
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Membrane optode for uranium(VI) preconcentration and colorimetric determination in real samples
Nader Hassana and
Alaa S. Amin *b
*b
aChemistry Department, Faculty of Science, Port Said University, Port Said, Egypt
bChemistry Department, Faculty of Science, Benha University, Benha, Egypt. E-mail: asamin2005@hotmail.com; Fax: +20 132222578; Tel: +20 552350996
First published on 2nd October 2017
Abstract
A membrane optode formed by physical inclusion of a chromophore, 2-(2-benzothiazolylazo)phenol (BTAP), and uranium(VI) anions into a plasticized cellulose triacetate (CTA) matrix was illustrated for preconcentration and colorimetric determination of U(VI) from aqueous samples. The optode was examined for uranium uptake in bicarbonate/carbonate medium (∼160 ng mL−1) at pH 6.5–7.0 in the presence of triethanolamine buffer (TEA). The inclusion of an anion-exchanger, dinonylnapthenic sulphonic acid (DNNS), was found to be necessary for the formation of a U(VI)–BTAP complex in the optode matrix, resulting in a distinct color change of the optode from orange to pink after uranium sorption. The composition of the optode was optimized by varying the amounts of different plasticizers and DNNS to obtain the minimum response time towards U(VI) in aqueous bicarbonate medium. The effects of different experimental parameters involved in the detection and quantification of U(VI) using the optode were optimized. The optode analytical performance was evaluated in terms of interference of cations and anions, reusability, equilibration time, detection limit, etc. and compared with the conventional spectrophotometric method for U(VI) using different chromophores. The optode developed in the present study was tested in real samples with good results compared to the graphite furnace atomic absorption spectroscopy (GF AAS) method.
Introduction
Natural radionuclides are released into surface and ground water from rocks and ores by dissolution and desorption during radioactive decay. Heavy metals and radionuclide contamination present a significant environmental problem1,2 as they are non-degradable and thus persistent.3–5 Release of heavy metals and radionuclides to the subsurface has been due to both natural and anthropogenic activities.4,6,7 Some chemolithotrophic and heterotrophic microorganisms are able to leach uranium and other radioactive elements from minerals in both acidic and alkaline water solutions.6 Uranium (U) is an important radionuclide contaminant in ground water, soils and subsurface sediments at nuclear weapons manufacturing and uranium mining sites, due to processing of uranium ore, mining, milling and tailing operations.8,9 The World Health Organization has reported that uranium is a carcinogen to humans and its concentration in aqueous solutions should not exceed 50 μg L−1.10The determination of trace amounts of uranium in environmental sites and in facilities of the nuclear industry is important for safety considerations. Atomic power plants continuously require uranium resources; therefore, 4.5 billion tons of total uranium in seawater can be recovered for atomic power utilization.11 Uranium is dissolved in seawater at a concentration of about 3.0 mg L−1 in the ionic form of uranyl tricarbonate ions.11 The World Health Organization (WHO), Health Canada and Australian drinking water guidelines have fixed the maximum uranium concentration in drinking water to be less than 9, 20 and 20 mg L−1, respectively.12,13 On the other hand, uranium and its compounds, like lead, are highly toxic, resulting in progressive or irreversible renal injury, and in acute cases may lead to kidney failure and death. The tolerable daily intake of uranium established by WHO based on Gilman's studies is 0.6 mg kg−1 of body weight per day.12–15
Thus, determination of uranium in environmental samples is very important. The methods including inductively coupled plasma atomic emission spectrometry (ICPAES),16 inductively coupled plasma-mass spectrometry (ICP-MS),17,18 ion chromatography (IC)19 and capillary zone electrophoresis (CZE),20 graphite furnace atomic absorption spectrometry (GF AAS),21 fluorimetry,22 spectrophotometry,23 neutron activation analysis24 and voltammetry25 have been used for determination of uranium. However, these methods require expensive equipments and higher running cost and their availability is limited. Alpha spectrometry has also been used for determination of uranium but it needs a number of preliminary separation steps for sample preparation.26
However, there are still difficulties due to increasing uranium in natural waters. Hence, the development of simple, inexpensive, reliable and practical methods for the determination of uranyl ions in water samples is of paramount importance. Neutron activation analysis (NAA),27 energy dispersive X-ray fluorescence (ED-XRF)28 and radiochemical methods, such as alpha spectrometry,29 gamma spectrometry30 and liquid scintillation counting,31 are suitable for the sensitive determination of uranyl ions. However, these methods are expensive, available mostly in equipped and specialized laboratories.32
Spectrophotometric methods, which can provide sufficient sensitivity, are cost-effective, common in laboratories and are the easiest techniques for the determination of uranyl ions.33,34 Several organic and inorganic reagents have been used for the spectrophotometric determination of uranyl ions. Among them, dibenzoylmethane (DBM) and Arsenazo III (AIII) have the ability to selectively measure very low concentrations of uranyl ions.35,36 However, AIII is the most sensitive reagent for the spectrophotometric determination of uranyl ions.37,38
Optical chemical sensors have drawn much attention in analytical chemistry, because of some advantages such as good sensitivity, selectivity, easy fabrication and low cost.39–44 Different strategies have been used to develop optical sensors for some target ions. Optical or visual test strips allow the simple detection of analyte by the naked eye, or by a portable spectrophotometer without any specific pretreatment.45
Membrane-based preconcentration method can be tailor-made for a specific analytical application. The analyte selective membrane can be converted to optical chemical sensor (optode) by immobilizing the indicator that responds to analyte in a concentration dependent manner.46–48 The membrane optodes have been prepared by immobilizing the indicator (ionophores, chromoionophores, and fluoroionophores) with or without extractant in the solid matrix using highly diversified methods.49–52 These optodes are based on the preconcentration of the analyte as a chromogenic species on a solid substrate and subsequent measurement of the absorbance/reflectance of the solid phase, without stripping the chromogenic species. The presence of extractant provides an increase in selectivity and sensitivity of optodes as compared to the corresponding solution spectrophotometry using same chromophore. The applicability of a fiber-optic-based laser-induced luminescence sensor for uranium (10−9–10−4 mol L−1) estimation in different samples has been studied.53 Collins et al. have developed a remote optical fiber flow-cell with Nafion-117 membrane and capillary electrophoresis method, both using Arsenazo III as an indicator, for sensing U(VI) in the ground water.54,55 However, these designs have not yet proven as a viable method for field applications due to complicated hardware and lower selectivity. The color changeable optode for uranium and thorium have been reported using non-specific Alizarin Red S56 and 4-(p-nitrophenylazo)-pyroctachol (NAP),57 respectively. The applications of these optodes require a careful evaluation of possible interferences in actual applications. Hexyphyrin (1.0.1.0.0.0) (isometyrin) has been found to give significant color change in the presence of UO22+, PuO22+, and NpO22+, which can be used for developing actinide sensor.58 However, isometyrin requires a week for complexation with actinides in dark.
In the present work, a colorimetric method for simultaneous preconcentration and determination of U(VI) anions from aqueous samples has been developed using a membrane optode formed by physical inclusion of a chromophore 2-(2-benzothiazolylazo)phenol (BTAP) into a plasticized cellulose triacetate matrix. The choice for BTAP was based on its high sensitivity and selectivity for uranium estimation by spectrophotometry.59 Two different plasticizers such as tris(2-ethylhexyl)phosphate (TEHP), and 2-nitrophenyloctyl ether (NPOE) have been used to plasticize cellulose triacetate. The dinonylnaphthalene sulphonic acid (DNNS), tri-iso-octyl amine (TiOA), and tricaprylylmethyl ammonium chloride (Aliquat-336) have been evaluated as the carriers for facilitating transfer of U(VI) from aqueous to the optode matrix. The carrier in the optode is necessary as immobilized BTAP does not interact with U(VI) present in the aqueous medium. The experimental parameters such as composition of optode, pH, interference of cations and anions on the determination of uranium, range of uranium concentration, response time, and detection limit have been studied and compared with the conventional spectrophotometry. Finally, the optode has been tested for quantitative determination of U(VI) in real sea water, soil and sediment samples.
Experimental
Reagents and apparatus
Analytical reagent grade chemicals and doubly distilled water were used throughout. 2-Nitrophenyloctyl ether, tricaprylylmethyl ammonium chloride (Aliquat-336), dinonylnapthenic sulphonic acid (DNNS), tri-isooctylamine, (TiOA), triethanolamine (TEA), and cellulose triacetate were obtained from Sigma-Aldrich (Steinheim, Switzerland). Tris(2-ethylhexyl)phosphate was obtained from Koch-Light Laboratories (Coinbrook Bucks, England). Uranyl nitrate, chloroform, and dichloromethane were obtained from Merck. A stock solution of uranium (10−3 mol L−1) was prepared by dissolving suitable quantities of UO2(NO3)2·6H2O in distilled water and the concentration of uranium was determined by Davies and Gray method.25 Working standard stock solutions of uranium were prepared by suitable dilution in the desired medium. Triethanolamine buffer was prepared by dissolving in distilled water and neutralizing with perchloric acid (pH 6.8).2-(2-Benzothiazolylazo)phenol (BTAP) used in this studies was prepared according to the procedure described previously (Fig. 1).59 An appropriate weight was dissolved in 100 mL of absolute ethanol (2 × 10−4 mol L−1). The solution was stable for more than one month.
 | ||
| Fig. 1 2-(2-Benzothiazolylazo)phenol structure. | ||
A microprocessor-based pH meter model Orion research model 601 A/digital ionalyzer was used for pH measurements. The thickness of the optode was measured by a digital micrometer (Mitutoy, Japan) with an accuracy of ±0.001 mm. UV-vis spectrophotometer model V 53 from JASCO (Tokyo, Japan) was used for recording the spectra and the absorbance measurements. The absorbance measurements were done by mounting the optode samples (3 cm × 1 cm) inside a quartz cuvette. The absorbance measurements of the optode samples were carried out with respect to air as well as blank optode sample.
Preparation of membrane optode
The membrane optodes were prepared by using the chromophore BTAP, extractant (DNNS/TiOA/Aliquat-336), plasticizer (TEHP/NPOE), and matrix forming polymer (CTA). Desired quantities of CTA, extractant, BTAP and plasticizer were dissolved separately in CHCl3. After complete dissolution, the casting solution was prepared by mixing the required volumes of the solutions containing known amounts of the components. The resulting casting solution was homogenized by ultrasonication for 3.0–5.0 min. The casting solution was poured in a Petri dish to allow the slow evaporation of CHCl3 and the formation of the homogeneous transparent membrane.Uptake experiments
The optode strips (3 cm × 1 cm) were equilibrated in sample solutions of desired medium (∼10−4 mol L−1 bicarbonate/carbonate solution in TEA buffer) and agitated with magnetic stirring bar at a rate of 500 rpm. TEA buffer (3.0 mL) was added in 10 mL (sample volume) to maintain a pH of 6.8 ± 0.2 unless stated otherwise. It should be noted that uranium precipitation was observed in the absence of bicarbonate medium. The membrane strips were taken out of the sample solutions and washed with a jet of distilled water. The color of optode strips changed from orange to pink depending on the uranium concentration in the sample solution. The change in absorbance of U(VI)–BTAP complex was monitored at λmax = 607 nm under different experimental conditions.Results and discussion
Composition of optode
Various combinations of the matrix forming polymer, plasticizer (TEHP/NPOE), chromophore BTAP, and the carrier (DNNS/TiOA/Aliquat-336) were studied to optimize the uranium uptake in the optode matrix from aqueous samples having pH range of 6.5–7.0. It is seen that BTAP immobilized in the optode could not form complex with U(VI) in absence of a carrier in the optode matrix. This seems to suggest that BTAP itself does not act as a carrier to facilitate the transfer of U(VI) from equilibrating solution to the optode matrix. Among the different carriers studied, DNNS was found to be efficient for facilitating the transfer of U(VI) from equilibrating solution to optode matrix. This may be attributed to the fact that the presence of bicarbonate/carbonate anions in the equilibrating solution helps in the formation of anionic uranium carbonate complex species. This anionic form of U(VI) could interact with the liquid anion exchanger DNNS present in the optode matrix to form an ion-pair. In addition, the DNNS may also be responsible to prevent leaching of U(VI)–BTAP anionic complex formed in the optode matrix.Spectral characteristics
BTAP is one of the most sensitive reagents for the spectrophotometric analysis of uranium present in various aqueous/organic phase samples. It forms 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stable complex with U(VI) in pH range of 6.5–7.0 (Fig. 2).59 The comparison of UV-vis spectra of optode samples equilibrated with different aqueous matrix having same concentration of U(VI) (100 ng mL−1) is shown in Fig. 1. It is seen from this figure that TEA buffer (pH 6.5–7.0) in the equilibrating solution enhanced the color change in optode sample corresponding to the formation of U(VI)–BTAP complex. In seawater matrix, the optode response towards U(VI) was significantly lower as compared to other aqueous matrices, and may not be useful for the detection and quantification of U(VI) in seawater. The change in absorbance spectra of optode samples, equilibrated with solutions having varying concentrations of U(VI) in TEA buffer is reported (Fig. 3). The comparison of spectra indicated that there was large bathochromic shift in absorbance of optode samples from 474 nm (blank) to 607 nm on equilibration with buffer solutions having U(VI) concentration ranging from 2.5 to 160 ng mL−1. The absorbance maxima at 585 nm is reported in the literature as a characteristic of U(VI)–BTAP complex.59 This indicated that the optode is responsive towards change in the uranium concentrations in the equilibrating solution. The absorbance of the blank optode sample (without uranium) is significantly lower as compared to that at 607 nm. Therefore, the change in the absorbance at 607 nm can be used for quantitative detection of U(VI) in the aqueous samples. It was also observed that pink color of U(VI)-loaded optode sample changes back to orange on immersing this optode in well-stirred 0.01 mol L−1 HNO3 for 10 min. This indicated that U(VI)–BTAP complex formed in the optode is broken at pH 2.0 to regenerate the BTAP in the optode.
:2 stable complex with U(VI) in pH range of 6.5–7.0 (Fig. 2).59 The comparison of UV-vis spectra of optode samples equilibrated with different aqueous matrix having same concentration of U(VI) (100 ng mL−1) is shown in Fig. 1. It is seen from this figure that TEA buffer (pH 6.5–7.0) in the equilibrating solution enhanced the color change in optode sample corresponding to the formation of U(VI)–BTAP complex. In seawater matrix, the optode response towards U(VI) was significantly lower as compared to other aqueous matrices, and may not be useful for the detection and quantification of U(VI) in seawater. The change in absorbance spectra of optode samples, equilibrated with solutions having varying concentrations of U(VI) in TEA buffer is reported (Fig. 3). The comparison of spectra indicated that there was large bathochromic shift in absorbance of optode samples from 474 nm (blank) to 607 nm on equilibration with buffer solutions having U(VI) concentration ranging from 2.5 to 160 ng mL−1. The absorbance maxima at 585 nm is reported in the literature as a characteristic of U(VI)–BTAP complex.59 This indicated that the optode is responsive towards change in the uranium concentrations in the equilibrating solution. The absorbance of the blank optode sample (without uranium) is significantly lower as compared to that at 607 nm. Therefore, the change in the absorbance at 607 nm can be used for quantitative detection of U(VI) in the aqueous samples. It was also observed that pink color of U(VI)-loaded optode sample changes back to orange on immersing this optode in well-stirred 0.01 mol L−1 HNO3 for 10 min. This indicated that U(VI)–BTAP complex formed in the optode is broken at pH 2.0 to regenerate the BTAP in the optode.
 | ||
| Fig. 2 The structure of the formed BTAP–UO2 complex. | ||
 | ||
| Fig. 3 UV-vis spectra of optode samples (1.0 cm × 3.0 cm) equilibrated with 10 mL of different aqueous samples spiked with 1.0 mg of U(VI) in bicarbonate medium. | ||
Optimization
The optode response depends on the rate of diffusion of anionic uranyl-carbonate species [UO2(CO3)(HCO3)−, UO2 (CO3)22−, or UO2(CO3)34−] existing in the aqueous phase to the optode interface, and then to form complex with the BTAP molecule present in the optode matrix. The primary role of DNNS appears to mediate the transfer of U(VI) from aqueous medium to optode matrix. In order to understand the effects of DNNS, the optode samples having varying amounts of DNNS were prepared by keeping same initial amounts of CTA, TEHP, and BTAP. The amount of BTAP in the optode was fixed to obtain the absorbance at 607 nm not exceeding 1.25%. The high amount of BTAP was found to degrade the optical quality of the optode. The amounts of BTAP and CTA used in preparation of the optodes are given in Table 1. The variation of U(VI) uptake as a function of DNNS amounts is shown in Fig. 4. As can be seen from Fig. 4, the U(VI) uptake in the optode samples was increased from 12 to 87% with increase in the DNNS concentration from 5.0 to 35% under identical conditions of the equilibrating solution. The optode samples were equilibrated for 15 h in well-stirred 10 mL buffer solution having U(VI) concentration 100 ng mL−1. The increase in U(VI) uptake with increase in DNNS concentration in the optode suggests that the DNNS enhances the U(VI) loading capacity of the optode. However, the kinetics of uranium uptake (15 h) was too slow to allow its use as a chemical sensor for wide ranging applications.| Composition | Response |
|---|---|
| CTA (45 wt%) + TEHP (40 wt%) + BTAP (0.2 wt%) | No color change |
| CTA (40 wt%) + TEHP (40 wt%) + BTAP (0.2 wt%) + DNNS (10 wt%) | No color change |
| CTA (30 wt%) + TEHP (40 wt%) + BTAP (0.2 wt%) + TiOA (10 wt%) | No color change |
| CTA (24 wt%) + TEHP (40 wt%) + BTAP (0.1 wt%) + DNNS (35 wt%) | Color changes from orange to pink |
 | ||
| Fig. 4 Variation of uranium uptake with DNNS concentration in the optode; Utotal = 1.0 μg; sample volume = 10 mL; duration = 15 h; pH 6.5–7.0 (TEA buffer). | ||
In this context, it is important to study the properties of plasticizer and their relative concentrations in the optode matrix. It was reported that increase in amount of plasticizer increases the diffusion rate of anions in the plasticized membranes.60 Therefore, the optode samples having varying amounts of two different plasticizers (e.g. TEHP and NPOE) were prepared and tested for their response time towards U(VI) ions in buffered carbonate aqueous medium.
The variation in absorbance of the optode samples, plasticized with varying amounts of TEHP and NPOE, was studied by equilibrating these with well-stirred 10 mL buffered sample solutions having 100 ng mL−1 U(VI) concentrations in bicarbonate/carbonate medium. The optode samples were taken out at regular time intervals to monitor the absorbance of U(VI)–BTAP complex at 607 nm as a function of equilibration time. The variations of absorbance at λmax in the optode samples as a function of equilibration time are shown in Fig. 5. This graph indicates the kinetics of sorption of anionic U(VI) species in the optode. As can be seen from Fig. 5, the rate of sorption of uranium anionic species increases with increase in amount of plasticizer in the optode. The matrix forming CTA chains in the liquid fraction of optode (plasticizer) matrix produces obstruction in the path of the moving ion-pair.
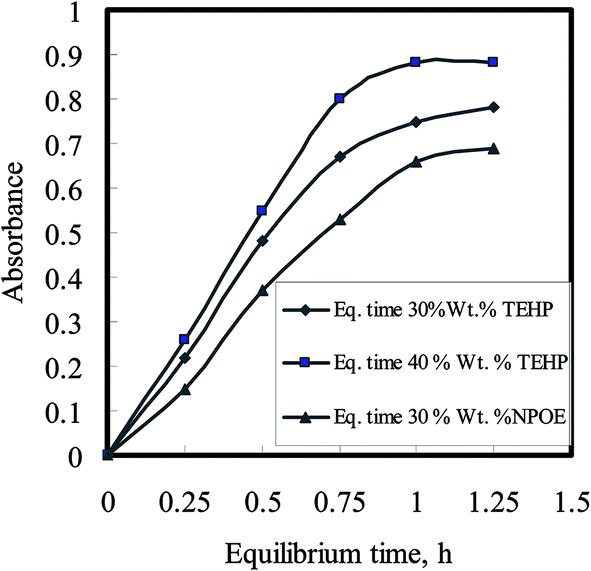 | ||
| Fig. 5 Uranium uptake kinetics as a function of plasticizer amount in the optode; Utotal = 1.0 μg; sample volume = 10 mL; pH 6.5–7.0 (TEA buffer). | ||
This obstruction is reduced with increase in the liquid fraction of the optode. Hence, the plasticizer, which is the major component of liquid phase, acts as a medium for diffusion transport of ions in the optode. The optode could not be plasticized more than 40 wt% of the plasticizer as mechanical strength of the optode was not enough to use it in well-stirred solution. Out of two plasticizer used in the preparation of optode, the optode sample with 40 wt% TEHP gave faster increase in absorbance corresponding to U(VI)–BTAP complex than the optode formed by NPOE. Although the equilibration time required for almost complete U(VI) sorption is longer than 3.0 h, the shorter equilibration time could be used for quantitative analysis of U(VI) as % uptake remains constant at fixed equilibration time. The equilibration time of 30 min could be used for constructing calibration graph for aqueous sample containing U(VI) greater than 160 ng mL−1. However, the detection limit for uranium would be better after 3.0 h equilibration of the optode sample as more than 95% of U(VI) sorption in the optode is achieved. Finally, the composition of optode was kept as: 24 wt% CTA + 35 wt% DNNS + 40 wt% TEHP + 0.1 wt% BTAP for uranium uptake studies with an equilibration time of 3.0 h.
The absorbance at 607 nm monitored for a long period over 18 h did not show the evidence of leaching of U(VI)–BTAP complex from the optode sample to equilibrating bicarbonate aqueous medium with TEA buffer. There was no drift in the absorbance when the film used for uranium uptake was exposed to light. No appreciable change in the optode absorbance value was observed when the film was dipped in de-ionized distilled water overnight. These observations suggest that the optode film was quite stable under the conditions of present studies.
Calibration and reproducibility
The response of the optode, in the form of change in absorbance at 607 nm, towards uranium concentration is up to 160 ng mL−1 after correcting for the blank. The blank absorbance at 607 nm was measured after equilibrating optode sample with blank solution at pH 6.5–7.0, without uranium. The absorbance linearly varies as a function of U(VI) concentration range of 2.5–160 ng mL−1. However, the calibration line did not pass through the zero. This may be due to the chemical changes produced on sorption of U(VI) in the matrix that might have changed the absorbance. These chemical changes may be counter ions or water content in the optode matrix. The water contents before and after U(VI) sorption in optode sample were found to be within 1.5 wt%. The minimum concentration of U(VI) required in the 10 mL equilibrating solution to produce distinct color change of optode (dimensions 3 cm × 1 cm) was found to be 160 ng mL−1. However, this detection limit of U(VI) concentration can be further enhanced by using larger volume of aqueous sample.The reproducibility of the optode membrane was determined using standard uranium solution (1000 μg mL−1 from Merck). Suitable aliquots were spiked from standard uranium solution in to the medium of uptake experiments, e.g., pH 6.6–7.0, TEA buffer, volume = 10 mL; and [U] = 100 ng mL−1. The optode sample was taken out for the absorbance measurements after equilibration for 3.0 h. The mean absorbance values with the relative standard deviation were found to be 1.33 ± 0.12 (n = 6). The small deviations in absorbance values suggest that optode membrane responses are reproducible under the conditions of this study.
In order to enhance the change in absorbance at 607 nm in the optode, the optode samples of different dimensions were equilibrated with buffered solutions having 1.0 μg of U(VI) in 10 mL in bicarbonate medium. As can be seen from Fig. 6, the absorbance increases from 0.07 to 0.3 by decreasing area of optode samples from 4.0 to 1.0 cm2. This can be attributed to increase in U(VI) concentration in the unit volume of the optode. Therefore, the possibility exist to improve U(VI) detection limit of optode by decreasing its volume.
 | ||
| Fig. 6 Variation in absorbance of the optode samples at 607 nm equilibrated with buffer solution containing 1.0 μg of U(VI). The area of optode was varied by changing length and keeping same breadth 1.0 cm. | ||
Interfering ions
The selectivity of the optode was tested by equilibrating optode samples with solutions containing different cations (Th4+, Eu3+, and Fe3+) and anions (F−, I−, NO3−, CO32−, and C2O42−). The absorbance spectra of optode samples did not change on its equilibration with solution containing 5.0–250 μg of Th4+. However, some of these ions were found to affect the U(VI) sorption in the optode. In order to examine the effects of these ions on U(VI) sorption in the optode, the uptake studies were carried out in the presence of micromolar concentrations of cations such as Th4+, Nd3+, Fe3+, and anions such as F−, C2O42− at pH 6.5–7.0 in TEA buffer with 100 ng mL−1 U(VI) concentration. In all cases except C2O42−, the absorbance variations in the optode samples were within ±5.0%, suggesting that presence of these cations/anions in the micro-molar concentrations will not significantly affect the U(VI) analysis. The tolerance limit was taken as 5.0% deviations in the mean absorbance values in U(VI)–BTAP complex at 607 nm in the absence of the competing cations/anions. In case of C2O42−, the absorbance was reduced to 11% which indicated that C2O42− ions interfere in the sorption of U(VI) in the optode. Addition of 1.0 mL of 1.0 × 10−3 mol L−1 ammonium molybdate eliminate the interference of C2O42− upto 500 fold molar excess.Comparison with solution spectrophotometry
The comparison of uranium determination by optode and spectrophotometry60 using chromophore BTAP is given. Unlike solution spectrophotometry, the presence of F− ions was not found to affect the formation of U(VI)–BTAP complex. Only bicarbonate/carbonate medium was found to be the appropriate medium for good response of optode towards U(VI). It is evident that the detection limit of U(VI) is significantly enhanced by using the optode (0.8 ng mL−1) than that which can be achieved with spectrophotometry (16 ng mL−1). The uranium determination by optode appears to be simple as it involves less sample manipulation and can be reused. Apart from this, the optode samples can be stored for longer period of time. This would be useful for preparation of standards for calibration plot or for visual colorimetric analysis of U(VI). The proposed method is more sensitive by 20 times than that by spectrophotometry.59A comparison of the proposed method with the previously reported methods for preconcentration and spectrophotometric determination of uranium62–65,67–69,71–73,75 in addition to that using ICP-AES and GFAAS61,70 FT-IR66 and Laser fluorimetric75 (Table 2) indicates that the proposed method is faster and simpler than the existing methods and that it provides a lower limit of detection. Although the procedures for xylenol orange74 using laser fluorimetry or pyrocatechol violet73 using spectrophotometry and polymer-based C18 cartridge75 have lower detection limits, the proposed method has more advantages through the sensitivity and interference point of view. To the best of our knowledge, this is the first report of using BTAP as chromophoric reagent for membrane optode for preconcentration and determination of uranium(VI).
| Chelating agent | Sorbent or micellar media | Analytical method | D.L. ng mL−1 | Ref. |
|---|---|---|---|---|
| o-Vanillin semicarbazone | Amberlite XAD-4 | ICP-AES and GFAAS | 100 | 61 |
| Dibenzoylmethane | Triton X-114 | Spectrophotometry | 11 | 62 |
| Arsenazo III | Silica gel | Spectrophotometry | 10 | 65 |
| 5,7-Dichloroquinoline-8-ol | Naphthalene | Spectrophotometry | 5.0 | 64 |
| 1,2-(Pyridylazo)-2-naphthol | Benzophenone | Spectrophotometry | 5.0 | 65 |
| Quinoline-8-ol | Chloromethylated polymeric resin | FT-IR spectroscopy | 50 | 66 |
| Arsenazo(III) | Silica | Spectrophotometry | 4.0 | 67 |
| Quinoline-8-ol | Amberlite XAD-4 | Spectrophotometry | 2.0 | 68 |
| 1,2-(Pyridylazo)-2-naphthol | Triton X-114 | Spectrophotometry | 1.1 | 69 |
| Quinoline-8-ol | Silica | ICP-AES | 1.0 | 70 |
| Dibromosuccinic acid | Amberlite XAD-4 | Spectrophotometry | 2.0 | 71 |
| Diarylazobisphenol | Activated carbon | Spectrophotometry | 5.0 | 72 |
| Pyrocatechol violet | Triton X-114 | Spectrophotometry | 0.06 | 73 |
| Xylenol orange | Silica | Laser fluorimetric | 0.05 | 74 |
| 2-(2-Benzothiazolylazo)-3-hydroxyphenol | Polymer-based C18 cartridge | Spectrophotometry | 0.6 | 75 |
| (BTAP) | Membrane optode | Spectrophotometry | 0.8 | This work |
Applications to real samples
In order to confirm the applicability of the proposed method, it has been applied to the determination of nanogram amounts of U(VI) in water and in real samples. The uptake experiment was done under specified experimental conditions such as pH 6.5–7.0 (with TEA buffer) and volume = 10 mL, using 0.5 mL of the effluent sample containing ±1.0 μg U(VI). The optode film (3 cm × 1 cm) was taken out for the absorbance measurements after exposure for 3.0 h. The mean absorbance values were found to be 0.87 ± 0.01. The calibration plot was constructed by adding known amount of U(VI) in buffered bicarbonate solution as described above.Seawater analysis
Results for the analysis of two seawater samples (Red Sea and Mediterranean Sea) are given in Table 3. Since a standard method for the determination of uranium in seawater has not been reported in literature, and inductively coupled plasma mass spectrometry (ICP-MS) as accepted independent method is not available, the accuracy of the method was examined by recovery study of the spiked samples. The recovery of the spikes added to seawater samples is given in Table 3. The quantitative recovery of the uranium spikes and relative standard deviation of 1.75% confirm the good precision and accuracy of the proposed optode method.| No. of samples | Uranium contentsa (ng mL−1) | |||
|---|---|---|---|---|
| Red Sea (El-Gharaa) | GFAAS method | Mediterranean sea (Alexandria) | GFAAS method | |
| a Mean ± S.D. (n = 6).b Theoretical value for t- and F-values for five degrees of freedom and 95% confidence limits are 2.57 and 5.05, respectively. | ||||
| 1 (t- and F-test)b | 2.37 ± 0.04 (1.17 and 2.66) | 2.37 ± 0.13 | 2.31 ± 0.05 (1.24 and 2.77) | 2.34 ± 0.13 |
| 2 (t- and F-test)b | 2.35 ± 0.04 (1.35 and 2.87) | 2.33 ± 0.09 | 2.29 ± 0.05 (1.14 and 2.59) | 2.29 ± 0.09 |
| 3 (t- and F-test)b | 2.38 ± 0.06 (1.28 and 2.57) | 2.39 ± 0.11 | 2.33 ± 0.04 (1.49 and 3.08) | 2.36 ± 0.11 |
| C = 2.367, RSD% = 1.75 | C = 2.363, RSD% = 2.60 | C = 2.31, RSD% = 1.35 | C = 2.33, RSD% = 2.7 | |
The performance of the proposed method was assessed by calculation of the t value (for accuracy) and F test (for precision)76 compared with GFAAS method. The mean values were obtained in Student's t and F tests at 95% confidence limits for five degrees of freedom. The results showed that the calculated values (Table 3) did not exceed the theoretical values. A wider range of determination, higher accuracy, more stability, and being less time consuming show the advantage of the proposed method over other method.
Analysis of soil and standard uranium ore samples
To verify applications and validations of the proposed method, one standard reference material, soil collected from farmland, and sediment collected from the Nile River were subjected to dissolution, preconcentration and determination. The results obtained for the standard reference material are shown in Table 4, and those for the soil sample and the sediment sample are shown in Table 5. The results obtained are in good agreement with the certified values, and the recoveries of spiked uranium were quantitative. Thus, these results indicated that the membrane optode method developed in the present work is accurate, simple, and low in cost for analyzing ore, soil, and sediment samples containing traces of uranium.| No. | U(VI) added/(μg g−1) | Uranium founda/(μg g−1) | Recovery/(%) | |
|---|---|---|---|---|
| Present method | Certifiedb | |||
| a Average and standard deviation from triplicate runs carried.b Certified values reported by Centre of China Reference Materials. | ||||
| 1 | None | 80.5 ± 0.50 | 79 | — |
| 2 | 30 | 111.3 ± 0.70 | 100.72 | |
| 3 | 60 | 139.6 ± 0.55 | 99.36 | |
| 4 | 90 | 172.2 ± 0.40 | 101.00 | |
| 5 | 120 | 202.0 ± 0.30 | 100.75 | |
| 6 | 150 | 227.8 ± 0.69 | 98.83 | |
| No. | Description of the sample | Uranium/(μg g−1) | Recovery/(%) | |
|---|---|---|---|---|
| Added | Founda | |||
| a Average and standard deviation from triplicate run. | ||||
| 1 | Soil sample from farmland nearby the Enshase City | — | 6.85 ± 0.30 | — |
| 5.0 | 11.8 ± 0.20 | 101.9 | ||
| 10 | 16.85 ± 0.25 | 101.3 | ||
| 2 | Sediment from Nile River nearby the Enshase City | — | 4.75 ± 0.15 | — |
| 6.0 | 10.70 ± 0.25 | 101.5 | ||
| 12 | 16.85 ± 0.20 | 100.5 | ||
Conclusions
A CTA-based optode has been developed for uranium preconcentration and its determination. The optode changes color due to uranium uptake in bicarbonate/carbonate medium (∼10−4 mol L−1) at pH 6.5–7.0 in the presence of TEA buffer. The intensity of color in the optode sample was found to be dependent on the uranium uptake from the sample solution as well as the composition of membrane. The composition of the optode was optimized as: 24 wt% CTA + 35 wt% DNNS + 40 wt% TEHP + 0.1 wt% BTAP, for uranium uptake studies with an equilibration time of 3.0 h. The detection limit of the optode film (dimension: 3 cm × 1 cm) was determined to be ∼0.8 ng mL−1 U(VI) for a 10 mL sample at pH 6.5–7.0 (in TEA buffer). The presence of micro-molar concentrations of cations such as such as Th4+, Nd3+, Fe3+, and of anions such as F−, I−, NO3−, and CO32−, etc. will not significantly affect the uranium analysis at pH 6.5–7.0 in TEA buffer. At higher concentration of these ions, the negative bias was observed in most cases except for Nd3+ ions. This indicated that performance of the optode is dependent on the concentrations of ions in aqueous samples. The optode film was quite stable under the conditions of present studies.Conflicts of interest
There are no conflicts to declare.References
- S. K. Papageorgiou, E. P. Kouvelos and F. K. Katsaros, Desalination, 2008, 224, 293 CrossRef CAS.
- A. Nakajima and T. Tsuruta, J. Radioanal. Nucl. Chem., 2004, 260, 13 CrossRef CAS.
- J. S. Chang and J. C. Huang, Biotechnol. Prog., 1998, 14, 735 CrossRef CAS PubMed.
- G. M. Gadd, Curr. Opin. Biotechnol., 2000, 11, 271 CrossRef CAS PubMed.
- R. Gupta, P. Ahuja, S. Khan, R. K. Saxena and H. Mohapatra, Curr. Sci., 2000, 78, 967 CAS.
- S. N. Groudev, P. S. Georgiev, I. I. Spasova and K. Komnitsas, Hydrometallurgy, 2001, 59, 311 CrossRef CAS.
- C. X. Chen, T. L. Chen, Y. J. Shi, X. W. Wu and X. Y. Chen, Colloids Surf., B, 2008, 61, 170–175 CrossRef PubMed.
- J. R. Spear, L. A. Figueroa and B. D. Honeyman, Appl. Environ. Microbiol., 2000, 66, 3711 CrossRef CAS PubMed.
- R. T. Anderson, H. A. Vrionis, I. Ortiz-Bernad, C. T. Resch, P. E. Long, R. Dayvault, K. Karp, S. Marutzky, D. R. Metzler, A. Peacock, D. C. White, M. Lowe and D. R. Lovley, Appl. Environ. Microbiol., 2003, 69, 5884 CrossRef CAS PubMed.
- J. S. Wang, X. J. Hu, J. Wang, Z. L. Bao, S. B. Xie and J. H. Yang, Biochem. Eng. J., 2010, 51, 19 CrossRef.
- S. H. Choi, M. S. Choi, Y. T. Park, K. P. Lee and H. D. Kang, Radiat. Phys. Chem., 2003, 67, 387 CrossRef CAS.
- A. P. Gilman, D. C. Villenuve, V. E. Secours, A. P. Yagminas, B. L. Tracy, J. M. Quinn, V. E. Valli, R. J. Willes and M. A. Moss, Toxicol. Sci., 1998, 41, 117 CAS.
- WHO, Guidelines for Drinking Water Quality, WHO, 3rd edn, 2003 Search PubMed.
- WHO, Guidelines for Drinking Water Quality, Addendum to Vol. 2. Health Criteria and other supporting information, WHO, Geneva, 2nd edn, 1998, WHO/EOS/98.1, p. 283 Search PubMed.
- P. Metilda, K. Prasad, R. Kala, J. M. Gladis, T. Prasada Rao and G. R. K. Naidu, Anal. Chim. Acta, 2007, 582, 147 CrossRef CAS PubMed.
- O. Fujino, S. Umetani, E. Ueno, K. Shigeta and T. Matsuda, Anal. Chim. Acta, 2000, 420, 65 CrossRef CAS.
- J. W. Ejnik, A. J. Carmichael, M. M. Hamilton, M. Mc Diarmid, K. Squibb, P. Boyd and W. Tardiff, Health Phys., 2000, 78, 143 CrossRef CAS.
- J. Avivar, L. Ferrer, M. Casas and V. Cerda, J. Anal. At. Spectrom., 2012, 27, 327 RSC.
- K. Grudpan, S. Laivoraungrath and P. Sooksamifi, Analyst, 1995, 120, 2107 RSC.
- B.-F. Lu, L.-B. Liu and J.-K. Cheng, Talanta, 1998, 47, 291 CrossRef.
- M. H. Khan and N. Yasmin, Pak. J. Biol. Sci., 2003, 6, 1276 CrossRef.
- K. B. Hong, K. W. Jung and K. H. Jung, Talanta, 1989, 36, 1095 CrossRef CAS PubMed.
- M. H. Khan, A. Ali and N. N. Khan, J. Radioanal. Nucl. Chem., 2001, 353, 353 CrossRef.
- A. R. Byrne and L. Benedik, Talanta, 1988, 35, 161 CrossRef CAS PubMed.
- W. Davies and W. Gray, Talanta, 1964, 11, 1203 CrossRef CAS.
- G. Koulouris, B. Slowikowski, R. Pilvio, T. Bostrom and M. Bickel, Appl. Radiat. Isot., 2000, 53, 279 CrossRef CAS PubMed.
- E. Montoya, P. Mendoza, P. Bedregal, O. Baltuano and I. Cohen, J. Radioanal. Nucl. Chem., 2012, 291, 175 CrossRef CAS.
- V. Natarajan, N. K. Porwal and S. V. Godbole, Indian J. Chem. Technol., 2012, 19, 399 CAS.
- G. Andreou, M. Efstathiou and I. Pashalidis, J. Radioanal. Nucl. Chem., 2012, 291, 865 CrossRef CAS.
- L. Dragounova and P. Rulik, Appl. Radiat. Isot., 2013, 81, 123 CrossRef CAS PubMed.
- Y. Yoon, S. Cho, K. Lee, K. Ko and K. Ha, J. Radioanal. Nucl. Chem., 2013, 296, 397 CrossRef CAS.
- P. A. Dimovasilis and M. I. Prodromidis, Sens. Actuators, B, 2011, 156, 689 CrossRef CAS.
- M. N. A. Lutfullah, N. Rahman and S. N. H. Azmi, J. Hazard. Mater., 2008, 155, 261 CrossRef CAS PubMed.
- M. Shamsipur, A. R. Ghiasvand and Y. Yamini, Anal. Chem., 1999, 71, 4892 CrossRef CAS PubMed.
- Z. Marczenko, Separation and spectrophotometric determination of elements, Ellis Harwood, London, 1986 Search PubMed.
- J. Minczewski, J. Chwastowska and R. Dybczynski, Separation and preconcentration methods in inorganic trace analysis, Ellis Harwood, London, 1982 Search PubMed.
- A. Niazi, J. Braz. Chem. Soc., 2006, 17, 1020 CrossRef CAS.
- A. Safavi and M. Bagheri, Anal. Chim. Acta, 2005, 530, 55 CrossRef CAS.
- I. Oehme and O. S. Wolfbies, Mikrochim. Acta, 1997, 126, 177 CrossRef CAS.
- S. Joannon, P. Telouk and C. Pin, Spectrochim. Acta, Part B, 1997, 52, 1783 CrossRef.
- I. M. Steinberg, A. Lobnik and O. S. Wolfbies, Sens. Actuators, B, 2003, 90, 230 CrossRef CAS.
- M. Lerchi, E. Bakker, B. Rusterholz and W. Simon, Anal. Chem., 1992, 64, 1534 CrossRef CAS.
- F. V. Bright, G. E. Poirier and G. M. Hieftje, Talanta, 1988, 35, 1138 CrossRef.
- G. J. Mohr and O. S. Wolbeis, Anal. Chim. Acta, 1994, 292, 41 CrossRef CAS.
- L. F. Capitan-Vallvey, M. D. Fernandez Ramos, P. Alvarez de Cienfuegos Galvez and J. Gomez, Talanta, 2005, 65, 239 CrossRef CAS PubMed.
- E. Bakker, P. B. Uhlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083 CrossRef CAS PubMed.
- E. Pretsch, P. B. Uhlmann and E. Bakker, Chem. Rev., 1998, 98, 1593 CrossRef.
- O. S. Wolfbeis, Anal. Chem., 2004, 78, 3269 CrossRef PubMed.
- Y. M. Scindia, A. K. Pandey, A. V. R. Reddy and S. B. Manohar, Anal. Chim. Acta, 2004, 515, 311 CrossRef CAS.
- S. Sodaye, R. Tripathi, A. K. Pandey and A. V. R. Reddy, Anal. Chim. Acta, 2004, 514, 159 CrossRef CAS.
- M. Plaschke, R. Czolk and H. J. Ache, Anal. Chim. Acta, 1995, 304, 107 CrossRef CAS.
- I. Murkovic and O. S. Wolfbeis, Sens. Actuators, B, 1997, 38–39, 246 CrossRef.
- P. T. Varineau, R. Duesing and L. E. Wangen, Appl. Spectrosc., 1991, 45, 1652 CrossRef CAS.
- G. E. Collins, Q. Lu, S. Abubeker and E. Vajs, Appl. Spectrosc., 2002, 56, 464 CrossRef CAS.
- G. E. Collins and Q. Lu, Anal. Chim. Acta, 2001, 436, 181 CrossRef CAS.
- A. Safavi and M. Bagheri, Anal. Chim. Acta, 2005, 530, 55 CrossRef CAS.
- A. Safavi and M. Sadeghi, Anal. Chim. Acta, 2006, 567, 184 CrossRef CAS.
- J. L. Sessler, P. J. Melfi, D. Seidel, A. E. V. Gordon, D. K. Ford, P. D. Palmer and C. D. Tait, Tetrahedron, 2004, 60, 11089 CrossRef CAS.
- A. S. Amin and E. H. El-Mossalamy, J. Trace Microprobe Tech., 2003, 21, 637 CrossRef CAS.
- S. Sodaye, G. Suresh, A. K. Pandey and A. Goswami, Radiochim. Acta, 2006, 94, 347 CrossRef CAS.
- V. K. Jain, A. Handa, S. S. Sait, P. Shrivastav and Y. K. Agarwal, Anal. Chim. Acta, 2001, 429, 237 CrossRef CAS.
- T. Madrakian, A. Afkhami and A. Mousavi, Talanta, 2007, 71, 610 CrossRef CAS PubMed.
- S. Sadeghi and E. Sheikhzadeh, J. Hazard. Mater., 2009, 163, 861 CrossRef CAS PubMed.
- M. Tarek, M. Zaki and W. H. Mahmoud, Talanta, 1988, 35, 253 CrossRef CAS PubMed.
- J. M. Gladis and T. P. Rao, Anal. Lett., 2002, 35, 501 CrossRef CAS.
- R. S. Praveen, P. Metilda, S. Daniel and T. P. Rao, Talanta, 2005, 67, 960 CrossRef CAS PubMed.
- P. Metilda, K. Sanghamitra, J. M. Gladis, G. R. K. Naidu and T. P. Rao, Talanta, 2005, 65, 192 CAS.
- J. M. Gladis and T. P. Rao, Anal. Bioanal. Chem., 2002, 373, 867 CrossRef CAS PubMed.
- M. E. F. Laespada, J. L. P. Pavon and B. M. Cordero, Analyst, 1993, 118, 209 RSC.
- S. Hirata, Y. Ishida, M. Aihara, K. Honda and O. Shikino, Anal. Chim. Acta, 2001, 438, 205 CrossRef CAS.
- F. Shemirani, R. R. Kozani and M. R. Jamali, Sep. Sci. Technol., 2005, 40, 2527 CrossRef CAS.
- A. M. Starvin and T. P. Rao, Talanta, 2004, 63, 225 CrossRef CAS PubMed.
- B. Cyriac and B. K. Balaji, Microchim. Acta, 2010, 171, 33 CrossRef CAS.
- S. Sadeghi, D. Mohammadzadeh and Y. Yamini, Anal. Bioanal. Chem., 2003, 375, 698 CrossRef CAS PubMed.
- A. S. Amin, Spectrosc. Lett., 2012, 45, 246 CrossRef CAS.
- J. C. Miller and J. N. Miller, Statistics for Analytical Chemistry, Ellis Horwood, Chichester, UK, 5th edn, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |