
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alternating and regioregular copolymers with high refractive index from COS and biomass-derived epoxides†
Lan-Fang Hu,
Yang Li,
Bin Liu,
Ying-Ying Zhang and
Xing-Hong Zhang *
*
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: xhzhang@zju.edu.cn
First published on 25th October 2017
Abstract
This study describes the catalytic formation of alternating and regioregular copolymers from carbonyl sulfide (COS) and epoxides along with eugenol-based glycidyl ether (EGE) and guaiacol-based glycidyl ether (GGE), derived from eugenol and guaiacol, respectively. The (salen)CrCl [salen = N,N′-bis(salicylidene) cyclohexanediimine] complex, accompanied with various organic bases, was highly active towards the EGE/COS, GGE/COS copolymerization and EGE/GGE/COS terpolymerization. The turnover of frequency (TOF) of the (salen)CrCl complex for the EGE/COS copolymerization was up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The number-average molecular weight (Mn) of the resultant EGE/COS copolymer was up to 62.2 kg mol−1. In the presence of 0.5–1.5 mol% chlorohydrin, which was a by-product of the synthetic process of EGE, α-Cl, ω-OH EGE/COS copolymers were obtained. This result suggests that chlorohydrin could act as a very efficient chain transfer agent for the copolymerization. The EGE/COS, GGE/COS, and EGE/GGE/COS copolymers were soluble in most of the common solvents and exhibited a high refractive index of more than 1.58 with high Abbe numbers of up to 40.4. This study provides an unprecedented and sustainable synthetic route for making soluble sulfur-rich polymers with high optical properties.
000 h−1. The number-average molecular weight (Mn) of the resultant EGE/COS copolymer was up to 62.2 kg mol−1. In the presence of 0.5–1.5 mol% chlorohydrin, which was a by-product of the synthetic process of EGE, α-Cl, ω-OH EGE/COS copolymers were obtained. This result suggests that chlorohydrin could act as a very efficient chain transfer agent for the copolymerization. The EGE/COS, GGE/COS, and EGE/GGE/COS copolymers were soluble in most of the common solvents and exhibited a high refractive index of more than 1.58 with high Abbe numbers of up to 40.4. This study provides an unprecedented and sustainable synthetic route for making soluble sulfur-rich polymers with high optical properties.
Introduction
Because of the limited petrochemical reserves and environmental issues, the use of biomass for synthesizing polymeric materials have received considerable attention in recent years.1 The resultant biomass-based polymers could be potentially used as elastomers,2 plastics,3 and resins,4 and in the composites5 that often have better biodegradability than most of the petroleum-based polymers. Among the most widely known biomass-derived chemicals, forestry products such as lignin,6,7 cellulose,8,9 and vegetable oils10,11 are of more interest due to their wide abundance and low-cost.12 It is well known that lignin is a major component of lignocellulosic biomass and accounts for 15–30% of the biomass. It is estimated that the entire terrestrial plants can synthesize 500 trillion tons of lignin per year; thus, a huge resource is available to be utilized. Moreover, lignin is a waste by-product in the paper and pulp industry.6 The direct chemical modification of the above biomass for producing materials has been widely studied.13,14 It is also a very promising route to prepare new materials by using the pure compounds from biomass as the building blocks. Herein, we have paid our attention to two lignin-derived monomers, guaiacol and eugenol (2-methoxy-4-(2-propenyl) phenol), for synthesizing polymers (Scheme 1). Guaiacol could be obtained from lignin via the abstraction, filtration, washing, and vacuum distillation process. Eugenol can be obtained either from the allylation of guaiacol or directly refined with high purity from natural essential oils, such as clove oils, nutmeg, and cinnamon.15,16 Up to date, the use of guaiacol and eugenol as the building blocks for synthesizing well-defined polymers is rarely reported.16,17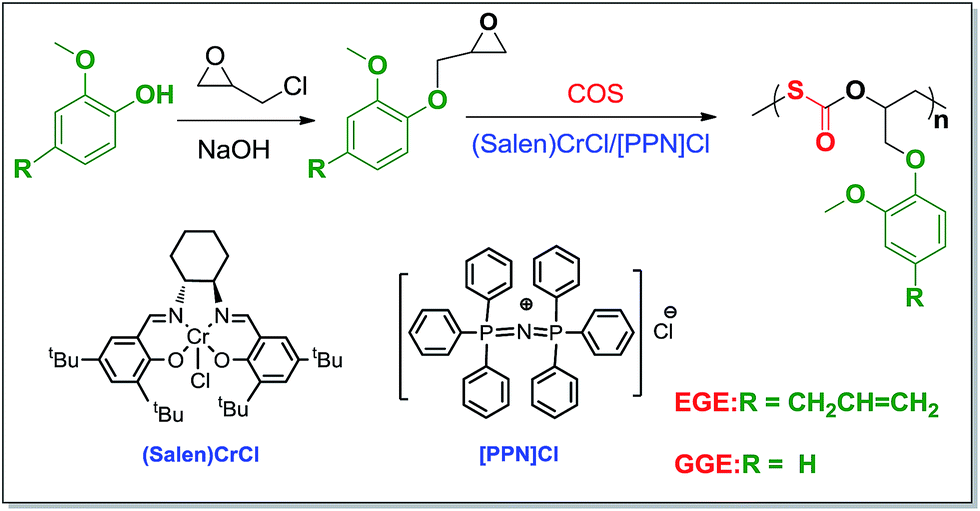 | ||
| Scheme 1 The synthetic route of COS and biomass-derived copolymers from guaiacol and eugenol by using the binary (salen)CrCl/[PPN]Cl system. Salen: N,N′-bis(salicylidene) cyclohexanediimine; EGE: eugenol-based glycidyl ether; GGE: guaiacol-based glycidyl ether. | ||
The abovementioned biomass-derived compounds, guaiacol and eugenol, could be converted to the epoxides by reacting them with epichlorohydrin (ECH), as shown in Scheme 1. On the other hand, recently, our group has discovered a sulfur-containing one-carbon (C1) building block, carbonyl sulfide (COS), which could copolymerize with various epoxides to afford poly(monothiocarbonate)s.18,19 Thus, we were inspired to synthesize sulfur-containing copolymers from COS with epoxides derived from guaiacol and eugenol. It would be a sustainable pathway for making polymeric materials because ECH can also be obtained from biomass20 and COS can be produced from the reaction of CO with sulfur that is exceptionally rich in nature and is the main by-product of the refinery.21 Concurrently, COS is an air pollutant that causes ozonosphere damage, acid rain, and the formation of sulfur aerosol that causes severe haze. Thus, it should be captured from the waste gas of the coal burning.19 Moreover, because the ring-opening polymerization of the cyclic monothiocarbonates and the condensation of thiols with phosgene are toxic processes,22 the copolymerization of COS with an epoxide is considered as a “greener” route for synthesizing poly(monothiocarbonate)s. In addition, the resultant poly(monothiocarbonate)s via this COS route are soluble in most of the common solvents and have a high refractive index (n) and tunable Abbe numbers (Vd) because of the introduction of carbon-sulfur bonds.22 These COS-based copolymers can thus be potentially used as the optical materials.
In the present study, we describe the first example of COS- and biomass-derived copolymers with a fully alternating degree and regioregular chain structure. These copolymers presented excellent optical properties. With unique features, α-Cl, ω-OH-terminated copolymers with relatively low molecular weights (MWs) were successfully prepared.
Results and discussion
Initially, EGE was synthesized with a yield of 84% by the reaction of eugenol with ECH (Scheme 1) according to the reported method.23 Although the synthesis of an epoxide using ECH is widely used and can be performed on a large scale, the uncompleted ring-closing reaction leads to the production of small amounts of chlorohydrin-1 in the final product as shown in Scheme 2. Purified by column chromatography and distillation for 1 time, EGE with ca. 1.5 mol% of chlorohydrin-1 was obtained, which was identified from the 1H NMR spectra of the EGE (Fig. 1) and the control chlorohydrin sample that was obtained from the direct reaction of EGE with hydrochloric acid (HCl) (Fig. S1†). White crystal EGE was obtained with ca. 0.5–1 mol% of chlorohydrin-1 by carrying out column separation twice, followed by distillation. The melting point of EGE was measured to be 38 °C by the capillary tube method. By the similar preparation process, GGE with 1.0–2.0 mol% of chlorohydrin-2 was obtained from the reaction of guaiacol with ECH (Fig. S2†). | ||
| Scheme 2 1-Chloro-3-(2-methoxy-4-prop-2-enylphenoxy) propan-2-ol (chlorohydrin-1) and 1-chloro-3-(2-methoxyphenoxy)-2-propyl alcohol (chlorohydrin-2) produced during the syntheses of EGE and GGE, respectively. | ||
 | ||
| Fig. 1 1H NMR spectrum of EGE with 0.5 mol% chlorohydrin-1. | ||
Because the purification process of both epoxides was time- and energy-consuming, EGE with 0.5 and 1.5 mol% of chlorohydrin-1 was directly employed in the copolymerization. The EGE/COS copolymer was successfully obtained by using the binary (salen)CrCl/[PPN]Cl catalyst in bulk at 40 °C. The results of the copolymerization under various conditions are listed in Table 1. EGE could be nearly completely converted to the copolymers with a monomer/catalyst ([EGE]/[Cr]) ratio of 1000:1 within 2.0 h; thus, the turnover frequency (TOF) was 500 h−1 as shown in entry 1 in Table 1. The resulting copolymer had a number-average molecular weight (Mn) of 18.3 kg mol−1 and a narrow polydispersity (PDI) of 1.27 obtained from the GPC result (Fig. 2). The GPC curves of these copolymers were bimodal, which was generally observed for the products of the CO2/epoxide copolymerization by using the same catalyst because the trace water in the system was involved in the chain transfer reaction, leading to the generation of new chains. Assuming that 1.5 mol% of chlorohydrin-1 participated in the chain transfer reaction totally, the calculated molecular weight of the resulting EGE/COS copolymer should be ca. 18.7 kg mol−1 based on the 100% EGE conversion, which was close to the value determined by the GPC method. This comparison suggested that chlorohydrin-1 was an efficient chain transfer agent for the EGE/COS copolymerization. The copolymer selectivity was >99%, revealed by the 1H NMR spectrum without any signals related to the cyclic monothiocarbonates. This selectivity was better than that of phenyl glycidyl ether (PGE)/COS copolymerization24 by using the same catalyst because more steric hindrance of EGE benefited the improvement of the copolymer selectivity.
| Entry | Cocatalyst | T (h) | Solv. | Conv.b (%) | TOFb (h−1) | Copolymer Selectivityb (%) | Mnc (kg mol−1) | Mw/Mnc |
|---|---|---|---|---|---|---|---|---|
| a The copolymerizations were performed in a 10 mL autoclave at 40 °C in bulk or 25 °C in solution (1.0 mL of solvent), [EGE]/[Cr]/[cocatalyst] = 1000:1:1, EGE 10.0 mmol; [chlorohydrin-1]/[EGE] = 1.5:100 (1:200 for entries 5 and 6); the alternating degree of all samples were >99%, determined by 1H NMR spectra of the purified copolymers.b Determined by 1H NMR spectrum of the crude product. TOF = turnover frequency of EGE to polymers, (mol EGE consumed)/(mol Cr h).c Determined by gel permeation chromatography in THF, calibrated with polystyrene standards.d [EGE]/[Cr] = 1500:1.e [EGE]/[Cr] = 2000:1.f 0 °C, EGE dissolved into COS immediately.g [EGE]/[Cr] = 100:1.h [EGE]/H2O = 50:1. The selected 1H NMR spectra of the substances in this Table can be seen in Fig. S7 and full 13C NMR spectrum of the copolymer (entry 1 in this Table) can be seen in Fig. S8. |
||||||||
| 1 | [PPN]Cl | 2.0 | — | >99 | 500 | >99 | 18.3 | 1.27 |
| 2 | [PPN]Cl | 0.5 | — | 94 | 1880 | >99 | 13.5 | 1.23 |
| 3 | [PPN]Cl | 0.2 | — | 93 | 4650 | >99 | 13.6 | 1.24 |
| 4 | [PPN]Cl | 0.1 | — | 89 | 8930 | >99 | 12.2 | 1.28 |
| 5d | [PPN]Cl | 0.1 | — | 80 | 12000 |
>99 | 62.2 | 1.65 |
| 6e | [PPN]Cl | 0.2 | — | 81 | 8100 | >99 | 49.1 | 1.55 |
| 7f | [PPN]Cl | 2.0 | — | 97 | 485 | >99 | 50.3 | 1.65 |
| 8 | [PPN]Cl | 2.0 | DCM | >99 | 500 | 98 | 15.8 | 1.31 |
| 9g | [PPN]Cl | 2.0 | DCM | >99 | 500 | 94 | 6.6 | 1.26 |
| 10h | [PPN]Cl | 2.0 | DCM | 32 | 160 | 71 | 2.5 | 1.28 |
| 11 | [PPN]Cl | 2.0 | THF | 93 | 465 | 71 | 9.9 | 1.24 |
| 12 | [PPN]Cl | 2.0 | Toluene | >99 | 500 | 98 | 16.8 | 1.18 |
| 13 | DBU | 0.1 | — | 74 | 7460 | >99 | 15.1 | 1.48 |
| 14 | DMAP | 0.5 | — | 29 | 580 | >99 | 16.2 | 1.24 |
| 15 | TBD | 0.1 | — | 11 | 1100 | >99 | 2.2 | 1.29 |
| 16 | DTMeAB | 0.5 | — | 50 | 1000 | >99 | 5.8 | 1.18 |
| 17 | TPPB | 0.1 | — | 13 | 1300 | >99 | 2.9 | 1.23 |
 | ||
| Fig. 2 Selected GPC curves of the purified EGE/COS copolymers in Table 1. | ||
Fig. 3 shows the 1H and 13C NMR spectra of the EGE/COS copolymer as shown in entry 1 in Table 1. No ether, carbonate and dithiocarbonate units were observed in 1H and 13C NMR spectra, which also confirmed the completely alternating structure as well as the complete depression of oxygen/sulfur exchange reaction (O/S ER) at 40 °C. Herein, the so-called O/S ER refers to the production of carbon dioxide and propylene sulfide that participated in the copolymerization mutually, resulting in the production of the mixed units in the chain.19,25 The peak a at 5.38 ppm and the peak b at 3.33 ppm (covered by the peak f), in Fig. 3A, were ascribed to the protons of methylene (CH2) and methine (CH) connecting with O and S atoms of the monothiocarbonate unit [–SC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O–], respectively.24 The integral area of the peaks a (OCH) and j (OCH3) were 1.00 and 3.00, respectively, which was in good agreement with the calculated value based on a fully alternating structure. The 13C NMR spectrum of the EGE/COS copolymer (Fig. 3B) suggested that the resulting copolymer had a tail-to-head (T–H) content of >99%, which was similar to that of the PGE/COS copolymer.24 Herein, the anionic sulfur of the growing chain predominantly attacked the less sterically crowded methylene carbon of EGE, resulting in a perfect regioselectivity.19 Due to two asymmetrical monomers used for the copolymerization, a special regio-isomer, i.e., a T–H diad, was formed through continuously attacking of propagating species to the methylene carbon of terminated epoxide, which has been well demonstrated in our previous study.18
O)O–], respectively.24 The integral area of the peaks a (OCH) and j (OCH3) were 1.00 and 3.00, respectively, which was in good agreement with the calculated value based on a fully alternating structure. The 13C NMR spectrum of the EGE/COS copolymer (Fig. 3B) suggested that the resulting copolymer had a tail-to-head (T–H) content of >99%, which was similar to that of the PGE/COS copolymer.24 Herein, the anionic sulfur of the growing chain predominantly attacked the less sterically crowded methylene carbon of EGE, resulting in a perfect regioselectivity.19 Due to two asymmetrical monomers used for the copolymerization, a special regio-isomer, i.e., a T–H diad, was formed through continuously attacking of propagating species to the methylene carbon of terminated epoxide, which has been well demonstrated in our previous study.18
 | ||
| Fig. 3 (A) 1H NMR and (B) 13C NMR spectra of the crude product (entry 1 in Table 1). | ||
The effect of the reaction time on the EGE/COS copolymerization by using the binary (salen)CrCl/[PPN]Cl catalyst were investigated and the results are listed in entries 2–6 in Table 1. The conversion of EGE was up to 89% within only 6 min at 40 °C, thus showing a high catalytic activity with a TOF of up to 8930 h−1. When EGE with 0.5 mol% of chlorohydrin-1 and a higher [EGE]/[Cr] molar ratio (1500:1) were employed under the same condition as entry 4 in Table 1, TOF was dramatically increased to 12000 h−1 (entry 5 in Table 1). Mn of the resultant copolymer was up to 62.2 kg mol−1, which was also close to the calculated value (ca. 56.0 kg mol−1) based on the assumption that chlorohydrin-1 was completely consumed. Further, on increasing the [EGE]/[Cr] molar ratio to 2000:1, the TOF decreased to 8100 h−1 because a relatively long reaction time (12 min) was used. Mn of the resulting copolymer was still high and up to 49.1 kg mol−1 (entry 6 in Table 1). Although 0.5 and 1.5 mol% chlorohydrin-1 were present in the reaction system, the binary (salen)CrCl/[PPN]Cl system still presented a remarkable high TOF as well as high MWs. In fact, the copolymerization of CO2 and biomass-based epoxides often led to the copolymers with low molecular weights due to the difficulty to remove such hydroxyl impurities in biomass-derived epoxides. In a recent study on biomass-based polycarbonate from limonene oxide and CO2, Greiner et al. successfully used sodium hydride and methyl iodide to completely eliminate the hydroxyl impurities for synthesizing high MW CO2 and biomass-derived polycarbonates (up to 109 kg mol−1).26 Herein, our system provided a sulfur-containing copolymer with high MW without further purification or other special operations for EGE.
The EGE/COS copolymerization was further investigated in bulk and in the presence of a solvent at low temperature (0 and 25 °C). The EGE/COS copolymerization at 0 °C in bulk afforded a copolymer with Mn of 50.3 kg mol−1, which was higher than that obtained at 25 °C in DCM (15.8 kg mol−1, entries 7 and 8 in Table 1). Low reaction temperature (0 °C) caused a lower TOF (486 h−1) compared with those at 25 and 40 °C. Furthermore, the copolymerization in DCM led to a slight loss of the copolymer selectivity (98%). Moreover, the decrease of the molar ratio of EGE to the catalyst (100:1) led to a decrease of the copolymer selectivity (94%, entry 9 in Table 1). When 2.0 mol% of water was introduced into the reaction system (entry 10 in Table 1), the copolymer selectivity and TOF decreased dramatically to 71% and 160 h−1, respectively. The resultant copolymer had a low Mn of 2.5 kg mol−1 with a single GPC curve due to the predominant occurrence of the chain transfer reaction caused by water. As compared with the copolymerization in DCM and toluene (entry 12, Table 1), the use of THF for the EGE/COS copolymerization led to a clear decrease of the copolymer selectivity (71%) at 25 °C. Thus, the solution copolymerization caused the production of the cyclic monothiocarbonate. It was proposed that the solvent may facilitate the backbiting route of the sulfur anion to produce cyclic monothiocarbonate.18
The type of the cocatalysts also had a strong influence on the EGE/COS copolymerization (entries 13–17 in Table 1). The binary (salen)CrCl/1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) system presented a highly catalytic activity (TOF: 7460 h−1) towards the copolymerization, while two relatively strong bases, 4-dimethylaminopyridine (DMAP) and 1,5,7-triazabicyclo [4.4.0]dec-5-ene (TBD), combined with the (salen)CrCl complex presented a low catalytic activity (580 and 1060 h−1, respectively), as shown in the results in entries 14 and 15 in Table 1. When the organic salts, dodecyltrimethylammonium bromide (DTMeAB) and tetraphenylphosphonium bromide (TPPB), were used as the cocatalysts, the catalytic activity of the (salen)CrCl complex were 1000 and 1260 h−1, respectively. However, these cocatalysts presented a high copolymer selectivity of more than 99%, revealed by their 1H NMR results.
As mentioned above, chlorohydrin acted as an efficient chain transfer agent for the copolymerization without significantly decreasing the catalytic activity of the catalyst system. To probe the efficiency of the chain transfer reaction of chlorohydrin-1, the EGE/COS copolymer with Mn of 6.6 kg mol−1 and PDI of 1.26 was characterized by MALDI-TOF-MS spectrometry (entry 9 in Table 1). In this instance, EGE with 1.5 mol% of chlorohydrin-1 was employed for the synthesis. One species of Cl-[EGE-COS]-EGE-OH + K+, i.e. a α-Cl, ω-OH copolymer, was observed as shown in Fig. 4A, indicating that the chain transfer reaction caused by chlorohydrin-1 was dominant. This result also confirmed that the resulting copolymer had a completely alternating structure. The observation of the sole monothiocarbonate unit meant that no O/S ER occurred during the copolymerization. I should be noted that in the previously reported CO2/epoxide copolymerization systems,27–29 the chain transfer reaction caused by the trace amounts of water (or the dihydroxyls from epoxide hydrolysis30) often produced the mixed products of a α-X, ω-OH copolymer (X: initiating anions) and a α-OH, ω-OH copolymer. Herein, the existence of chlorohydrin-1 in EGE dramatically depressed the chain transfer reaction of the propagating species caused by the trace amounts of water. However, when considerable amounts of exogenous water (2.0 mol%) were purposely introduced into the copolymerization system (1.5 mol% of chlorohydrin-1, entry 10 in Table 1), the resulting copolymer presented one main species of the α-OH, ω-OH copolymer with one dithiocarbonate unit as shown in Fig. 4B. The presence of such dithiocarbonate unit was also confirmed by FT-IR spectra of the copolymer (Fig. S3†), indicating the occurrence of O/S ER reaction to a slight extent.18 However, the α-Cl, ω-OH copolymer could be minimized as shown in Fig. 4B.
 | ||
| Fig. 4 MALDI-TOF mass spectra of the purified EGE/COS copolymers of entry 9 (A) and entry 10 (B) in Table 1. | ||
For comparison purpose, the copolymerization of EGE containing 2.0 mol% of chlorohydrin-1 with COS was performed either in bulk or in the presence of 2.0 mol% exogenous water in toluene (Table S1 and Fig. S6†). The α-Cl, ω-OH copolymers were mainly produced (Fig. S4†). This result meant that the chain transfer reaction caused by water could be dramatically inhibited when an equivalent chlorohydrin-1 existed in the reaction system. No dithiocarbonate unit was observed in the FT-IR spectra (Fig. S5†), indicating no occurrence of O/S ER reaction.18 Therefore, using the epoxide-derived chlorohydrin as the chain transfer agent for synthesizing the α-Cl, ω-OH copolymer is an effective method even with the presence of some amount of water in the copolymerization system, which makes its large-scale production process easier.
As an analogue of EGE, GGE was also successfully copolymerized with COS by using the same catalyst system. The results of the solution copolymerization of GGE/COS are briefly summarized in Table S2† and the 1H NMR spectra of the copolymers could be seen in Fig. S9.† The results showed that GGE could be completely consumed in a wide temperature range of 20–60 °C within 2 h. The copolymer selectivity was 95–98% and Mns of the resultant copolymers were up to 22.3 kg mol−1 with relatively narrow PDIs of 1.2–1.4. The bulk GGE/COS copolymerization resulted in the production of ca. 2.0% polyether at 40 °C (entry 2 in Table 2) because GGE had a smaller steric hindrance than that of EGE. The GGE/COS copolymer had a tail-to-head (T–H) content of >99% (Fig. S13B†). Based on the above results of the EGE/COS and GGE/COS copolymerization, the terpolymerization of EGE, GGE, and COS was performed and the results are listed in Table 2. The 1H and 13C NMR spectra of the polymers could be seen in Fig. S10–13.† TOFs of the terpolymerization with various EGE/GGE molar ratios (4:1, 1:1, and 1:4) were slightly lower than that of the EGE/COS copolymerization, which could be possibly due to the higher content of chlorohydrin-2 in GGE (2.0 mol%). The molar ratios of two units of [EGE-COS]/[GGE-COS] in the resulting copolymers were 85:15, 54:46, and 21:79 that were bigger than the corresponding initial feeding ratios of EGE/GGE of 4:1, 1:1, and 1:4, respectively (Fig. S10†). Clearly, EGE was more active toward the copolymerization than GGE.
| Entry | [EGE]/[GGE] | Conv.b (%) | TOF (h−1) | Copolymer Selectivityb (%) | [EGE-COS]/[GGE-COS] | Mnc (kg mol−1) | Mw/Mnc | Tgd (°C) | Td,5wt%e (°C) | ndf | Vdf |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a The reactions were performed in a 10 mL autoclave at 40 °C for 2 h. (GGE + EGE) 10 mmol, 0.5 mol% of chlorohydrin-1 in EGE, 2.0 mol% of chlorohydrin-2 in GGE, [GGE + EGE]/[Cr]/[PPN]Cl = 1000:1:1.b Determined by 1H NMR spectroscopy. TOF = turnover frequency of epoxides to products, (mol GGE and EGE consumed)/(mol Cr h).c Determined by gel permeation chromatography in THF, calibrated with polystyrene standards.d Determined by differential scanning calorimetry (DSC).e Decomposition temperature (Td,5wt%) determined by thermogravimetric analysis (TGA) at 5 wt% weight loss of the polymer.f The refractive index, Vd is the Abbe number, determined by spectroscopic ellipsometer, Vd was calculated by (nd−1)/(nF−nC), where nC, nd, and nF are the refractive indices of the polymer film at 656.3, 587.6, and 486.1 nm, respectively. |
|||||||||||
| 1 | 1:0 |
>99 | 500 | >99 | 1:0 |
51.9 | 1.54 | 22.6 | 252.1 | 1.588 | 32.5 |
| 2 | 4:1 |
94 | 470 | >99 | 85:15 |
27.6 | 1.35 | 17.0 | 195.8 | 1.589 | 34.5 |
| 3 | 1:1 |
93 | 465 | >99 | 54:46 |
25.4 | 1.28 | 18.3 | 169.7 | 1.591 | 36.0 |
| 4 | 1:4 |
78 | 390 | >99 | 21:79 |
13.6 | 1.22 | 16.2 | 165.0 | 1.592 | 37.7 |
| 5 | 0:1 |
82 | 410 | 98 | 0:1 |
15.8 | 1.36 | 25.7 | 152.6 | 1.605 | 40.4 |
The thermal properties of the above-synthesized polymers were tested (Fig. S14–15†). Glass transition temperature (Tg) of the EGE/COS copolymer was 22.6 °C and lower than that of the GGE/COS copolymer (25.7 °C) because the EGE/COS copolymer had a residual allyl group that could plasticize the copolymer. The initial thermal decomposition temperature (Td,5wt%) of the GGE/COS copolymer was 152.6 °C, which was lower than that of the EGE/COS copolymer (252.1 °C). The large steric effect of the EGE/COS copolymer may inhibit the backbiting process for the decomposition. Td,5wt%s of the terpolymers were in the range of 165.0–195.8 °C and increased with the increase in the EGE/GGE molar ratios. Specifically, Tgs of the terpolymers were slightly increased with the increase in the EGE/GGE molar ratios but lower than those of the EGE/COS copolymer and the GGE/COS copolymer, suggesting a random chain structure of the terpolymers.
The obtained EGE(GGE)/COS copolymers were transparent and highly soluble in some common organic solvents, such as toluene, DCM, chloroform, and THF. These copolymers were fabricated to obtain a thin film in a spin coater. The refractive index (nd, n value at the wavelength of 587.6 nm) of the EGE/COS copolymer and the GGE/COS copolymer (entries 1 and 5, Table 2) was measured to be 1.588 and 1.605, respectively by spectroscopic ellipsometer (Fig. 5) and higher than those of the COS/PO copolymer (1.524), the COS/CHO copolymer (1.548)22 and very close to that of the COS/PGE copolymer (1.602). Herein, the aryl group in the copolymer contributed much to the optical properties, while the allyl group in the EGE/COS copolymer weakened the nd value. In comparison, nds of the commercially available plastics such as polystyrene (PS) and aromatic polycarbonate (PC) are 1.59 and 1.58, respectively. The Vd values of PS and PC are 31 and 30, respectively, which are the minimum Vd values required for making visual optical materials (generally, 30–60). Herein, the EGE/COS and GGE/COS copolymers had higher Vds (32.5 and 40.4, respectively) than those of PS and PC, indicating that both copolymers can have potential applications in visual optical materials. Interestingly, nd values of the terpolymers were in the range of 1.589–1.592 and close to that of the EGE/COS copolymer (1.588), while Vd values were from 34.5 to 37.7 with the increase in the GGE content in the copolymer. This could be attributed to the change of the polarizability of the C–S bond in each repeated unit due to the random terpolymerization.22 In particular, only a slight variation of nds of all copolymers, listed in Table 2, was observed in the wavelength range of 600–1400 nm, which was beneficial to their applications. These low thermal-stable and high optical-active copolymers are promising scarifying polymeric materials in the case of reusable devices.
 | ||
| Fig. 5 The refractive indices of the EGE/COS copolymer, GGE/COS copolymer, and EGE/GGE/COS terpolymers determined by spectroscopic ellipsometer. | ||
Conclusions
In summary, we have successfully prepared biomass- and COS-derived copolymers through the copolymerization of two biomass-derived epoxides with COS by using the binary (salen)CrCl/[PPN]Cl catalyst. The copolymerization exhibited high TOF of up to 12000 h−1 at 40 °C and the products have a perfectly alternating degree of 100%, regio-selectivity with a T–H content >99%, and high Mn of up to 62.2 kg mol−1. Specifically, it is a powerful technique to exclusively synthesize the α-Cl, ω-OH poly(monothiocarbonate)s with low Mns by directly using EGE(GGE) monomers with ca.1.5–2.0 mol% of chlorohydrin without the loss of activity and selectivity. Such α-Cl, ω-OH copolymers could be potentially used to synthesize a diblock copolymer via the previously reported methods.31 Moreover, the EGE/COS copolymer and EGE/GGE/COS terpolymers have residual double bonds, which could be used for further functionalization. The resulting copolymers exhibited high refractive index (n) of more than 1.58 and suitable Vd (32.5–40.4), representing a new family of high refractive index polymers.
Experimental
Materials and methods
All water-sensitive syntheses and manipulations were carried out in a double-manifold Schlenk vacuum line under an argon atmosphere or a nitrogen-filled glovebox. Following purification, the materials were stored in a nitrogen-filled glovebox prior to use unless otherwise specified. Carbon disulfide (99.9%, ACS Grade) was purchased from the APK (Shanghai) Gas Company LTD. and used directly. Guaiacol, eugenol, (R,R)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanedi-aminochromium(III) chloride ((salen)CrCl), bis(triphenyl-phosphoranylidene) ammonium chloride ([PPN]Cl), tetraphenyl-phosphonium bromide (TPPB), dodecyltrimethylammonium bromide (DTMeAB), 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and 1,5,7-triazabicyclo [4.4.0]dec-5-ene (TBD) were purchased from Aladdin Reagent Company (Shanghai) and used as received. Dichloromethane (DCM) and toluene were purchased from Aladdin Reagent Company (Shanghai) and distilled over CaH2. Tetrahydrofuran (THF) was distilled from sodium/benzophenone mixture before used.Characterization
1H and 13C nuclear magnetic resonance (NMR) spectra of the polymers were recorded on a Bruker Advance DMX 400 MHz spectrometer. Chemical shift values were referenced to TMS as an internal standard at 0.0 ppm for 1H NMR (400 MHz) and against CDCl3 at 77.16 ppm for 13C NMR (101 MHz). Molecular weights and molecular weight distributions of the polymers were determined with a PL-GPC220 chromatograph (Polymer Laboratories Ltd.) equipped with an HP 1100 pump from Agilent Technologies. The GPC columns were eluted with THF at 1.0 mL min−1 at 40 °C. The sample concentration was 0.5 wt% and the injection volume was 50 μL. The calibration was performed using monodisperse polystyrene standards covering the molecular weight range from 580 to 460000 Da. Infrared spectra were measured by using a Brucker Vector 22 FT-IR spectrophotometer. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectra were collected on a Bruker UltraFLEX MALDI-TOF mass spectrometer in the linear mode. Potassium trifluoroacetate was used as the cationization agent. Dithranol (DIT) was used as a matrix. Glass transition temperature (Tg) of the polymer was determined by differential scanning calorimetry (DSC) on a TA DSC-Q200 instrument. The sample was heated in two cycles from room temperature to 120 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere. Tg was determined from the second run. Thermogravimetric analysis (TGA) was carried out on a Perkin-Elmer Pyris 1 instrument under the N2 atmosphere at a heating rate of 10 °C min−1 from room temperature to 600 °C. The refractive index (n) was measured by Spectroscopic Ellipsometer.
Synthesis of EGE and GGE
EGE was synthesized according to the previously reported method.32 Eugenol (0.1 mol) and ECH (200 mL) were stirred in a round-bottomed flask at 120 °C. NaOH aqueous solution (0.23 mol) was added dropwise over a period of 3.0 h. The reaction was carried out for another 2.5 h. Following this, the mixture was filtered to remove salt and concentrated with a rotary evaporator to remove the excess of ECH and water. The crude product went through the silica column eluted with DCM. The white solid obtained was distilled over CaH2 under reduced pressure. Yield: 84%. 1H NMR (400 MHz, CDCl3) δ 6.86 (d, J = 8.0 Hz, 1H), 6.70 (d, J = 9.7 Hz, 2H), 5.95 (dd, J = 16.9, 10.1 Hz, 1H), 5.15–4.99 (m, 2H), 4.21 (dd, J = 11.4, 3.5 Hz, 1H), 4.02 (dd, J = 11.4, 5.5 Hz, 1H), 3.85 (s, 3H), 3.42–3.35 (m, 1H), 3.33 (d, J = 6.7 Hz, 2H), 2.91–2.84 (m, 1H), 2.72 (dd, J = 4.9, 2.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 149.53 (s), 146.29 (s), 137.53 (s), 133.83 (s), 120.47 (s), 115.70 (s), 114.35 (s), 112.38 (s), 70.43 (s), 55.84 (s), 50.24 (s), 44.93 (s), 39.80 (s).GGE was synthesized from guaiacol via the similar method. Yield: 82%. 1H NMR (400 MHz, CDCl3) δ 7.01–6.85 (m, 4H), 4.24 (dd, J = 11.4, 3.6 Hz, 1H), 4.05 (dd, J = 11.4, 5.5 Hz, 1H), 3.87 (s, 3H), 3.44–3.35 (m, 1H), 2.93–2.86 (m, 1H), 2.74 (dd, J = 4.9, 2.7 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 149.65 (s), 148.00 (s), 121.96 (s), 120.96 (s), 114.24 (s), 111.95 (s), 70.23 (s), 55.88 (s), 50.24 (s), 44.98 (s).
The copolymerization of COS and EGE (GGE)
All reactions were performed in the glovebox using a 10 mL or 30 mL stainless-steel autoclave equipped with a magnetic stirrer and a barometer. In a typical experiment, the autoclave was first dried in an oven for 24 h and transferred into the glovebox. The (salen)CrCl, cocatalyst, and EGE were added into the autoclave successively. The autoclave was pressurized to the appropriate pressure with COS and the reaction mixture was stirred at 0–40 °C for 0.1–2.0 h. After the copolymerization, the autoclave was cooled down. An aliquot portion was then taken from the crude product for the determination of the molar ratio of different linkages and species by 1H NMR. Following this, the crude product was dissolved in dichloromethane (2 mL) and precipitated from methanol (20 mL) three times. The yellow precipitate was collected and dried under vacuum at 40 °C for overnight.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the Distinguished Young Investigator Fund of Zhejiang Province (LR16B040001) and the National Science Foundation of the People's Republic of China (no. 21474083).Notes and references
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- M. S. Ganewatta, W. Ding, M. A. Rahman, L. Yuan, Z. Wang, N. Hamidi, M. L. Robertson and C. Tang, Macromolecules, 2016, 49, 7155–7164 CrossRef CAS.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS.
- M. P. Elizabeth Grillo Fernandes and E. Chiellini, Biomacromolecules, 2004, 5, 1200–1205 CrossRef PubMed.
- S. Zhou, Y. Xue, A. Sharma and X. Bai, ACS Sustainable Chem. Eng., 2016, 4, 6608–6617 CrossRef CAS.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- D. Shen, R. Xiao, S. Gu and K. Luo, RSC Adv., 2011, 1, 1641–1660 RSC.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- U. Biermann, U. Bornscheuer, M. A. Meier, J. O. Metzger and H. J. Schafer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- A. M. Crump, M. A. Sefton and K. L. Wilkinson, Food Chem., 2014, 162, 261–263 CrossRef CAS PubMed.
- A. C. A. G. Milczarek, Anal. Chem., 1999, 71, 1055–1061 CrossRef PubMed.
- J. Deng, B. Yang, C. Chen and J. Liang, ACS Sustainable Chem. Eng., 2015, 3, 599–605 CrossRef CAS.
- M. Luo, X.-H. Zhang, B.-Y. Du, Q. Wang and Z.-Q. Fan, Macromolecules, 2013, 46, 5899–5904 CrossRef CAS.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- K. Karan, A. K. Mehrotra and L. A. Behie, Ind. Eng. Chem. Res., 1998, 37, 4609–4616 CrossRef CAS.
- M. Luo, X. H. Zhang, B. Y. Du, Q. Wang and Z. Q. Fan, Polym. Chem., 2015, 6, 4978–4983 RSC.
- E. Zago, E. Dubreucq, J. Lecomte, P. Villeneuve, F. Fine, H. Fulcrand and C. Aouf, New J. Chem., 2016, 40, 7701–7710 RSC.
- M. Luo, X. H. Zhang and D. J. Darensbourg, Polym. Chem., 2015, 6, 6955–6958 RSC.
- X.-H. Zhang, F. Liu, X.-K. Sun, S. Chen, B.-Y. Du, G.-R. Qi and K. M. Wan, Macromolecules, 2008, 41, 1587–1590 CrossRef CAS.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- X.-H. Zhang, R.-J. Wei, X.-K. Sun, J.-F. Zhang, B.-Y. Du, Z.-Q. Fan and G.-R. Qi, Polymer, 2011, 52, 5494–5502 CrossRef CAS.
- X.-K. Sun, X.-H. Zhang, R.-J. Wei, B.-Y. Du, Q. Wang, Z.-Q. Fan and G.-R. Qi, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2924–2934 CrossRef CAS.
- M. Luo, Y. Li, Y.-Y. Zhang and X.-H. Zhang, Polymer, 2016, 82, 406–431 CrossRef CAS.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- A. Kumar and R. A. Gross, Macromolecules, 2002, 35, 7606–7611 CrossRef CAS.
- L. R. Madivada, R. R. Anumala, G. Gilla, M. Kagga and R. Bandichhor, Org. Process Res. Dev., 2012, 16, 1660–1664 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08958a |
| This journal is © The Royal Society of Chemistry 2017 |