
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA short synthesis of 7-amino alkoxy homoisoflavonoides†
Ali Asadipoura,
Yaghoub Pourshojaei *ab,
Khalil Eskandari*a and
Alireza Foroumadic
*ab,
Khalil Eskandari*a and
Alireza Foroumadic
aDepartment of Medicinal Chemistry, Faculty of Pharmacy & Pharmaceutics Research Center, Institute of Neuropharmacology, Kerman University of Medical Sciences, Kerman, Iran. E-mail: y.pourshoja@kmu.ac.ir; khalileskandari@yahoo.com
bNeuroscience Research Center, Institute of Neuropharmacology, Kerman University of Medical Sciences, Kerman, Iran
cDrug Design and Development Research Center, Tehran University of Medical Sciences, Tehran, Iran
First published on 18th September 2017
Abstract
The synthesis of novel derivatives of homoisoflavonoids as potentially interesting medicinally important heterocycles in an efficient catalytic two step route is introduced. In the first step, 7-aminoalkoxychromane-4-ones are synthesized via reaction between 7-hydroxychroman-4-one and aminoethylchlorides in the presence of potassium carbonate as a Brønsted base catalyst. In the next step, obtained 7-aminoalkoxychromane-4-ones are reacted with a wide range of arylaldehydes in the presence of hydrochloric acid as Brønsted acid catalyst to obtain homoisoflavonoids. Target products are medicinally very important heterocycles, because their analogs show cytotoxic activities towards human cancer cell lines.
Introduction
Homoisoflavonoids are a class of flavonoid-type natural products which have been isolated from numerous plants,1 notably in the Hyacinthaceae family.2 Their chemical structures consist of two aromatic carbocycles (phenyl) and one non-aromatic heterocycle (dihydropyran-4-one). The homoisoflavonoids can be divided into five categories: the 3-benzyl-4-chromanones (A), the 3-hydroxy-3-benzyl-4-chromanones (B), the 3-benzyl-chrom-2-en-4-ones (C), the scillascillins (D), and the 3-benzylidene-4-chromanones (E or Z) (E) (Fig. 1).3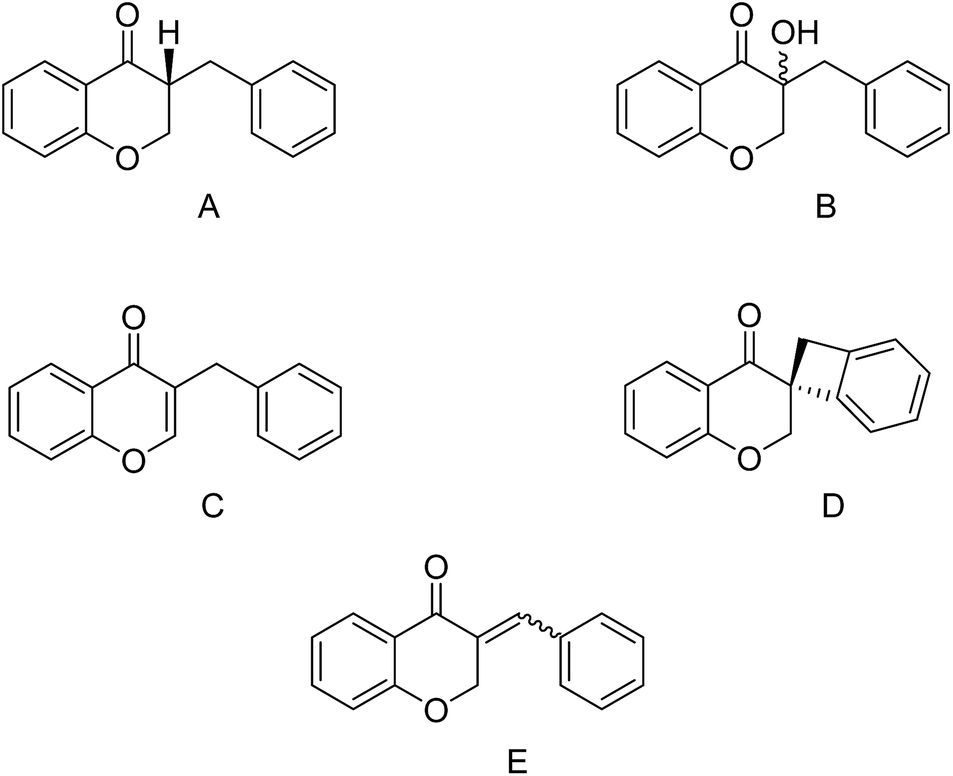 | ||
| Fig. 1 Chemical structure classification of homoisoflavonoids. | ||
To date, a wide range of biological behaviors of homoisoflavonoids, their synthesis and biosynthesis have been studied and reported in the literature.1,4 These types of compounds have shown many biological activities such as antiangiogenic,5 antifungal,6 hypocholesterolemic,7 antimutagenic,8 anti-inflammatory,9 and antiviral activities.10,11
Among natural products containing homoisoflavonoid scaffold, Bonducellin (isolated from Caesalpinia bonducella and Caesalpinia pulcherrima)12 acts as inhibitor of multidrug resistance efflux pump.13 Also, isobonducellin, (the Z-isomer of Bonducellin), and 8-methoxybonducellin (isolated from C. pulcherrima)14,15 have been shown anti-oxidant activity.16 In addition, Sappanone A (isolated from the heartwood of Caesalpinia) is another homoisoflavonoid type compounds which exhibits antioxidant activity (Fig. 2).17
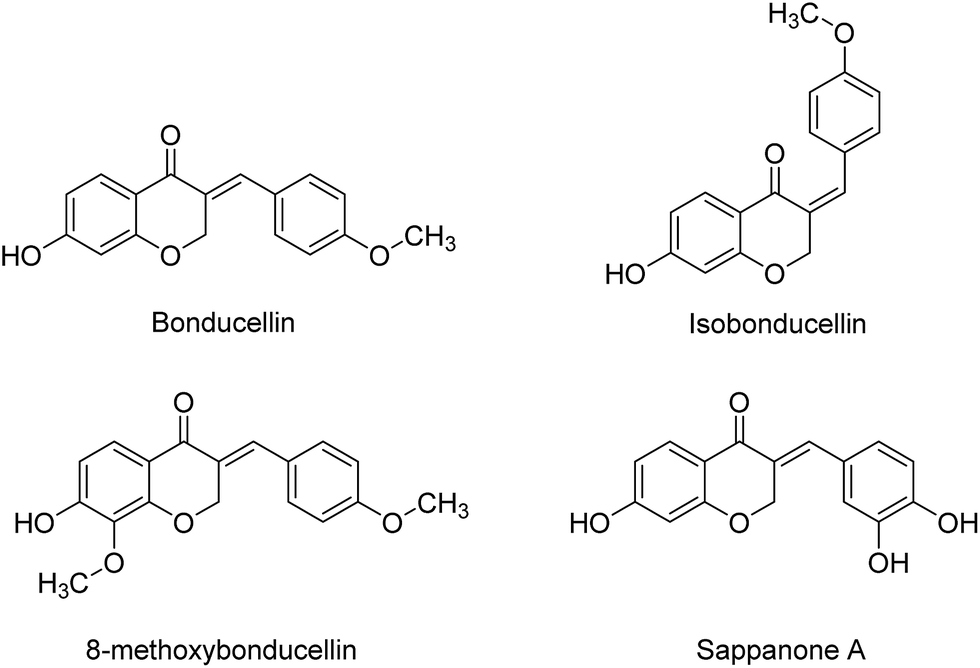 | ||
| Fig. 2 Some selected homoisoflavonoids having pharmacological activity. | ||
Considering above and existence the great interest in synthesis of important pharmacological active heterocycles,18 the synthesis of homoisoflavonoids has also attracted the interest of chemists and pharmaceutics.
Nowadays, there are numerous successful attempts to the laboratory synthesis of these kinds of compounds. In general, there are two protocols for the synthesis of these compounds. In the first approach, homoisoflavonoids are obtained from condensation reaction between 4-chromanones and arylaldehydes in methanol as solvent and by passing HCl gas as Brønsted acid catalyst or by using piperidine as a Brønsted base catalyst19,20 followed by isomerisation of the double bond in the presence of Pd/C at 250 °C. In second ones homoisoflavonoids are obtained by hydrogenation of chalcones followed by one carbon extension by the use of ethyl formate/sodium21 or methanesulfonyl chloride/DMF.22 Based on all the above, herein, we wish to report an efficient route to the synthesis of a wide range of novel homoisoflavonoids by rely on first mentioned protocol. By this achievement the scope of homoisoflavonoids as medicinally important molecules was developed.
Results and discussion
Following to our continuous attempts to develop the new and green strategies for the synthesis of newly prepared organic compounds23 especially new chemical intermediates and heterocycles,24,25 and also alternative multi-steps routes to the synthesis of medicinally important and biological active heterocycles,26 in this study, our attention, is turned towards the synthesis of new homoisoflavonoid derivatives. For this purpose, we first had to prepare 7-hydroxychroman-4-one 4 via a two-step reported method17 including the Friedel–Crafts acylation and then intramolecular cyclization (Scheme 1). As can be seen from Scheme 1, at first, 2′,4′-dihydroxy-3-chloro propiophenone 3 is synthesized from acylation reaction between resorcinol (1) and 3-chloropropionic acid (2), in the presence of CF3SO3H as catalyst. Then, 2′,4′-dihydroxy-3-chloro propiophenone 3 in the presence of NaOH, undergoes intramolecular cyclization to obtain 7-hydroxychroman-4-one 4.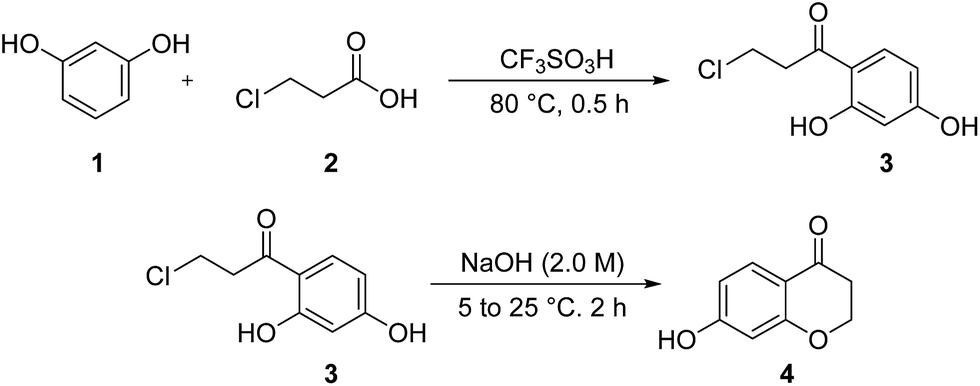 | ||
| Scheme 1 Alternative two steps route to the synthesis of 7-hydroxychroman-4-one. | ||
In the next step, 7-hydroxychroman-4-one 4 via a reaction which is well-known as Williamson ether synthesis is reacted with appropriate aminoethylchlorides 5 to obtain 7-aminoalkoxychroman-4-ones 6. The role of KI in this reaction is activation of aminoethylchloride to nucleophilic attack. Also K2CO3 has been used as Brønsted catalyst to activation of phenolic OH. After preparation, purification, and characterization of 7-aminoalkoxychroman-4-ones 6, they are employed in the reaction with a wide range of arylaldehydes 7 containing both electron donor and electron withdrawing groups via Knoevenagel condensation reaction to give novel homoisoflavonoid derivatives 8 (Scheme 2). The role of HCl in this reaction is inevitable and acts as Brønsted acid catalyst to activation of both 7-aminoalkoxychroman-4-ones and aryl aldehydes to condense together.
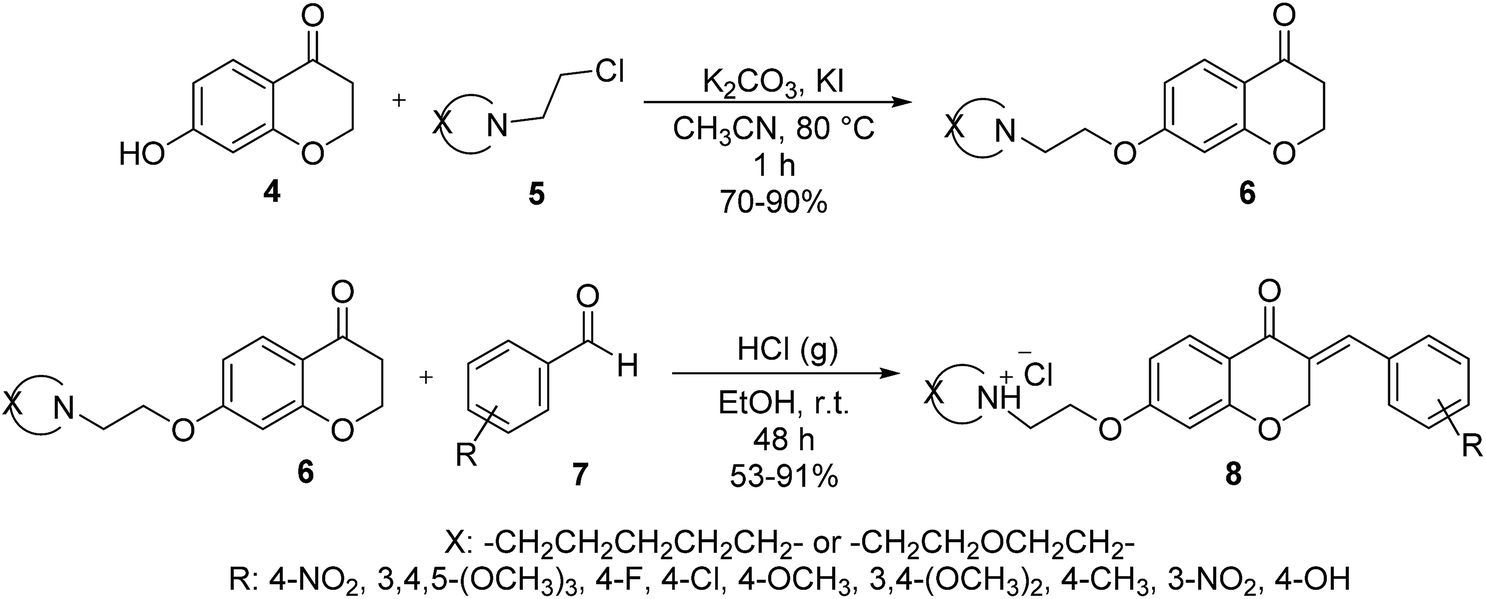 | ||
| Scheme 2 Cascade two step routes to the synthesis of homoisoflavonoid derivatives. | ||
By this achievement, a broad range of novel homoisoflavonoids as important potentially interesting biological active products were synthesized and the scope of these compounds were developed (Table 1).
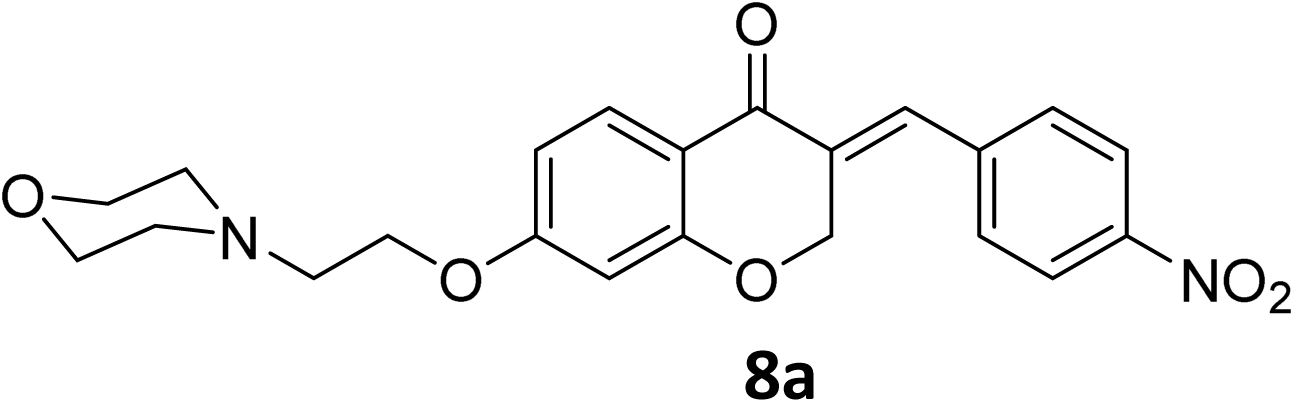
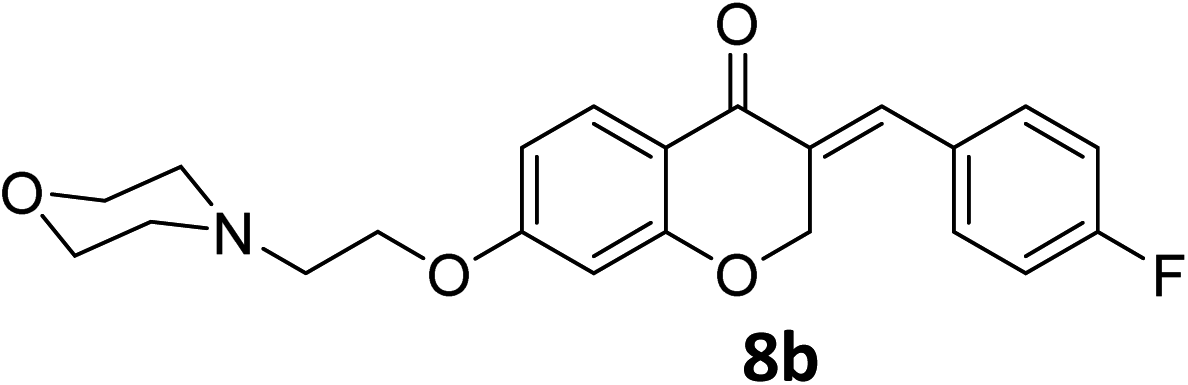
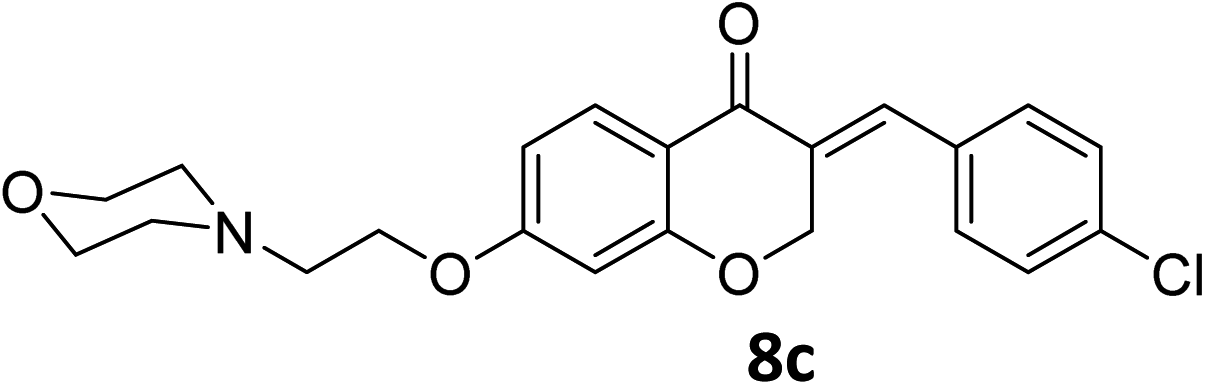
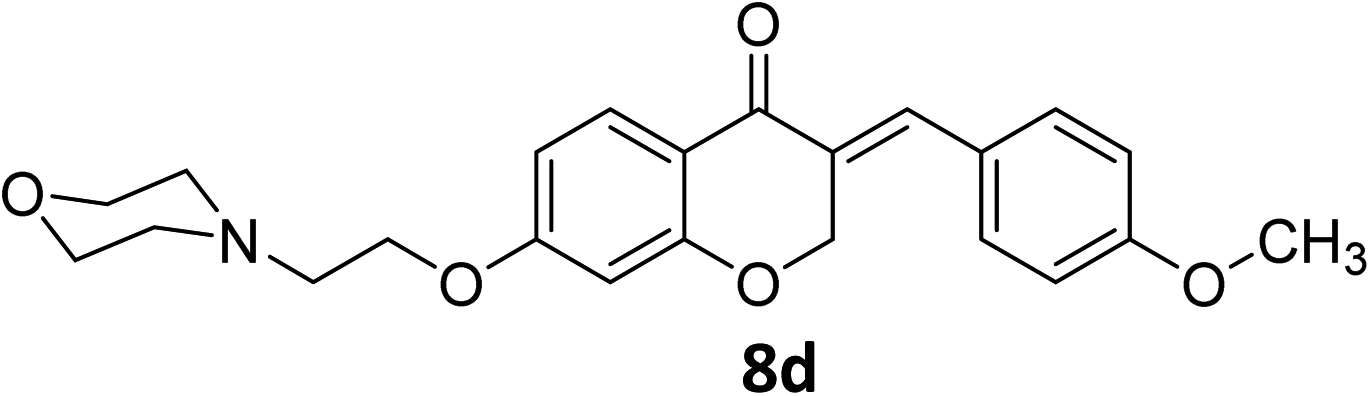
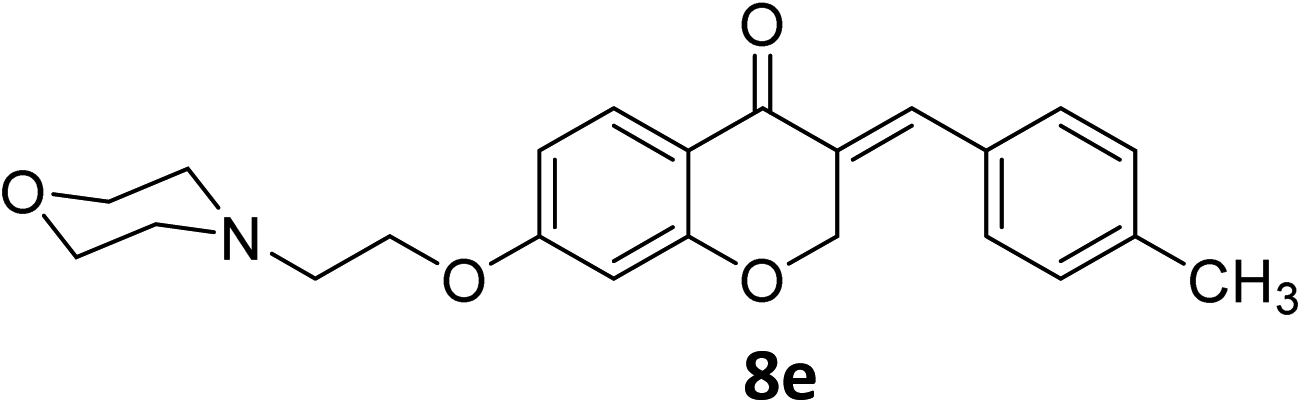
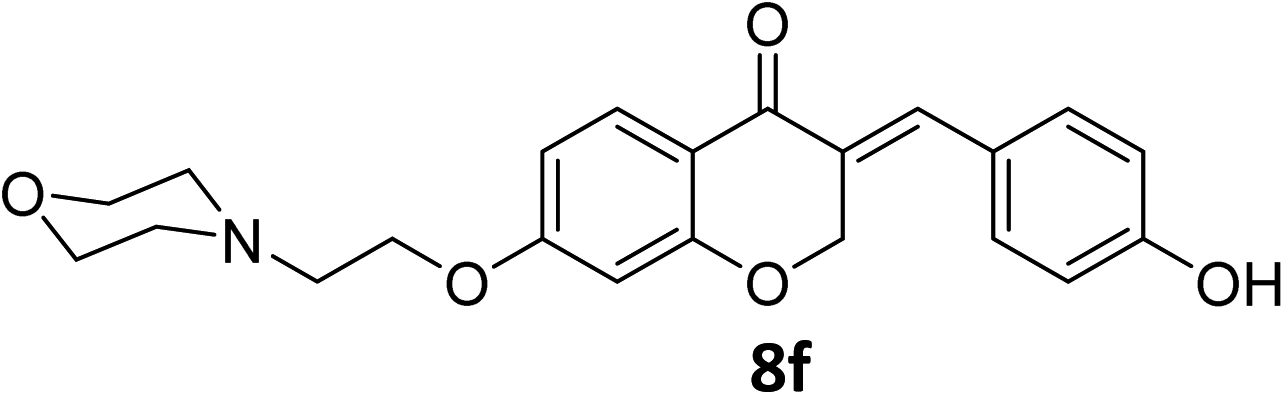
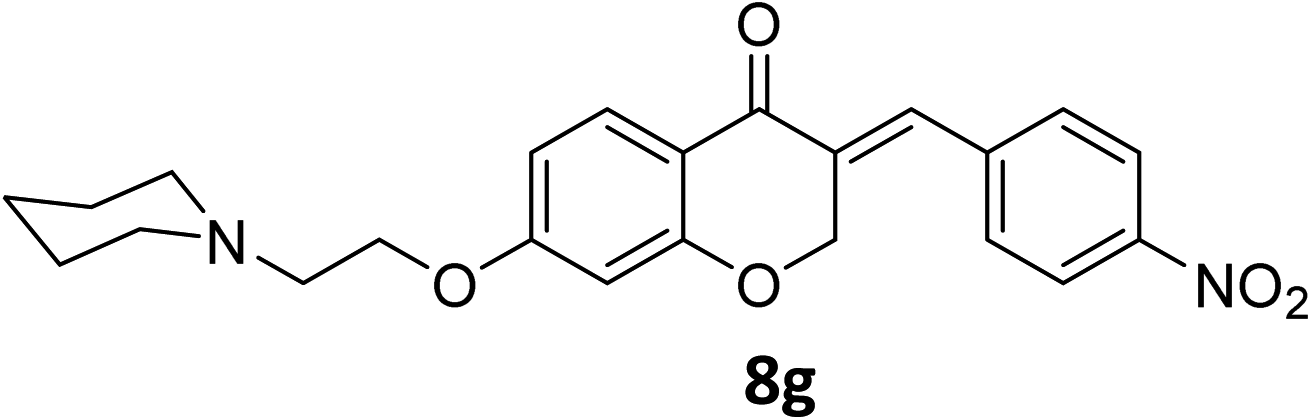
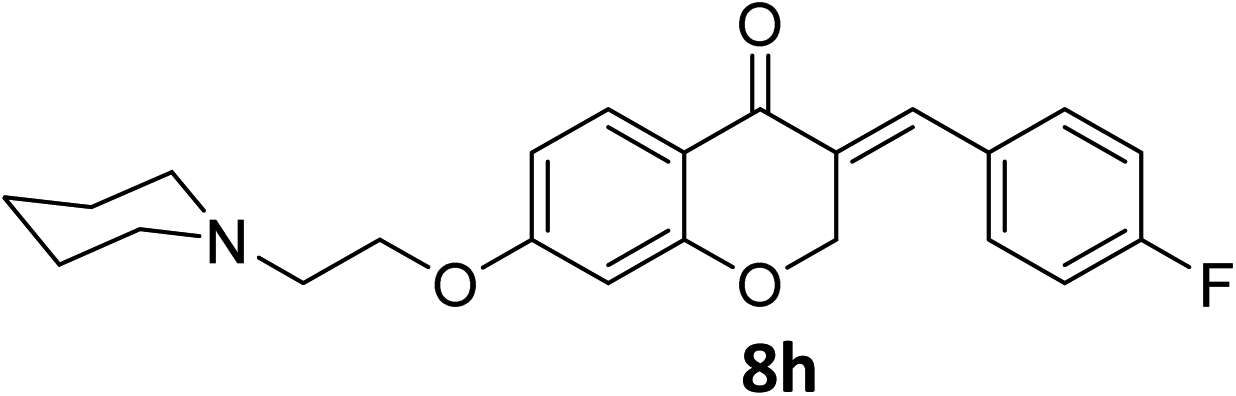
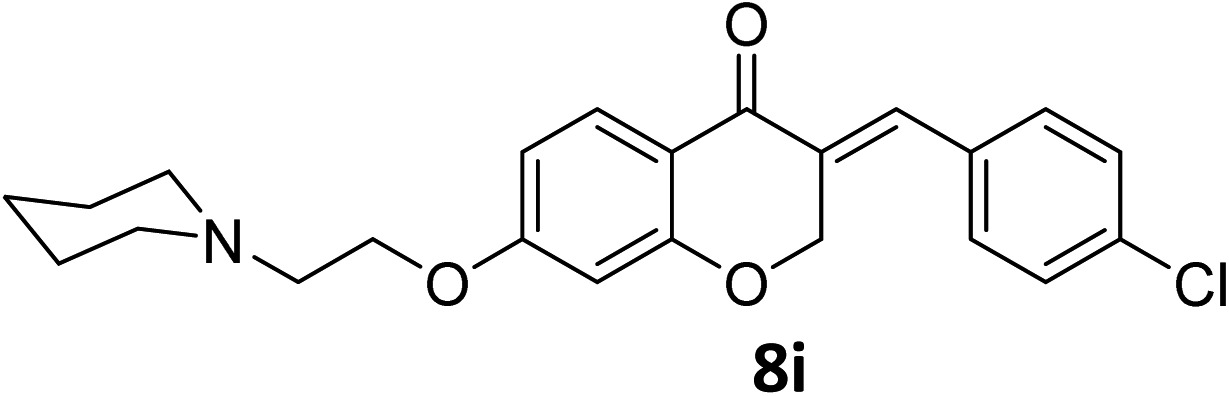
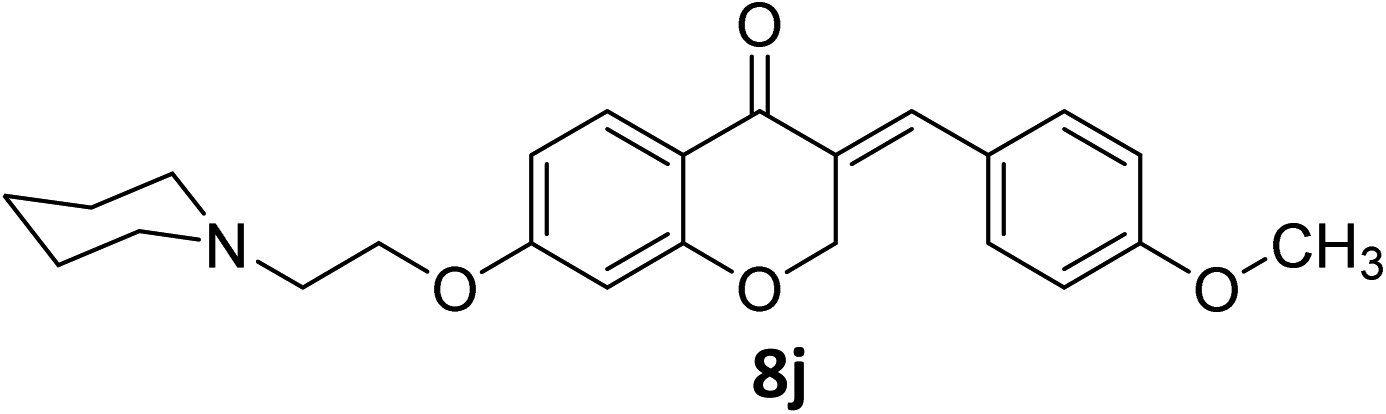
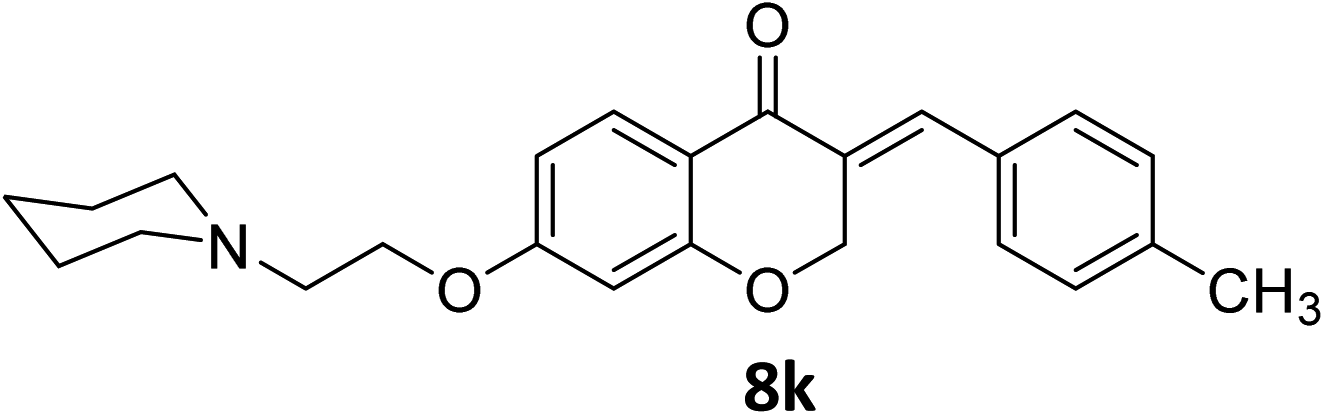
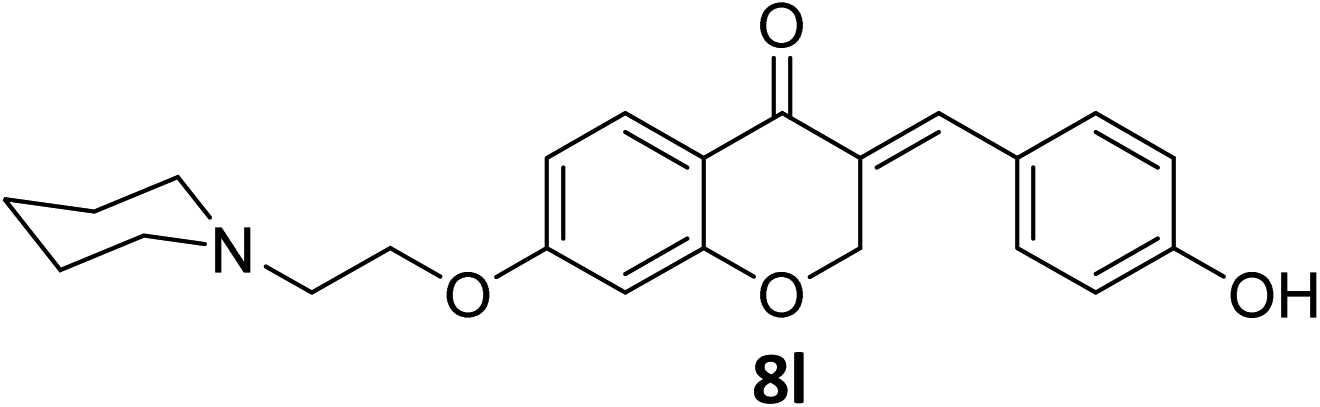
It was clearly observed that the process can tolerate both electron-withdrawing and electron-donating groups on the phenyl ring. All target products were fully characterized by spectral data (H NMR, C NMR, and IR), along with elemental analysis. The data of all products were in good agreement with the expected structures (see ESI†). Also, the numbering of atoms in the target compounds for interpretation of NMR data is indicated in Fig. 3.
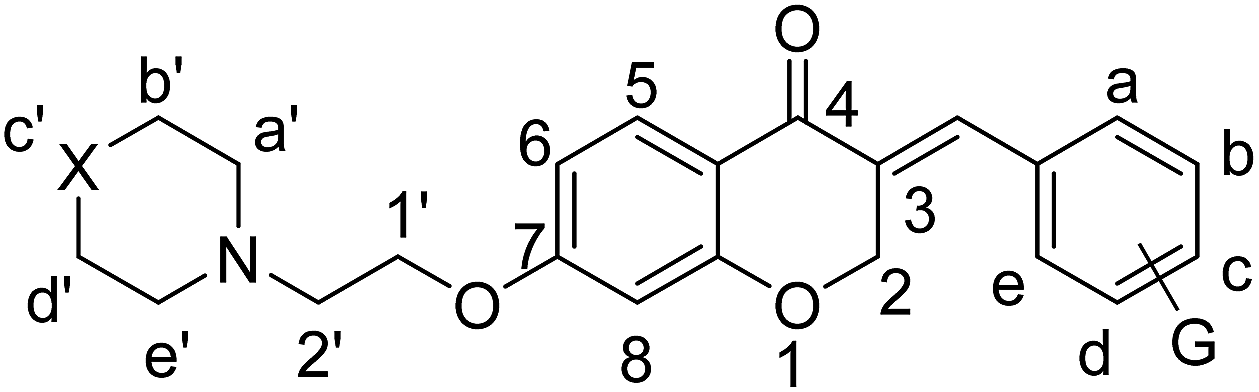 | ||
| Fig. 3 Atom numbering refers to the order of real atoms in interpretation of NMR data. | ||
In another investigation, we examined other conditions such as using various solvents including H2O, CHCl4, CH3CN, dioxane, toluene, DMF, DMSO in different temperatures at r.t., 40 °C, 60 °C, 80 °C, 100 °C, and 120 °C, and also the effect of other homogeneous and heterogeneous basic and acidic catalysts such as SSC, K2CO3, NaOH, KOH, piperidine, and triethylamine, ZnO, MgCl2, FeCl3, CF3COOH, CH3COOH, H3PO4 on model reaction (8j) was evaluated. The results obtained from this experiment show that only using HCl(g) at room temperature in ethanol provides best conditions to obtain our products with highest yields. Because of our target products have containing amine side chain, the use of HCl(g) in our procedure cause to obtain amine containing HCl as salt products which leads to precipitate target products and subsequently results to improve the yield of reaction.
As all of our synthesized products are novel compounds, for comparison of our current approach to others available, we selected compound 8j as model and compared its obtaining method with other homoisoflavonoids containing 4-OR substituted on benzylidene moiety under different conditions (Scheme 3). The results show that best and more efficient condition to the synthesis of homoisoflavonoids is provided by using HCl in ethanol at room temperature which leads to achieve the product with highest yield than others.27–34
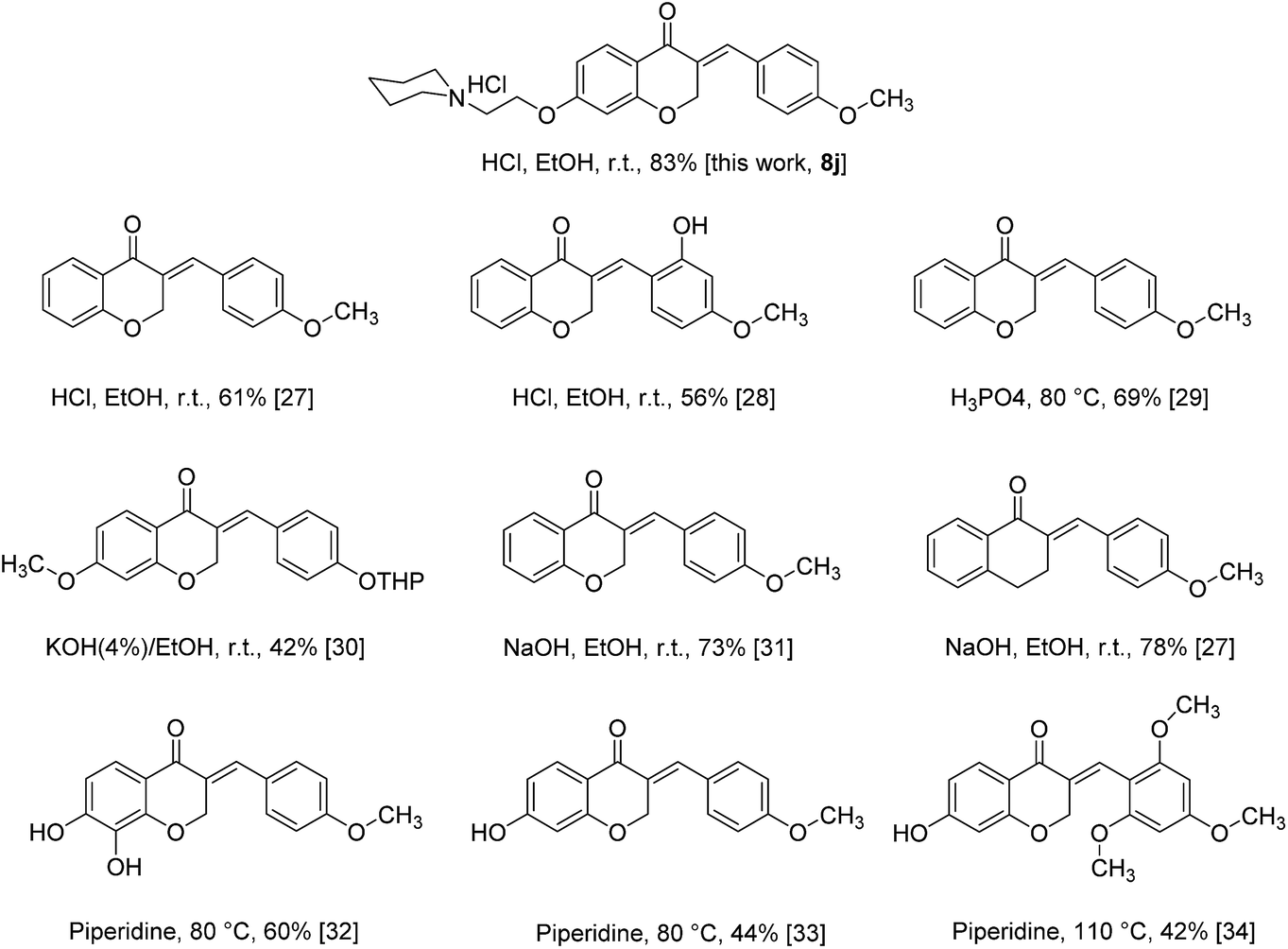 | ||
| Scheme 3 Comparison of homoisoflavonoids synthesis under different conditions with our current protocol. | ||
Based on the above, by this achievement, the scope of homoisoflavonoids as very medicinally important compounds was developed. Rapid formation and easily purification of target products, needing a little of HCl(g) to accelerate reaction can be countered as other advantages of this protocol. However, this protocol tolerate some disadvantages such as using HCl(g) as hazardous material, the excessing use of HCl(g) lead to obtain unfavorable products, and working with HCl(g) needs to observe more safety principles in the laboratory.
Experimental
All reagents, starting materials, and solvents applied in this work were purchased from commercial Merck AG, Aldrich, and Acros Organics chemical companies. Melting points of compounds were determined on Kolfer hot stag apparatus and are uncorrected. IR spectra were taken using Nicolet FT-IR Magna 550 spectrographs on KBr plates. The NMR spectra were recorded on Bruker Avance 400 or 500 MHz, FT-NMR spectrometers, Germany. 1H NMR data are reported in the following order: chemical shifts (δ) in part per million (ppm) down field from TMS as internal standard; approximate coupling constant values (J) in Hertz (HZ); spin multiplicities (s, singlet; d, doublet; t, triplet; m, multiplet; q, quartet; br, broad; quin, quintet). Thin-layer chromatography (TLC) was performed on pre-coated Silica Gel F254 plates for controlling the reaction progress.Typical procedure to the synthesis of 2′,4′-dihydroxy-3-chloro propiophenone 335
Trifluoromethanesulfonic acid (100 g, 0.67 mol) was added to a stirred mixture of resorcinol (1) (20.0 g, 0.182 mol) and 3-chloropropionic acid (2) (20.0 g, 0.184 mol) at room temperature. It was allow to solution reaching 80 °C for 30 min, and then cooled to room temperature over 15 min. In the following step, the solution was poured into chloroform (400 mL), and then slowly poured into water (400 mL). Afterwards, the layers were separated from each other. The aqueous phase was extracted with 2 × 200 mL of CHCl3. The combined organic phases were washed with brine, dried over Na2SO4, and finally filtered. The highly viscous product was obtained by concentration in vacuum as an orange semisolid (24.1 g, 66%). 1H-NMR δ 12.56 (lH, s), 7.63 (lH, d, J = 7.6 Hz), 6.37–6.46 (2H, m), 3.92 (2H, t, J = 6.3 Hz), 3.41 (2H, t, J = 6.3 Hz).Typical procedure to the synthesis of 7-hydroxychroman-4-one 435
2′,4′-Dihydroxy-3-chloro propiophenone 3 (24.1 g, 0.12 mol) was added to a stirred solution of NaOH (2 M, 1.0 L) at 5 °C. Then the solution was allowed to warm around room temperature over 2 h. Afterwards, the reaction mixture was re-cooled to 5 °C, and by the use of H2SO4 (6 M, 100 mL), the pH was adjusted to 2. At the end, the mixture was extracted three times with EtOAc (3 × 200 mL), washed with brine, dried over Na2SO4, and then filtered. The crude product was obtained by concentration in vacuum as a tan solid. The crude product was softened by hexanes, filtrated, and then recrystallized by water to give colorless crystals (18.4 g, 61%): mp: 143 °C; 1H-NMR δ 7.84 (lH, d, J = 9.4 Hz), 6.73 (1H, brd s), 6.55 (1H, dd, J = 9.4, 1.8 Hz), 6.43 (1H, d, J = 1.8 Hz), 4.54 (2H, t, J = 6.5 Hz), 2.78 (2H, t, J = 6.5 Hz); IR (cm−1): 3160, 1660, 1596, 1451, 1370, 1254.General procedure for the synthesis of 7-(2-aminoethoxy)chroman-4-ones 623b
To a mixture of 7-hydroxychroman-4-one 1 (1 mmol) and appropriate aminoethyl chloride hydrochloride (1.1 mmol) in acetonitrile (10 mL) in a single-neck round bottom flask, potassium carbonate (3 mmol) and a catalytic amount of potassium iodide (5% mol) were added. The mixture was stirred under reflux for 10–14 h and regularly monitored for reaction progress by TLC using 10% methanol/dichloromethane as the solvent system. After cooling down to room temperature, the entire content was filtered under suction and the solid inorganic salts were washed with acetonitrile (3 × 10 mL). The resulting filtrate was evaporated to dryness under reduced pressure and residue was purified by silica column chromatography (1–9% methanol in dichloromethane, v/v as eluent) to afford compounds 6 in 70–90% yields.General procedure for the synthesis of E-7-(2-aminoethoxy)-3-aryliden-2,3-dihydrochromen-4-one hydrochloride derivatives 8
A solution of 7-aminoalkoxychroman-4-ones 6 (1 mmol), and appropriate arylaldehyde (1 mmol) in EtOH (5 mL) was stirred at room temperature for 1 min, under passing a steam of HCl gas. After the time, the insertation of HCl was blockade, and the mixture was stirred overnight at room temperature to precipitate product. Subsequently, filter-off the precipitated product and crystallized from EtOH to give compounds 8. In some cases which the crystals need to further purification, silica gel column chromatography with ethyl acetate/n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as eluent was applied to obtain crystalline pure products.
:2) as eluent was applied to obtain crystalline pure products.
Typical procedure for the synthesis of E-7-(2-morpholino-ethoxy)-3-(4-nitrobenzyliden)-2,3-dihydrochromen-4-one hydrochloride 8a
To a stirred mixture of 7-morpholinoethoxychroman-4-one 6 (0.277 g, 1 mmol) in EtOH (5 mL) at room temperature equipped with bypassing HCl(g) valve, 4-nitrobenzaldehyde 7a (0.151 g, 1 mmol) was added. Afterwards, it was permitted to HCl(g) to insert in reaction mixture for 1 min. When inserting of HCl to the mixture was completed, it was allowed to the mixture to remain overnight at room temperature to precipitate product. Then, the mixture was filtered, and obtained crude product was recrystallized in boiling EtOH to afford crystalline pure 8a. The obtained product was characterized by IR, 1H NMR, 13C NMR and elemental analysis (C, H and N).Spectral data of product 8
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1613 (CC alkene); 1H NMR (CDCl3, 500 MHz) δ: 8.31 (d, 2H, H-a, J = 8 Hz), 7.97 (d, 1H, H-6, J = 8 Hz), 7.83 (s, 1H, H-8), 7.46 (d, 2H, H-b, J = 8 Hz), 6.68 (d, 1H, H-5, J = 8 Hz), 6.24 (s, 1H, vinylic-H), 5.28 (s, 2H, H-2), 4.16 (br s, 2H, H-1′), 3.74 (br, 4H, H-b′), 2.82 (br s, 2H, H-2′), 2.58 (br s, 4H, H-a′); 13C NMR (125 MHz, CDCl3) δ: 180.1, 165.5, 163.1, 147.7, 140.9, 133.9, 133.7, 130.4, 129.8, 123.9, 115.7, 115.5, 111.2, 101.5, 67.4, 66.8, 66.4, 57.2, 54.0. Anal. calcd for C22H22N2O6·HCl (446.88): C, 59.13; H, 5.19; N, 6.27. Found: C, 59.18; H, 5.21; N, 6.29.O), 1614 (CC alkene); 1H NMR (DMSO-d6, 400 MHz), δ: 11.56 (s, 1H, NH), 7.87 (d, 1H, H-5, J = 8.8 Hz), 7.72 (s, 1H, H-8), 7.53 (dd, 1H, H–Ar, J = 5.6 and 3.2 Hz), 7.34 (m, 2H, H–Ar), 6.79 (dd, 1H, H-6, J = 8.8 and 2.4 Hz), 6.67 (d, 1H, vinylic-H, J = 2.2 Hz), 5.41 (d, 2H, H-2, J = 2.2 Hz), 4.55 (t, 2H, H-1′, J = 5 Hz), 3.96–3.82 (br m, 4H, H-b′), 3.56 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.20 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 164.3, 163.0, 161.8, 135.4, 133.2, 133.1, 130.9, 129.6, 116.4, 116.2, 116.0, 111.4, 102.4, 68.1, 63.6, 63.2, 55.0, 52.0. Anal. calcd for C22H22FNO4·HCl (419.87): C, 62.93; H, 5.52; N, 3.34. Found: C, 62.91; H, 5.50; N, 3.34.O), 1613 (CC alkene); 1H NMR (DMSO-d6, 400 MHz), δ: 11.47 (br s, 1H, NH), 7.87 (d, 1H, H-5, J = 8.8 Hz), 7.71 (s, 1H, H-8), 7.55 (d, 2H, H-a, J = 8.4 Hz), 7.50 (d, 2H, H-b, J = 8.4 Hz), 6.79 (dd, 1H, H-6, J = 8.8 and 2.4 Hz), 6.67 (d, 1H, vinylic-H, J = 2.2 Hz), 5.41 (d, 2H, H-2, J = 2.2 Hz), 4.55 (t, 2H, H-1′, J = 4.8 Hz), 3.93–3.86 (br d, 4H, H-b′), 3.55 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.19 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.1, 164.3, 163.0, 135.1, 134.7, 133.2, 132.5, 131.7, 129.7, 129.3, 116.0, 111.4, 102.4, 68.1, 63.6, 63.3, 55.0, 52.1. Anal. calcd for C22H22ClNO4·HCl (436.33): C, 60.56; H, 5.31; N, 3.21. Found: C, 60.57; H, 6.19; N, 3.34.O), 1663 (CC alkene); 1H NMR (DMSO-d6, 400 MHz) δ: 11.51 (s, 1H, NH), 7.85 (d, 1H, H-5, J = 8.8 Hz), 7.68 (s, 1H, H-8), 7.44 (d, 2H, H-a, J = 8.4 Hz), 7.07 (d, 2H, H-b, J = 8.4 Hz), 6.77 (dd, 1H, H-6, J = 8.8 and 2 Hz), 6.65 (d, 1H, vinylic-H, J = 2 Hz), 5.43 (d, 2H, H-2, J = 2 Hz), 4.54 (br s, 2H, H-1′), 3.92 (br s, 4H, H-b′), 3.82 (s, 3H, –OMe), 3.55 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.19 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 164.1, 162.8, 136.4, 132.2, 132.1, 129.6, 129.1, 128.9, 126.8, 116.2, 114.8, 111.2, 102.3, 68.3, 67.9, 63.6, 63.2, 55.9, 55.0, 52.1. Anal. calcd for C23H25ClNO5·HCl (431.91): C, 63.96; H, 6.07; N, 3.24. Found: C, 63.97; H, 6.12; N, 3.31.O), 1603 (CC alkene); 1H NMR (DMSO-d6, 400 MHz), 11.38 (s, 1H, NH), 7.86 (d, 1H, H-5, J = 8.8 Hz), 7.69 (s, 1H, H-8), 7.35 (d, 2H, H-a, J = 8.4 Hz), 7.32 (d, 2H, H-b, J = 8.4 Hz), 6.77 (dd, 1H, H-6, J = 8.8 and 2.4 Hz), 6.66 (d, 1H, vinylic-H, J = 2 Hz), 5.42 (d, 2H, H-2, J = 2 Hz), 4.53 (t, 2H, H-1′, J = 5 Hz), 3.92 (br s, 4H, H-b′), 3.55 (t, 2H, H-2′, J = 5 Hz), 3.45 (br s, 2H, H-a′), 3.20 (br s, 2H, H-a′), 2.37 (s, 3H, –CH3). 13C NMR (100 MHz, DMSO-d6) δ: 180.3, 164.2, 162.9, 140.1, 136.5, 131.5, 130.8, 130.3, 129.9, 129.6, 116.1, 111.3, 102.4, 68.2, 63.6, 63.2, 55.0, 52.1, 21.5. Anal. calcd for C23H25NO4·HCl (415.91): C, 66.42; H, 6.30; N, 3.37. Found: C, 66.66; H, 6.39; N, 3.39.O), 1582 (CC alkene); 1H NMR (DMSO-d6, 500 MHz), 11.54 (s, 1H, NH), 10.22 (s, 1H, OH), 7.83 (d, 1H, H-5, J = 8.8 Hz), 7.63 (s, 1H, H-8), 7.32 (d, 2H, H-a, J = 8.6 Hz), 6.90 (d, 2H, H-b, J = 8.6 Hz), 6.76 (dd, 1H, H-6, J = 8.8 and 2.35 Hz), 6.64 (d, 1H, vinylic-H, J = 2 Hz), 5.41 (d, 2H, H-2, J = 2 Hz), 4.54 (br s, 2H, H-1′), 3.96–3.81 (br m, 4H, H-b′), 3.56–3.44 (br s, 4H, H-a′), 3.22 (t, 2H, H-2′, J = 9.2 Hz). 13C NMR (125 MHz, DMSO-d6) δ: 179.8, 163.5, 162.3, 159.3, 159.1, 136.4, 132.7, 129.0, 127.4, 124.8, 115.8, 110.6, 101.8, 67.9, 63.1, 62.7, 54.5, 51.6. Anal. calcd for C22H23NO5·HCl (417.88): C, 63.23; H, 5.79; N, 3.35. Found: C, 63.61; H, 5.88; N, 3.37.O), 1609 (CC alkene); 1H NMR (DMSO-d6, 500 MHz) δ: 10.55 (br s, 1H, NH), 8.31 (d, 2H, H-a, J = 6.75 Hz), 7.87 (d, 1H, H-5, J = 8.8 Hz), 7.79 (s, 1H, H-8), 7.72 (d, 2H, H-b, J = 6.75 Hz), 6.79 (d, 1H, H-6, J = 8.8 Hz), 6.67 (s, 1H, vinylic-H), 5.42 (s, 2H, H-2), 4.51 (br s, 2H, H-1′), 3.48 (br s, 4H, H-a′), 2.99 (br s, 2H, H-2′), 1.78–1.37 (br m, 6H, H-c′ and H-b′). 13C NMR (125 MHz, DMSO-d6) δ: 180.1, 164.8, 163.1, 133.9, 133.7, 130.4, 130.0, 129.6, 123.9, 123.5, 115.9, 111.0, 101.5, 67.5, 65.7, 55.3, 53.8, 22.6, 22.2. Anal. calcd for C23H24N2O5 HCl (444.91): C, 62.09; H, 5.66; N, 6.30. Found: C, 62.26; H, 5.79; N, 6.34.O), 1616 (CC alkene); 1H NMR (CDCl3, 300 MHz) δ: 12.53 (s, 1H, NH), 7.97 (br s, 1H, H-5), 7.80 (s, 1H, H-8), 7.30–7.14 (m, 4H, H-a and H-b), 6.63 (br s, 1H, H-6), 6.45 (br s, 1H, vinylic-H), 5.31 (s, 2H, H-2), 4.68 (br s, 2H, H-1′), 3.66–3.50 (br s, 4H, H-a′), 2.84 (br s, 2H, H-2′), 1.92–1.47 (br m, 6H, H-b′ and H-c′). 13C NMR (75 MHz, CDCl3) δ: 180.8, 164.8, 163.3, 163.0 (d, J = 112.4), 136.1, 135.9, 132.0, 131.9, 130.4, 130.1, 116.1, 115.8, 110.7, 102.2, 67.9, 63.6, 55.0, 52.0, 23.0, 22.0. Anal. calcd for C23H24FNO3·HCl (417.9): C, 66.10; H, 6.03; N, 3.35. Found: C, 66.26; H, 6.14; N, 3.37.O), 1583 (CC alkene); 1H NMR (DMSO-d6, 500 MHz) δ: 10.84 (br s, 1H, NH), 7.84 (d, 1H, H-5, J = 8.8 Hz), 7.69 (s, 1H, H-8), 7.55 (d, 2H, H-a, J = 7.5 Hz) 7.48 (d, 2H, H-b, J = 7.5 Hz), 6.77 (d, 1H, H-6, J = 8.8 Hz), 6.65 (s, 1H, vinylic-H), 5.39 (s, 2H, H-2), 4.51 (br s, 2H, H-1′), 3.47 (br s, 4H, H-a′), 2.98 (br s, 2H, H-2′), 1.78–1.37 (br m, 6H, H-c′ and H-b′). 13C NMR (125 MHz, DMSO-d6) δ: 179.6, 163.9, 162.6, 134.7, 134.3, 132.8, 132.1, 131.2, 129.2, 128.8, 115.5, 111.0, 101.8, 67.6, 62.9, 54.3, 52.5, 23.3, 21.2. Anal. calcd for C23H24ClNO3·HCl (434.36): C, 63.60; H, 5.80; N, 3.22. Found: C, 63.66; H, 6.09; N, 3.34.O), 1663 (CC alkene); 1H NMR (DMSO-d6, 400 MHz) δ: 11.51 (s, 1H, NH), 7.85 (d, 1H, H-5, J = 8.8 Hz), 7.68 (s, 1H, H-8), 7.44 (d, 2H, H-a, J = 8.4 Hz), 7.07 (d, 2H, H-b, J = 8.4 Hz), 6.77 (dd, 1H, H-6, J = 8.8 and 2 Hz), 6.65 (d, 1H, vinylic-H, J = 2 Hz), 5.43 (d, 2H, H-2, J = 2 Hz), 4.54 (br s, 2H, H-1′), 3.92 (br s, 4H, H-b′), 3.82 (s, 3H, OMe), 3.55 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.19 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 164.1, 162.8, 136.4, 132.2, 132.1, 129.6, 129.1, 128.9, 126.8, 116.2, 114.8, 111.2, 102.3, 68.3, 67.9, 63.6, 63.2, 55.9, 55.0, 52.1. Anal. calcd for C24H27ClNO4·HCl (431.91): C, 63.96; H, 6.07; N, 3.24. Found: C, 63.97; H, 6.12; N, 3.31.O), 1611 (CC alkene); 1H NMR (DMSO-d6, 500 MHz) δ: 11.06 (br s, 1H, NH), 7.84 (d, 1H, H-5, J = 8.6 Hz), 7.68 (s, 1H, H-8), 7.34 (d, 2H, H-a, J = 7.6 Hz), 7.30 (d, 2H, H-b, J = 7.6 Hz), 6.76 (d, 1H, H-6, J = 8.6 Hz), 6.64 (s, 1H, vinylic-H), 5.41 (s, 2H, H-2), 4.53 (br s, 2H, H-1′), 3.47 (br s, 2H, H-a′), 3.39 (br s, 2H, H- a′), 2.98 (t, 2H, H-2′, J = 10.40 Hz), 2.35 (s, 3H, CH3), 1.84–1.34 (br m, 6H, H-b′ and H-c′). 13C NMR (125 MHz, DMSO-d6) δ: 180.3, 164.2, 162.9, 140.1, 136.5, 131.5, 130.8, 130.3, 129.9, 129.6, 116.1, 111.3, 102.4, 68.2, 63.6, 63.2, 55.0, 52.1, 21.5. Anal. calcd for C24H27NO3·HCl (413.94): C, 69.64; H, 6.82; N, 3.38. Found: C, 66.63; H, 6.69; N, 3.34.O), 1605 (CC alkene); 1H NMR (300 MHz, DMSO-d6) δ: 10.80 (s, 1H, NH), 10.24 (s, 1H, –OH), 7.83 (d, 1H, H-5, J = 8.7 Hz), 7.62 (s, 1H, H-8), 7.32 (d, 2H, H-a, J = 7.5 Hz), 6.90 (d, 2H, H-b, J = 7.5 Hz), 6.75 (d, 1H, H-6, J = 8.7 Hz), 6.62 (s, 1H, vinylic-H), 5.41 (s, 2H, H-2), 4.50 (br s, 2H, H-1′), 3.44 (br s, 4H, H-a′), 2.97 (br s, 2H, H-2′), 1.78–1.36 (br m, 6H, H-b′, H-c′). 13C NMR (75 MHz, DMSO-d6) δ: 180.2, 164.0, 162.8, 159.7, 136.9, 136.8, 133.1, 129.6, 127.8, 125.2, 116.2, 116.2, 111.1, 102.2, 68.4, 63.2, 54.8, 53.0, 22.7, 21.6. Anal. calcd for C23H25NO4·HCl (415.91): C, 66.42; H, 6.30; N, 3.37. Found: C, 66.56; H, 6.34; N, 3.39.
O), 1613 (CC alkene); 1H NMR (CDCl3, 500 MHz) δ: 8.31 (d, 2H, H-a, J = 8 Hz), 7.97 (d, 1H, H-6, J = 8 Hz), 7.83 (s, 1H, H-8), 7.46 (d, 2H, H-b, J = 8 Hz), 6.68 (d, 1H, H-5, J = 8 Hz), 6.24 (s, 1H, vinylic-H), 5.28 (s, 2H, H-2), 4.16 (br s, 2H, H-1′), 3.74 (br, 4H, H-b′), 2.82 (br s, 2H, H-2′), 2.58 (br s, 4H, H-a′); 13C NMR (125 MHz, CDCl3) δ: 180.1, 165.5, 163.1, 147.7, 140.9, 133.9, 133.7, 130.4, 129.8, 123.9, 115.7, 115.5, 111.2, 101.5, 67.4, 66.8, 66.4, 57.2, 54.0. Anal. calcd for C22H22N2O6·HCl (446.88): C, 59.13; H, 5.19; N, 6.27. Found: C, 59.18; H, 5.21; N, 6.29.O), 1614 (CC alkene); 1H NMR (DMSO-d6, 400 MHz), δ: 11.56 (s, 1H, NH), 7.87 (d, 1H, H-5, J = 8.8 Hz), 7.72 (s, 1H, H-8), 7.53 (dd, 1H, H–Ar, J = 5.6 and 3.2 Hz), 7.34 (m, 2H, H–Ar), 6.79 (dd, 1H, H-6, J = 8.8 and 2.4 Hz), 6.67 (d, 1H, vinylic-H, J = 2.2 Hz), 5.41 (d, 2H, H-2, J = 2.2 Hz), 4.55 (t, 2H, H-1′, J = 5 Hz), 3.96–3.82 (br m, 4H, H-b′), 3.56 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.20 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 164.3, 163.0, 161.8, 135.4, 133.2, 133.1, 130.9, 129.6, 116.4, 116.2, 116.0, 111.4, 102.4, 68.1, 63.6, 63.2, 55.0, 52.0. Anal. calcd for C22H22FNO4·HCl (419.87): C, 62.93; H, 5.52; N, 3.34. Found: C, 62.91; H, 5.50; N, 3.34.O), 1613 (CC alkene); 1H NMR (DMSO-d6, 400 MHz), δ: 11.47 (br s, 1H, NH), 7.87 (d, 1H, H-5, J = 8.8 Hz), 7.71 (s, 1H, H-8), 7.55 (d, 2H, H-a, J = 8.4 Hz), 7.50 (d, 2H, H-b, J = 8.4 Hz), 6.79 (dd, 1H, H-6, J = 8.8 and 2.4 Hz), 6.67 (d, 1H, vinylic-H, J = 2.2 Hz), 5.41 (d, 2H, H-2, J = 2.2 Hz), 4.55 (t, 2H, H-1′, J = 4.8 Hz), 3.93–3.86 (br d, 4H, H-b′), 3.55 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.19 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.1, 164.3, 163.0, 135.1, 134.7, 133.2, 132.5, 131.7, 129.7, 129.3, 116.0, 111.4, 102.4, 68.1, 63.6, 63.3, 55.0, 52.1. Anal. calcd for C22H22ClNO4·HCl (436.33): C, 60.56; H, 5.31; N, 3.21. Found: C, 60.57; H, 6.19; N, 3.34.O), 1663 (CC alkene); 1H NMR (DMSO-d6, 400 MHz) δ: 11.51 (s, 1H, NH), 7.85 (d, 1H, H-5, J = 8.8 Hz), 7.68 (s, 1H, H-8), 7.44 (d, 2H, H-a, J = 8.4 Hz), 7.07 (d, 2H, H-b, J = 8.4 Hz), 6.77 (dd, 1H, H-6, J = 8.8 and 2 Hz), 6.65 (d, 1H, vinylic-H, J = 2 Hz), 5.43 (d, 2H, H-2, J = 2 Hz), 4.54 (br s, 2H, H-1′), 3.92 (br s, 4H, H-b′), 3.82 (s, 3H, –OMe), 3.55 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.19 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 164.1, 162.8, 136.4, 132.2, 132.1, 129.6, 129.1, 128.9, 126.8, 116.2, 114.8, 111.2, 102.3, 68.3, 67.9, 63.6, 63.2, 55.9, 55.0, 52.1. Anal. calcd for C23H25ClNO5·HCl (431.91): C, 63.96; H, 6.07; N, 3.24. Found: C, 63.97; H, 6.12; N, 3.31.O), 1603 (CC alkene); 1H NMR (DMSO-d6, 400 MHz), 11.38 (s, 1H, NH), 7.86 (d, 1H, H-5, J = 8.8 Hz), 7.69 (s, 1H, H-8), 7.35 (d, 2H, H-a, J = 8.4 Hz), 7.32 (d, 2H, H-b, J = 8.4 Hz), 6.77 (dd, 1H, H-6, J = 8.8 and 2.4 Hz), 6.66 (d, 1H, vinylic-H, J = 2 Hz), 5.42 (d, 2H, H-2, J = 2 Hz), 4.53 (t, 2H, H-1′, J = 5 Hz), 3.92 (br s, 4H, H-b′), 3.55 (t, 2H, H-2′, J = 5 Hz), 3.45 (br s, 2H, H-a′), 3.20 (br s, 2H, H-a′), 2.37 (s, 3H, –CH3). 13C NMR (100 MHz, DMSO-d6) δ: 180.3, 164.2, 162.9, 140.1, 136.5, 131.5, 130.8, 130.3, 129.9, 129.6, 116.1, 111.3, 102.4, 68.2, 63.6, 63.2, 55.0, 52.1, 21.5. Anal. calcd for C23H25NO4·HCl (415.91): C, 66.42; H, 6.30; N, 3.37. Found: C, 66.66; H, 6.39; N, 3.39.O), 1582 (CC alkene); 1H NMR (DMSO-d6, 500 MHz), 11.54 (s, 1H, NH), 10.22 (s, 1H, OH), 7.83 (d, 1H, H-5, J = 8.8 Hz), 7.63 (s, 1H, H-8), 7.32 (d, 2H, H-a, J = 8.6 Hz), 6.90 (d, 2H, H-b, J = 8.6 Hz), 6.76 (dd, 1H, H-6, J = 8.8 and 2.35 Hz), 6.64 (d, 1H, vinylic-H, J = 2 Hz), 5.41 (d, 2H, H-2, J = 2 Hz), 4.54 (br s, 2H, H-1′), 3.96–3.81 (br m, 4H, H-b′), 3.56–3.44 (br s, 4H, H-a′), 3.22 (t, 2H, H-2′, J = 9.2 Hz). 13C NMR (125 MHz, DMSO-d6) δ: 179.8, 163.5, 162.3, 159.3, 159.1, 136.4, 132.7, 129.0, 127.4, 124.8, 115.8, 110.6, 101.8, 67.9, 63.1, 62.7, 54.5, 51.6. Anal. calcd for C22H23NO5·HCl (417.88): C, 63.23; H, 5.79; N, 3.35. Found: C, 63.61; H, 5.88; N, 3.37.O), 1609 (CC alkene); 1H NMR (DMSO-d6, 500 MHz) δ: 10.55 (br s, 1H, NH), 8.31 (d, 2H, H-a, J = 6.75 Hz), 7.87 (d, 1H, H-5, J = 8.8 Hz), 7.79 (s, 1H, H-8), 7.72 (d, 2H, H-b, J = 6.75 Hz), 6.79 (d, 1H, H-6, J = 8.8 Hz), 6.67 (s, 1H, vinylic-H), 5.42 (s, 2H, H-2), 4.51 (br s, 2H, H-1′), 3.48 (br s, 4H, H-a′), 2.99 (br s, 2H, H-2′), 1.78–1.37 (br m, 6H, H-c′ and H-b′). 13C NMR (125 MHz, DMSO-d6) δ: 180.1, 164.8, 163.1, 133.9, 133.7, 130.4, 130.0, 129.6, 123.9, 123.5, 115.9, 111.0, 101.5, 67.5, 65.7, 55.3, 53.8, 22.6, 22.2. Anal. calcd for C23H24N2O5 HCl (444.91): C, 62.09; H, 5.66; N, 6.30. Found: C, 62.26; H, 5.79; N, 6.34.O), 1616 (CC alkene); 1H NMR (CDCl3, 300 MHz) δ: 12.53 (s, 1H, NH), 7.97 (br s, 1H, H-5), 7.80 (s, 1H, H-8), 7.30–7.14 (m, 4H, H-a and H-b), 6.63 (br s, 1H, H-6), 6.45 (br s, 1H, vinylic-H), 5.31 (s, 2H, H-2), 4.68 (br s, 2H, H-1′), 3.66–3.50 (br s, 4H, H-a′), 2.84 (br s, 2H, H-2′), 1.92–1.47 (br m, 6H, H-b′ and H-c′). 13C NMR (75 MHz, CDCl3) δ: 180.8, 164.8, 163.3, 163.0 (d, J = 112.4), 136.1, 135.9, 132.0, 131.9, 130.4, 130.1, 116.1, 115.8, 110.7, 102.2, 67.9, 63.6, 55.0, 52.0, 23.0, 22.0. Anal. calcd for C23H24FNO3·HCl (417.9): C, 66.10; H, 6.03; N, 3.35. Found: C, 66.26; H, 6.14; N, 3.37.O), 1583 (CC alkene); 1H NMR (DMSO-d6, 500 MHz) δ: 10.84 (br s, 1H, NH), 7.84 (d, 1H, H-5, J = 8.8 Hz), 7.69 (s, 1H, H-8), 7.55 (d, 2H, H-a, J = 7.5 Hz) 7.48 (d, 2H, H-b, J = 7.5 Hz), 6.77 (d, 1H, H-6, J = 8.8 Hz), 6.65 (s, 1H, vinylic-H), 5.39 (s, 2H, H-2), 4.51 (br s, 2H, H-1′), 3.47 (br s, 4H, H-a′), 2.98 (br s, 2H, H-2′), 1.78–1.37 (br m, 6H, H-c′ and H-b′). 13C NMR (125 MHz, DMSO-d6) δ: 179.6, 163.9, 162.6, 134.7, 134.3, 132.8, 132.1, 131.2, 129.2, 128.8, 115.5, 111.0, 101.8, 67.6, 62.9, 54.3, 52.5, 23.3, 21.2. Anal. calcd for C23H24ClNO3·HCl (434.36): C, 63.60; H, 5.80; N, 3.22. Found: C, 63.66; H, 6.09; N, 3.34.O), 1663 (CC alkene); 1H NMR (DMSO-d6, 400 MHz) δ: 11.51 (s, 1H, NH), 7.85 (d, 1H, H-5, J = 8.8 Hz), 7.68 (s, 1H, H-8), 7.44 (d, 2H, H-a, J = 8.4 Hz), 7.07 (d, 2H, H-b, J = 8.4 Hz), 6.77 (dd, 1H, H-6, J = 8.8 and 2 Hz), 6.65 (d, 1H, vinylic-H, J = 2 Hz), 5.43 (d, 2H, H-2, J = 2 Hz), 4.54 (br s, 2H, H-1′), 3.92 (br s, 4H, H-b′), 3.82 (s, 3H, OMe), 3.55 (br s, 2H, H-2′), 3.47 (br s, 2H, H-a′), 3.19 (br s, 2H, H-a′). 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 164.1, 162.8, 136.4, 132.2, 132.1, 129.6, 129.1, 128.9, 126.8, 116.2, 114.8, 111.2, 102.3, 68.3, 67.9, 63.6, 63.2, 55.9, 55.0, 52.1. Anal. calcd for C24H27ClNO4·HCl (431.91): C, 63.96; H, 6.07; N, 3.24. Found: C, 63.97; H, 6.12; N, 3.31.O), 1611 (CC alkene); 1H NMR (DMSO-d6, 500 MHz) δ: 11.06 (br s, 1H, NH), 7.84 (d, 1H, H-5, J = 8.6 Hz), 7.68 (s, 1H, H-8), 7.34 (d, 2H, H-a, J = 7.6 Hz), 7.30 (d, 2H, H-b, J = 7.6 Hz), 6.76 (d, 1H, H-6, J = 8.6 Hz), 6.64 (s, 1H, vinylic-H), 5.41 (s, 2H, H-2), 4.53 (br s, 2H, H-1′), 3.47 (br s, 2H, H-a′), 3.39 (br s, 2H, H- a′), 2.98 (t, 2H, H-2′, J = 10.40 Hz), 2.35 (s, 3H, CH3), 1.84–1.34 (br m, 6H, H-b′ and H-c′). 13C NMR (125 MHz, DMSO-d6) δ: 180.3, 164.2, 162.9, 140.1, 136.5, 131.5, 130.8, 130.3, 129.9, 129.6, 116.1, 111.3, 102.4, 68.2, 63.6, 63.2, 55.0, 52.1, 21.5. Anal. calcd for C24H27NO3·HCl (413.94): C, 69.64; H, 6.82; N, 3.38. Found: C, 66.63; H, 6.69; N, 3.34.O), 1605 (CC alkene); 1H NMR (300 MHz, DMSO-d6) δ: 10.80 (s, 1H, NH), 10.24 (s, 1H, –OH), 7.83 (d, 1H, H-5, J = 8.7 Hz), 7.62 (s, 1H, H-8), 7.32 (d, 2H, H-a, J = 7.5 Hz), 6.90 (d, 2H, H-b, J = 7.5 Hz), 6.75 (d, 1H, H-6, J = 8.7 Hz), 6.62 (s, 1H, vinylic-H), 5.41 (s, 2H, H-2), 4.50 (br s, 2H, H-1′), 3.44 (br s, 4H, H-a′), 2.97 (br s, 2H, H-2′), 1.78–1.36 (br m, 6H, H-b′, H-c′). 13C NMR (75 MHz, DMSO-d6) δ: 180.2, 164.0, 162.8, 159.7, 136.9, 136.8, 133.1, 129.6, 127.8, 125.2, 116.2, 116.2, 111.1, 102.2, 68.4, 63.2, 54.8, 53.0, 22.7, 21.6. Anal. calcd for C23H25NO4·HCl (415.91): C, 66.42; H, 6.30; N, 3.37. Found: C, 66.56; H, 6.34; N, 3.39.Conclusions
In summary, for the first time, we presented an efficient route to the synthesis of novel homoisoflavonoids as potentially interesting pharmacological active compounds via a cascade four steps reaction from 7-aminoalkoxychroman-4-ones and arylaldehydes. Medicinally and pharmacologically point of view, with respect to the importance of homoisoflavonoids, by this achievement, the scope of this class of compounds was developed. Also this protocol may be attracted the interest for further investigations and synthetic manipulations to the synthesis and development of medicinally important heterocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge from Pharmaceutics Research Center, Institute of Neuropharmacology, Kerman University of Medical Sciences for partial support of this work.Notes and references
- K. Du-Toit, S. E. Drewes and J. Bodenstein, Nat. Prod. Res., 2010, 24, 457 CrossRef CAS PubMed.
- K. Du-Toit, E. E. Elgorashi, S. F. Malan, D. A. Mulholland, S. E. Drewes and J. Van-Staden, S. Afr. J. Bot., 2007, 73, 236 CrossRef CAS.
- D. A. Mulholland, S. L. Schwikkard and N. R. Crouch, Nat. Prod. Rep., 2013, 30, 1165 RSC.
- B. M. Abegaz, J. Mutanyatta and M. Nindi, Nat. Prod. Commun., 2007, 2, 475 CAS.
- B. Lee, W. Sun, H. Lee, H. Basavarajappa, R. S. Sulaiman, K. Sishtla, X. Fei, T. W. Corson and S. Y. Seo, Bioorg. Med. Chem. Lett., 2016, 26, 4277 CrossRef CAS PubMed.
- T. Nakib, V. Bezjak, M. J. Meegan and R. Chandy, Eur. J. Med. Chem., 1990, 25, 455 CrossRef.
- B. S. Kirkiacharian, M. Gomis and P. Koutsourakis, Eur. J. Med. Chem., 1989, 24, 309 CrossRef.
- M. E. Wall, M. C. Wani, G. Manikumar, H. Taylor and R. McGivney, J. Nat. Prod., 1989, 52, 774 CrossRef CAS.
- A. Gupta and S. R. Chaphalkar, J. HerbMed Pharmacol., 2016, 5, 120 Search PubMed.
- N. Askarian-Dehkordi, H. Shirzad, L. Salimzadeh, B. Zamanzad, R. Meshkate and M. Kiyani, J. HerbMed Pharmacol., 2014, 3, 125 Search PubMed.
- N. Desideri, S. Olivieri, M. L. Stein, R. Sgro, N. Orsi and C. Conti, Antiviral Chem. Chemother., 1997, 8, 545 CrossRef CAS.
- K. K. Purushothaman, K. Kalyani, K. Subramaniam and S. P. Shanmughanathan, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1982, 21, 383 Search PubMed.
- D. D. McPherson, G. A. Cordell, D. D. Soejarto, J. M. Pezzuto and H. H. S. Fong, Phytochemistry, 1983, 22, 2835 CrossRef CAS.
- K. V. N. S. Srinivas, Y. K. Rao, I. Mahender and B. Das, Phytochemistry, 2003, 63, 789 CrossRef CAS PubMed.
- P. Zhao, Y. Iwamoto, I. Kouno, Y. Egami and H. Yamamoto, Phytochemistry, 2004, 65, 2455 CrossRef CAS PubMed.
- S. K. Roy, U. C. Agrahari, R. Gautam, A. Srivastava and S. M. Jachak, Nat. Prod. Res., 2012, 26, 690 CrossRef CAS PubMed.
- A. Foroumadi, A. Samzadeh-Kermani, S. Emami, G. Dehghan, M. Sorkhi, F. Arabsorkhi, M. R. Heidari, M. Abdollahi and A. Shafiee, Bioorg. Med. Chem. Lett., 2007, 17, 6764 CrossRef CAS PubMed.
- (a) K. Eskandari and M. Rafieian-Kopaei, Chem. Heterocycl. Compd., 2016, 52, 158 CrossRef CAS; (b) C. Li and F. Zhang, Tetrahedron Lett., 2017, 58, 1572 CrossRef CAS; (c) H. Nagarajaiah, A. Mukhopadhyay and J. Narasimha-Moorthy, Tetrahedron Lett., 2016, 57, 5135 CrossRef CAS.
- S. Malhotra, V. K. Sharma and V. S. Parmar, J. Chem. Res., 1988, 179 CAS.
- L. Farkas, A. Gottsegen and M. Nogradi, Tetrahedron, 1970, 26, 2787 CrossRef CAS.
- F. A. Davis and B. C. Chen, J. Org. Chem., 1993, 58, 1751 CrossRef CAS.
- A. C. Jain and P. Paliwal, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1989, 28, 416 Search PubMed.
- (a) K. Eskandari, B. Karami, M. Farahi and V. Mouzari, Tetrahedron Lett., 2016, 57, 487 CrossRef CAS; (b) Y. Pourshojaei, A. Gouranourimi, S. Hekmat, A. Asadipour, S. Rahmani-Nezhad, A. Moradi, H. Nadri, F. Homayouni-Moghadam, S. Emami and A. Foroumadi, Eur. J. Med. Chem., 2015, 97, 181 CrossRef CAS PubMed; (c) S. Rahmani-Nezhad, L. Khosravani, M. Saeedi, K. Divsalar, L. Firoozpour, Y. Pourshojaei, Y. Sarrafi, H. Nadri, A. Moradi, M. Mahdavi, A. Shafiee and A. Foroumadi, Synth. Commun., 2015, 45, 751 CrossRef; (d) S. Khodabakhshi, B. Karami, K. Eskandari and M. Farahi, Tetrahedron Lett., 2014, 55, 3753 CrossRef CAS; (e) L. Poorali, B. Karami, K. Eskandari and M. Azizi, J. Chem. Sci., 2013, 125, 591 CrossRef CAS.
- (a) K. Eskandari, B. Karami and S. Khodabakhshi, J. Chem. Res., 2014, 38, 600 CrossRef CAS; (b) K. Eskandari, B. Karami, S. Khodabakhshi and M. Farahi, Lett. Org. Chem., 2015, 12, 38 CrossRef CAS; (c) B. Karami, R. Ferdosian and K. Eskandari, J. Chem. Res., 2014, 38, 41 CrossRef CAS.
- (a) K. Eskandari and B. Karami, Monatsh. Chem., 2016, 147, 2119 CrossRef CAS; (b) B. Karami, K. Eskandari, Z. Zare and S. Gholipour, Chem. Heterocycl. Compd., 2014, 49, 1715 CrossRef CAS; (c) K. Eskandari and B. Karami, Comb. Chem. High Throughput Screening, 2016, 19, 728 CrossRef CAS.
- (a) F. Mehrabi, Y. Pourshojaei, A. Moradi, M. Sharifzadeh, L. Khosravani, R. Sabourian, S. Rahmani-Nezhad, M. Mohammadi-Khanaposhtani, M. Mahdavi, A. Asadipour, H. R. Rahimi, S. Moghimi and A. Foroumadi, Future Med. Chem., 2017, 9, 659 CrossRef CAS PubMed; (b) S. Rahmani-Nezhad, M. Safavi, M. Pordeli, S. Kabudanian, L. Khosravani, Y. Pourshojaei, M. Mahdavi, S. Emami, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2014, 86, 562 CrossRef CAS PubMed; (c) N. Azizi, F. Aryanasab and M. R. Saidi, Tetrahedron Lett., 2009, 50, 32 CrossRef; (d) M. R. Saidi, Y. Pourshojaei and F. Aryanasab, Synth. Commun., 2009, 39, 1109 CrossRef CAS.
- T. A. Nakibl, V. Bezjakl, M. J. Meeganz and R. Chandyl, Eur. J. Med. Chem., 1990, 25, 455 CrossRef.
- N. Desideri, A. Bolasco, R. Fioravanti, L. P. Monaco, F. Orallo, M. Yanez, F. Ortuso and S. Alcaro, J. Med. Chem., 2011, 54, 2155 CrossRef CAS PubMed.
- N. Desideri, S. Olivieri, M. L. Stein, R. Sgro, N. Orsin and C. Conri, Antiviral Chem. Chemother., 1997, 8, 545 CrossRef CAS.
- S. W. Yee, L. Jarno, M. S. Goma, C. Elford, L. L. Ooi, M. P. Coogan, R. McClelland, R. I. Nicholson, B. A. J. Evans, A. Brancale and C. Simons, J. Med. Chem., 2005, 48, 7123 CrossRef CAS PubMed.
- X. Zong, J. Cai, J. Chen, C. Sun, L. Lib and M. Ji, RSC Adv., 2015, 5, 24814 RSC.
- V. Siddaiah, M. Maheswara, C. V. Rao, S. Venkateswarlub and G. V. Subbaraju, Bioorg. Med. Chem. Lett., 2007, 17, 1288 CrossRef CAS PubMed.
- Y. Sun, J. Chen, X. Chen, L. Huang and X. Li, Bioorg. Med. Chem., 2013, 21, 7406 CrossRef CAS PubMed.
- Y. Jacquot, C. Byrne, A. Xicluna and G. Leclercq, Med. Chem. Res., 2013, 22, 681 CrossRef CAS.
- K. Koch and M. S. Biggers, J. Org. Chem., 1994, 59, 1216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08990b |
| This journal is © The Royal Society of Chemistry 2017 |