2007, 89, 189–195 CrossRef.
- S. Nagib and K. Inoue, Hydrometallurgy, 2000, 56, 269–292 CrossRef CAS.
- E. C. Barrett, E. H. Nenniger and J. Dziewinski, Hydrometallurgy, 1992, 30, 59–68 CrossRef CAS.
- K. Binnemans and P. T. Jones, J. Sust. Metall., 2017, 3, 570–600 CrossRef.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- A. P. Abbott, A. Al-Bassam, A. Goddard, R. C. Harris, G. Jenkin, F. Nisbett and M. Wieland, Green Chem., 2017, 19, 2225–2233 RSC.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945–949 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89(8), 4595–4603 CrossRef PubMed.
- J. S. Casas and J. Sordo, Lead: Chemistry, Analytical Aspects, Environmental Impact and Health Effects, Elsevier, 2011, pp. 26–27 Search PubMed.
- Environmental Agency, Waste Sampling and Testing for Disposal to Landfill, Environment Agency Horizon house, Bristol, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction patterns of matte and slag. See DOI: 10.1039/c7ra09150h |
| This journal is © The Royal Society of Chemistry 2017 |