
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of polyurethane/polyvinyl alcohol hydrogel and its performance enhancement via compositing with silver particles†
Xiuqi Liab,
Yanjiao Jiangbc,
Fu Wangb,
Zengjie Fanc,
Haining Wangb,
Caihong Tao*a and
Zhaofeng Wang *b
*b
aSchool of Chemical and Biological Engineering, Lanzhou Jiaotong University, Lanzhou, 730070, PR China. E-mail: taoch@mail.lzjtu.cn
bState Key Laboratory of Solid Lubrication, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, Gansu 730000, China. E-mail: zhfwang@licp.cas.cn
cSchool of Stomatology, Lanzhou University, Lanzhou, 730000, China
First published on 2nd October 2017
Abstract
In this study, polyurethane/polyvinyl alcohol hydrogel was prepared by chemical cross-linking. Silver particles were further composited in the hydrogel via an in situ synthesis technique. The chemical structure and porous morphology were determined by Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy and field-emission scanning electron microscopy. The swelling abilities, mechanical behaviors, tribological properties, biocompatibilities and antibacterial properties of the hydrogels were investigated. The results suggest that the incorporation of silver particles in polyurethane/polyvinyl alcohol hydrogels could enhance the Young's modulus, tensile strength and elongation, as well as reduce the friction coefficient, while maintaining hydrogels with a moderate water absorption ability and good biocompatibility. Moreover, the addition of silver particles could endow the polyurethane/polyvinyl alcohol hydrogel with prominent antibacterial performance, suggesting that polyurethane/polyvinyl alcohol/silver composite hydrogels could be applied as dressings for wound healing.
Introduction
As the first defense line of the human body, the skin can protect human tissues and organs by preventing invasions from external pathogenic microbial, physical and chemical activities.1,2 When a large area of skin is injured, wound exposure results in bacterial infection, leading to serious complications.3 Therefore, it is required to cover wound dressings to promote wound healing. At present, the medical dressings, such as dry gauze and oil gauze, employed in clinics just play a passive protective role based on the barrier effect.4 However, in order to efficiently promote wound healing, there is an urgent need to develop some novel dressing materials with an active healing function. For this purpose, a variety of polymeric dressing materials such as foams, films, hydrocolloids, alginates, and hydrogels have been currently verified to promote wound healing effectively.5Hydrogel, as a wet and soft material, has received wide attention in tissue engineering,6 drug delivery carriers,7 actuators,8 sensors9 and bio-imaging,10 and even optoelectronic devices.11 To date, extensive research has focused on their mechanical weakness that seriously limits the related practical applications, while different structural hydrogels including double-network (DN) hydrogel,12,13 topological (TP) hydrogel,14 and nanocomposite (NC) hydrogel,15,16 have been synthesized to improve their mechanical properties. In addition, hydrogels have also drawn great attention as effective wound dressings because they are capable of providing a moist environment at the wound–dressing interface, allowing oxygen to diffuse, absorbing excess exudates, and preventing body fluid loss. At the same time, the easy functionalization of hydrogels also opens up the opportunity for loading antibacterial drugs, achieving initiative therapy for wounds.17–19
Polyurethane (PU), which has urethane linkages within the backbone, can be prepared by polyaddition polymerization using isocyanate and polyol. Because of their excellent biocompatibility, low biological toxicity, desirable mechanical properties, and high molecular design freedom, various polyurethane-based materials have been investigated for biomedical applications.20–22 For example, Zhou et al. prepared pH-sensitive polyurethanes using caprolactone derivatives in the soft segment and L-lysine ethyl ester diisocyanate in the hard segment, which can be potential materials as biodegradable and biocompatible multifunctional carriers for active intracellular drug delivery.23 PU-based hydrogels were also employed for applications in wound healing. For example, Yoo et al. prepared polyurethane/polyethylene glycol (PU/PEG) hydrogels by polyaddition reaction in an emulsion system.24 The PU/PEG hydrogels exhibited high water absorption and water vapor transmission rates, and were confirmed to have better wound healing efficacy of a full thickness rat wound model than that of gauze without any significant adverse reactions.
In this study, the waterborne polyvinyl alcohol (PVA) molecules were designed to crosslink with PU. To further enhance the performance of the PU/PVA hydrogel, silver nanoparticles were introduced by an in situ synthesis technique. The as-prepared PU/PVA/Ag nanocomposite hydrogels were confirmed to have a uniform porous morphology, desirable mechanical properties, excellent biocompatibility, and outstanding antibacterial activities against E. coli and S. aureus. Moreover, the PU/PVA/Ag hydrogels show a relatively low coefficient of friction, in the range of 0.2 to 0.35, indicating that the hydrogel dressings could be applied in wounds located at various sites of human skin without hampering daily actions.
Experimental
Materials
Hexamethylene diisocyanate (HMDI), PVA (Mw = 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) and trisodium citrate dehydrate (Na3C6H5O7·2H2O) were purchased from Aladdin (China); polycaprolactone diol (PCL, Mw = 2000 Da) was obtained from Sigma-Aldrich. Dimethyl sulfoxide (DMSO, solvent) was purchased from Lianlong Bohua (Tianjin) Pharmaceutical Chemical Co., Ltd. (China). Silver nitrate (AgNO3) was purchased from Sinopharm Chemical Reagent Co. Ltd. (China).
000 Da) and trisodium citrate dehydrate (Na3C6H5O7·2H2O) were purchased from Aladdin (China); polycaprolactone diol (PCL, Mw = 2000 Da) was obtained from Sigma-Aldrich. Dimethyl sulfoxide (DMSO, solvent) was purchased from Lianlong Bohua (Tianjin) Pharmaceutical Chemical Co., Ltd. (China). Silver nitrate (AgNO3) was purchased from Sinopharm Chemical Reagent Co. Ltd. (China).
Preparation of PU/PVA hydrogels
PVA and PCL were dried in a vacuum oven at a pressure of 50 mbar and a temperature of 80 °C, 24 h before the commencement of the experiments. Then, 2.5 mmol of PCL was dissolved in 100 mL of DMSO and poured into a three-necked reactor purged with nitrogen gas (N2). The reactor was immersed into an oil bath at 80 °C. After that, 5 mmol of HMDI was dissolved in 8.4 mL of DMSO, and the HMDI/DMSO solution was added dropwise into the reactor with stirring for 10 h to prepare urethane prepolymers (PPU). The synthetic route of PPU is presented in Scheme S1 (ESI†). At the same time, 5.84 g of PVA powder was dissolved in 48.66 mL DMSO, and the PVA/DMSO solution was obtained at 90 °C with stirring. After cooling to room temperature, the PPU solution was poured into PVA and stirred evenly, and then the mixture was poured into the mold for crosslinking at room temperature for 12 h. Then, the samples were immersed in 70% ethanol solution for 24 h to remove the residual solvents and unreacted monomers, and PU/PVA hydrogels were obtained.Loading of silver particles into PU/PVA hydrogels
The PU/PVA hydrogels were immersed in distilled water for 24 h and then transferred to a silver nitrate solution (0.5 mmol L−1, 1 mmol L−1, 5 mmol L−1, 10 mmol L−1) for another 24 h. The silver ion loaded PU/PVA hydrogels were subsequently transferred into a beaker containing 10 mmol L−1 of trisodium citrate solution, allowing for 24 h of reduction at room temperature. During this process, the hydrogels turned brown in colour, indicating the formation of silver nanoparticles. The sample was then immersed in distilled water for 48 h to remove unreacted reagents. According to the concentration of the employed silver nitrate solution, the nanocomposite hydrogels are denoted as PU/PVA/Ag-0.5, PU/PVA/Ag-1, PU/PVA/Ag-5 and PU/PVA/Ag-10. The synthetic routes of PU/PVA/Ag are illustrated in Scheme 1.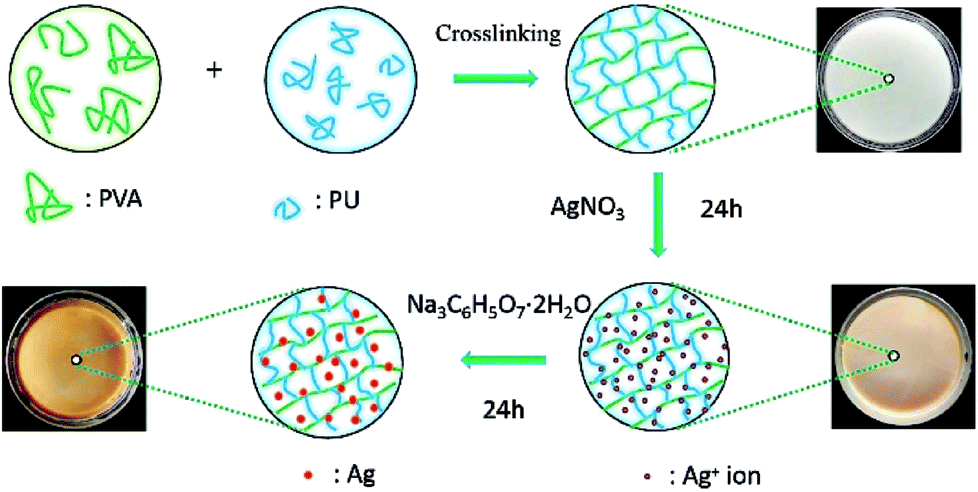 | ||
| Scheme 1 Synthesis of PU/PVA/Ag hydrogel networks. | ||
Biocompatibility test
The to-be-tested PU/PVA and PU/PVA/Ag hydrogels with a size of 5 mm × 5 mm × 1 mm were sterilized in 75% ethanol for 30 min and irradiated with ultraviolet (UV) radiation for 12 h and used for cell cultures. L929 fibroblast cells (obtained from the American Type Culture Collection, Rockville, MD, USA) were employed to evaluate the biocompatibility of PU/PVA and PU/PVA/Ag hydrogels. The L929 cells were cultured in Dulbecco's modified Eagle's medium supplemented with fetal bovine serum, penicillin–streptomycin (100 UI mL−1) and D-glucose (4.5 g L−1) under 5% aseptic CO2 and at 37 °C. The culture medium was refreshed every two days. Single cell suspensions were prepared by trypsin digestion when the cells were filled with the medium. Then, PU/PVA and PU/PVA/Ag hydrogels were placed into a 24-well plate and seeded with 104 cells mL−1 concentration on each well. Within a given period (1, 2 and 4 days), 100 μL of MTT solution was injected into each well, and then the cells were cultured for another 4 h. After the completion of the culture, the blue formazan reaction product was dissolved by adding 750 μL of DMSO, while the upper solvent was discarded. The resultant mixed solution was transferred into a 96-well plate, and its absorbance was measured using a microplate reader (Bio-Rad iMark) three times, and the average absorbance value was calculated.Antibacterial activity evaluation
The colony counts method was applied to detect the antibacterial activity of PU/PVA and PU/PVA/Ag nanocomposite hydrogels. The bacterial suspension liquid of common pathogens during the logarithmic phase (Gram-negative Escherichia coli denoted as E. coli and Gram-positive Staphylococcus aureus denoted as S. aureus) with a concentration of 1.0 × 106 CFU mL−1 was prepared. After centrifugation at 8000 rpm for 10 min, the suspension liquid was split into the removed supernatant and precipitate, which was diluted to the original concentration with sterilized physiological saline. The samples of the control and experimental groups were put into Eppendorf tubes with 1 mL bacterial suspension (106 CFU mL−1). Then, the Eppendorf tubes were incubated in a shaker at 250 rpm for 24 h at 37 °C. The suspension interacting with the samples was diluted to a concentration of 106 CFU mL−1 using a tenfold gradient dilution method. Then, 100 μL of the suspension was taken out and coated on the culture medium. After that, the culture medium was incubated for 24 h at 37 °C to form colony units, and then the number of colonies on each plate was counted. The samples of each experimental group and the control group were tested three times in parallel to ensure the accuracy of the experiment. The antibacterial rate formula is as follows:where R (%) is the antibacterial rate, where the value takes four significant digits, B is the number of colonies recovered in the control panel after 24 h, and C is the number of colonies recovered in the experimental group panel after 24 h.
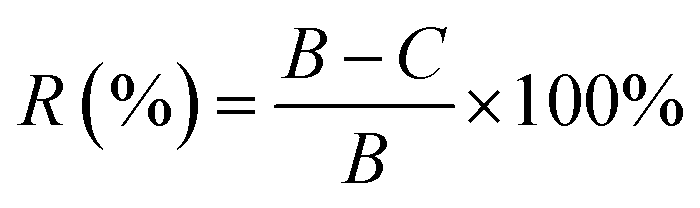
Characterizations
The chemical composition and structural information were determined by Fourier transform infrared spectroscopy (FTIR; Nicolet iS10; 4000–400 cm−1; resolution of 1 cm−1) and X-ray photoelectron spectroscopy (XPS; PHI-5702; equipped with a monochromated Al-Kα radiation source with a chamber pressure of 3 × 10−8 Torr). The porous morphology was examined using field-emission scanning electron microscopy (SEM; JSM-6701F) at an accelerating voltage of 5 kV. The mechanical properties of the hydrogels (length 70 mm × width 10 mm × thickness 1 mm) were tested on a microcomputer-controlled electronic universal testing machine (WDT-5, Tianshui Hongshan Testing Machine Co., Ltd, China). The stretching speed was kept at 30 mm min−1. The friction testing of a stainless ball (10 mm in diameter) against the PU/PVA/Ag hydrogels was performed on a ball-on-disc tribometer (CSM Instruments, Switzerland), with a reciprocating sliding frequency of 2 Hz and an applied normal load of 1 N.Result and discussion
Fig. 1 presents the FTIR spectra of PVA, PU and PU/PVA. For PU, characteristic peaks at 3345 cm−1 (N–H stretching), 2865–2950 cm−1 (C–H stretching), 1725 cm−1 (free C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1625 cm−1 (hydrogen-bonded CO stretching), 1471 cm−1 and 1367 cm−1 (C–H bending), and 1244 cm−1 and 1182 cm−1 (C–O–C stretching) can be observed.25–27 From the FTIR spectrum of PVA, peaks at 3410 cm−1 (O–H stretching), 2897–2954 cm−1 (C–H stretching), 1738 cm−1 (residual acetate CH3CO), 1248–1452 cm−1 (C–H bending) and 1005–1144 cm−1 (C–O–C stretching) were found.28,29 For the sample of PU/PVA, the broad peak around 3334 cm−1 should be ascribed to N–H and O–H stretching. The reduced intensity of –OH compared with PVA should come from the chemical reaction between the isocyanate peak (NCO) group of PU and –OH of PVA. To further confirm the above chemical crosslinking, the FTIR spectra of PU and PU/PVA from 2000 to 2200 cm−1 was magnified and compared as shown in Fig. 1b and c. It is obviously found that the NCO stretching peak at ca. 2095 cm−1 disappears in PU/PVA.25 Moreover, the peak of hydrogen-bonded CO stretching at 1625 cm−1 becomes weaker for PU/PVA. The mobility of molecular chains was restrained by the reaction between –OH and –NCO, which may prevent the formation of hydrogen-bonded CO in the PU/PVA hydrogel. The abovementioned results suggest that PU and PVA were well crosslinked, and the PU/PVA hydrogel was formed.
O stretching), 1625 cm−1 (hydrogen-bonded CO stretching), 1471 cm−1 and 1367 cm−1 (C–H bending), and 1244 cm−1 and 1182 cm−1 (C–O–C stretching) can be observed.25–27 From the FTIR spectrum of PVA, peaks at 3410 cm−1 (O–H stretching), 2897–2954 cm−1 (C–H stretching), 1738 cm−1 (residual acetate CH3CO), 1248–1452 cm−1 (C–H bending) and 1005–1144 cm−1 (C–O–C stretching) were found.28,29 For the sample of PU/PVA, the broad peak around 3334 cm−1 should be ascribed to N–H and O–H stretching. The reduced intensity of –OH compared with PVA should come from the chemical reaction between the isocyanate peak (NCO) group of PU and –OH of PVA. To further confirm the above chemical crosslinking, the FTIR spectra of PU and PU/PVA from 2000 to 2200 cm−1 was magnified and compared as shown in Fig. 1b and c. It is obviously found that the NCO stretching peak at ca. 2095 cm−1 disappears in PU/PVA.25 Moreover, the peak of hydrogen-bonded CO stretching at 1625 cm−1 becomes weaker for PU/PVA. The mobility of molecular chains was restrained by the reaction between –OH and –NCO, which may prevent the formation of hydrogen-bonded CO in the PU/PVA hydrogel. The abovementioned results suggest that PU and PVA were well crosslinked, and the PU/PVA hydrogel was formed.
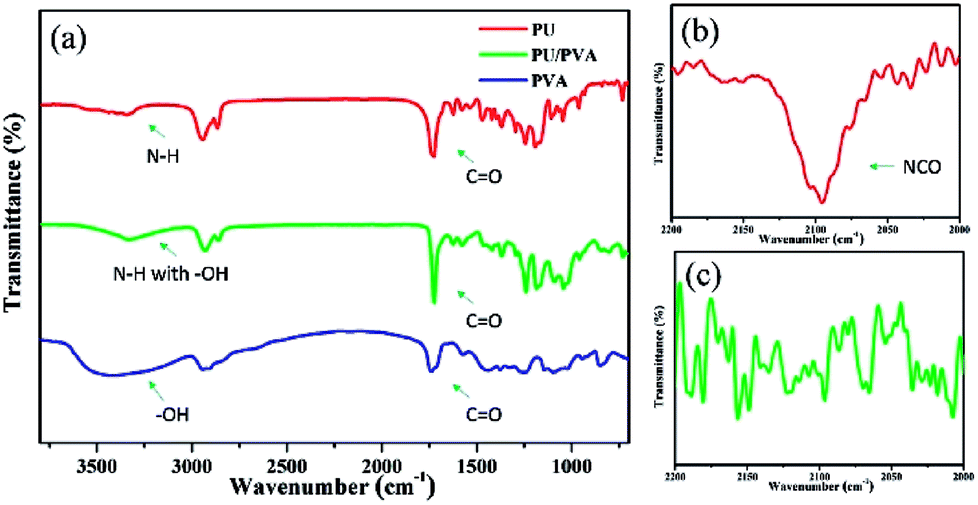 | ||
| Fig. 1 (a) FTIR spectra of PU, PVA and PU/PVA hydrogels; (b and c) are the magnified FTIR spectra from 2000 to 2200 cm−1 of PU and PU/PVA, respectively. | ||
Based on the formation of PU/PVA, silver particles were further composited via the solution reduction method at room temperature. The solution reduction method has been widely adopted to prepare silver nanoparticles for various uses.30 Fig. 2 shows the XPS Ag 3d spectra of PU/PVA and PU/PVA/Ag series to confirm the existence of silver. With the increase in the Ag concentration, the XPS Ag 3d spectrum becomes gradually remarkable, which involves two peaks at around 373.9 and 367.9 eV, corresponding to Ag 3d3/2 and Ag 3d5/2, respectively. The energy difference is calculated to be 6.0 eV, confirming that metallic silver (Ag0) has been immobilized in the PU/PVA hydrogels.31
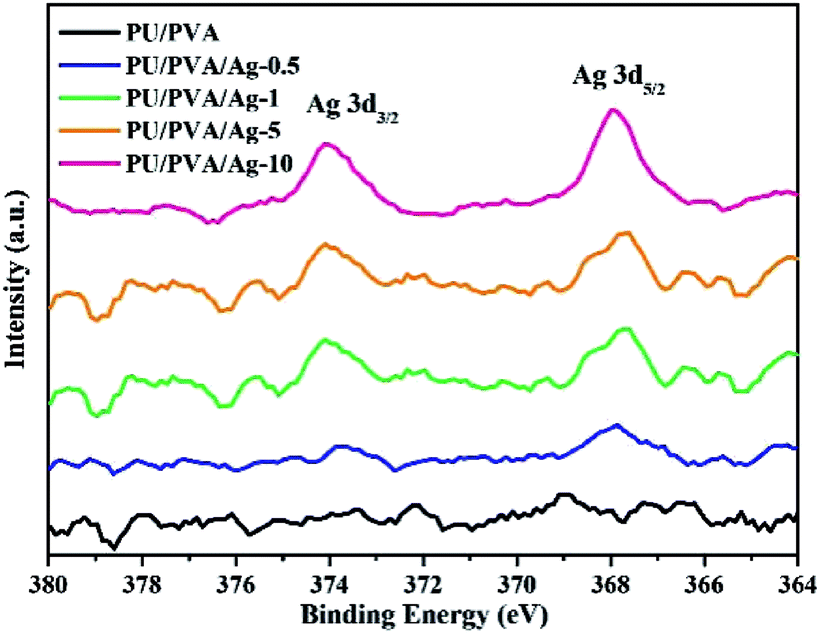 | ||
| Fig. 2 High-resolution XPS Ag 3d spectra of PU/PVA and the PU/PVA/Ag composite hydrogels. | ||
Fig. 3 shows the SEM images of PU/PVA and PU/PVA/Ag to observe their microstructure. It can be found that the hydrogels exhibit a porous structure. Such a structure is advantageous for wound healing because it could allow the supply of oxygen, absorption of the exudate, and maintenance of a moist environment. The pore diameter of PU/PVA is estimated to be 5–10 μm, which is gradually decreased along with an increase in Ag. The decreased pore diameter of hydrogels after compositing Ag should be related to the binding of silver nanoparticles with electron-rich O and N atoms of the polyurethane chains, which could produce additional cross-linking.32,33
 | ||
| Fig. 3 SEM images of the cross-sections of (a) PU/PVA; (b) PU/PVA/Ag-0.5; (c) PU/PVA/Ag-1; (d) PU/PVA/Ag-5 and (e) PU/PVA/Ag-10 hydrogels. | ||
The swelling capacity of the hydrogels plays an important role in the wound healing process and other biomedical applications. Better wound dressings should have adequate water absorption capacity for the sake of absorbing wound exudates.34 As shown in Fig. 4, the swelling degree of PU/PVA and PU/PVA/Ag hydrogels increases with extended soaking time and reaches equilibrium after 6 h of soaking. The order of the swelling capacity of hydrogels is PU/PVA > PU/PVA/Ag-0.5 > PU/PVA/Ag-1 > PU/PVA/Ag-5 > PU/PVA/Ag-10. The lowered swelling capacity of PU/PVA/Ag hydrogels should be ascribed to the decreased pore diameter of the hydrogels as revealed in Fig. 3. The higher crosslinks limit the infiltration of water in the hydrogels network for swelling.32,33 Even so, the PU/PVA/Ag hydrogels show a water absorption capacity of 150–250%.
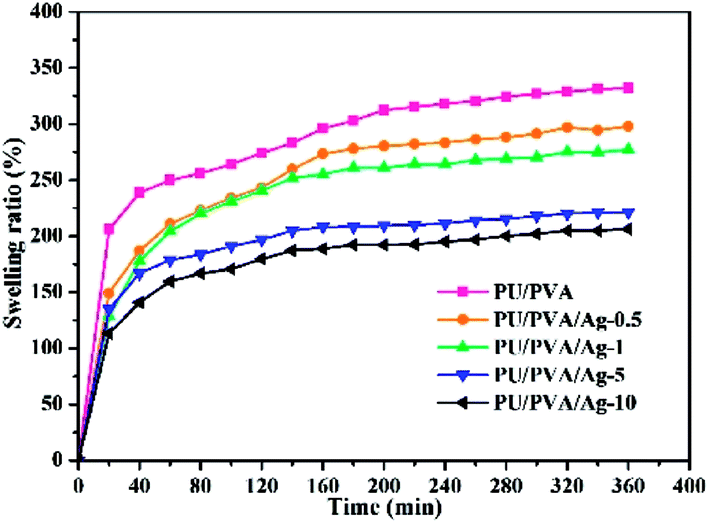 | ||
| Fig. 4 Swelling ability of PU/PVA and PU/PVA/Ag hydrogels soaked in deionized water for various times. | ||
Since skin is a motorial tissue accompanying various motions, the mechanical behaviour investigation of wound dressings is required to ensure its daily activities. Fig. 5 depicts the stress–strain curves of PU/PVA and PU/PVA/Ag nanocomposite hydrogels with different silver loadings. The corresponding summary of Young's modulus, tensile strength and elongation at break are given in Table S1 (ESI†). With the introduction of Ag and further increasing its concentration, the Young's modulus is improved from 0.12 to 0.34 MPa, while the tensile strength and elongation at break are significantly improved from 0.05 to 0.52 MPa and from 200% to 621%, respectively. The enhanced mechanical activities of PU/PVA/Ag hydrogels should also originate from the additional crosslink between Ag and the PU chains.32,33 In order to further characterize the mechanical properties of PU/PVA hydrogels, we carried out tensile loading–unloading tests at a constant speed of 30 mm min−1 to evaluate the energy dissipation. Apparent hysteresis loops are observed in the loading–unloading curves of PU/PVA/Ag-10 hydrogel at different strains in Fig. 5b, indicating that the PU/PVA/Ag-10 hydrogel dissipates energy effectively.35 It is found that the PU/PVA/Ag-10 hydrogel could dissipate energy of about 282.4 kJ m−3 at a strain of 400%. Fig. 5c shows the loading–unloading tests of PU/PVA hydrogel and PU/PVA/Ag nanocomposite hydrogels at the same strain (200%). Although the apparent hysteresis loops are observed in all hydrogels, the hysteresis loop of PU/PVA/Ag-10 hydrogel is the largest. The corresponding dissipation energy of PU/PVA/Ag-10, PU/PVA/Ag-5, PU/PVA/Ag-1, PU/PVA/Ag-0.5, and PU/PVA hydrogels are 39.87, 33.97, 30.13, 30.01, and 18.36 kJ m−3, respectively. It indicates that the introduction of Ag could make the PU/PVA/Ag hydrogels dissipate energy more efficiently than the PU/PVA hydrogel, causing excellent mechanical properties such as extensibility and toughness.36 It is noteworthy that the energy dissipation is irreversible during the stretching process as seen in Fig. 5d. It is found that the first cycle shows a significant hysteresis loop, while the hysteresis becomes negligible and overlaps during the next nine loading–unloading processes. This indicates that the network structure of hydrogels has an irreversible damage.37
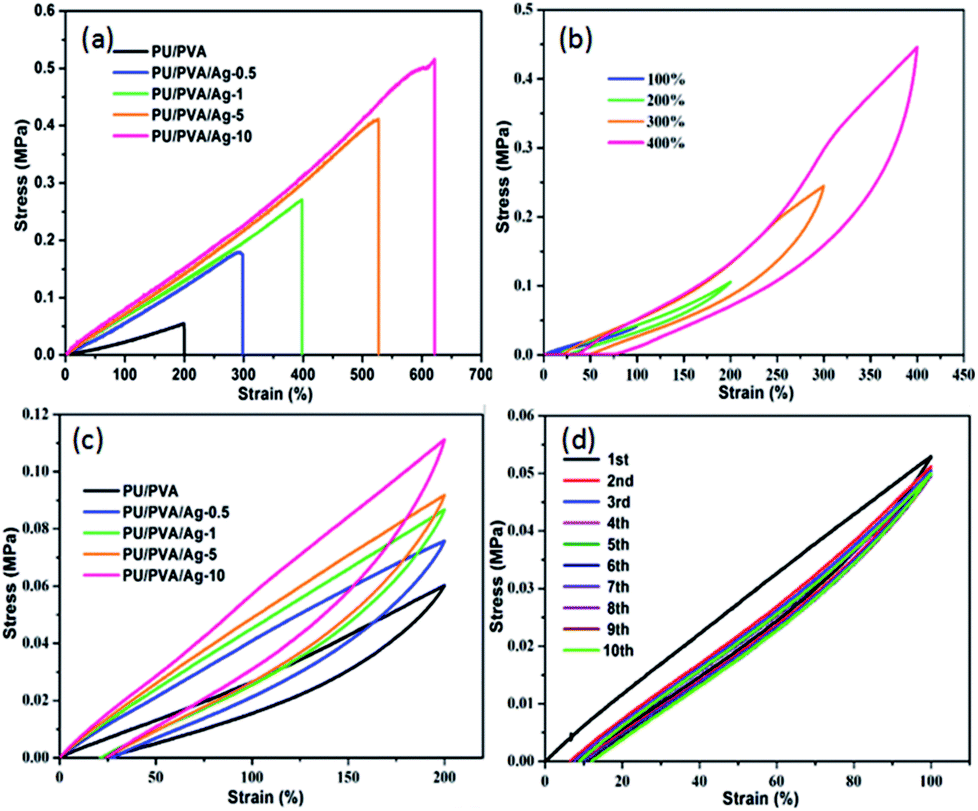 | ||
| Fig. 5 (a) Tensile stress–strain curves; (b) tensile loading–unloading tests of PU/PVA/Ag-10 hydrogel under different strains (100%, 200%, 300%, 400%); (c) tensile loading–unloading tests of PU/PVA hydrogel and PU/PVA/Ag nanocomposite hydrogels under 200% strain; (d) ten cyclic tensile loading–unloading tests of PU/PVA/Ag-10 hydrogel. | ||
Fig. 6 shows the friction coefficient of PU/PVA and PU/PVA/Ag nanocomposite hydrogels. For the PU/PVA, PU/PVA/Ag-0.5, PU/PVA/Ag-1 and PU/PVA/Ag-5 hydrogels, the friction coefficient decreased along with an increase in the Ag concentration. The nanoscale polishing effect should be responsible for the reduction of the friction coefficient. With an appropriate size, hardness and volume content, nanoparticles could polish the counterpart surface to a very fine scale, which could reduce the related shear stress and thus decrease the friction coefficient.38 In addition to the nanoscale polishing effect, the rolling effect of nanoparticles should also exist, which has been reported to reduce both the frictional coefficient and the wear rate.39 When the Ag concentration is further increased to form PU/PVA/Ag-10, its friction coefficient exhibits an increasing trend, which should be aroused by the enhanced aggregation degree of Ag nanoparticles in the composite hydrogels. The tribological tests suggest that the PU/PVA/Ag nanocomposite hydrogels possess a moderate friction coefficient from 0.2 to 0.35, which is close to the friction coefficient of human skin as described in literature.40
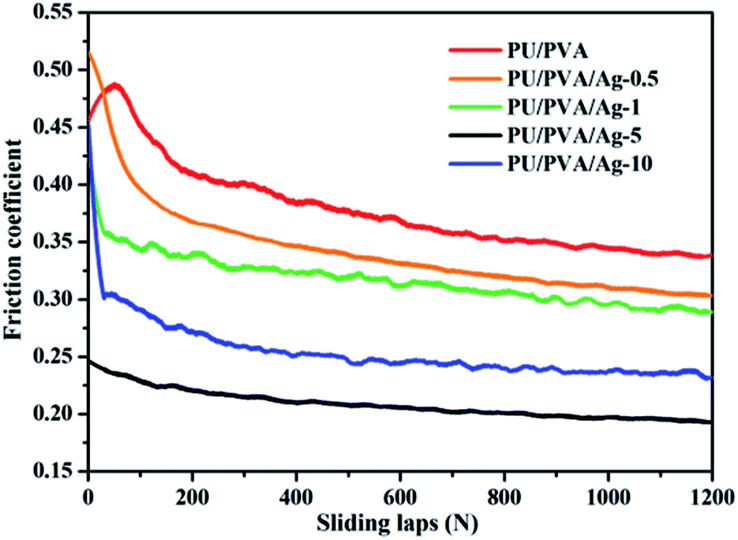 | ||
| Fig. 6 The friction coefficients of PU/PVA hydrogels and PU/PVA/Ag nanocomposite hydrogels. | ||
In addition to the porous structure and good mechanical and tribological properties, biocompatibility is another criterion to estimate the feasibility of wound dressing. Biocompatibility is often assessed using the MTT assay that is commonly used to analyse the possible harmful effects induced in cells by materials, with which the cellular mitochondria activity upon exposure to materials is quantified.41,42 For this purpose, PU/PVA/Ag nanocomposite hydrogels were selected as the experimental groups, and the PU/PVA hydrogel was selected as the control group, where L929 cells were seeded directly onto the surface of the samples. As shown in Fig. 7, the cell viability of the PU/PVA/Ag-0.5, PU/PVA/Ag-1 and PU/PVA/Ag-5 nanocomposite hydrogels tends to be close to or higher than that of the control group. The PU/PVA/Ag-10 nanocomposite hydrogels exhibit lower cell viability in 1 day and 2 day incubations, but show higher cell viability than the control group in 4 day incubations. In brief, the cell viability of the PU/PVA/Ag-10 nanocomposite hydrogels is lower than that of the other experimental groups, which is probably attributed to the aggregation of Ag nanoparticles, causing slight harmful effects to cell viability. While the cell viability of the remaining experimental groups is better, it indicates that PU/PVA/Ag-0.5, PU/PVA/Ag-1 and PU/PVA/Ag-5 have no cytotoxicity and good biocompatibility.
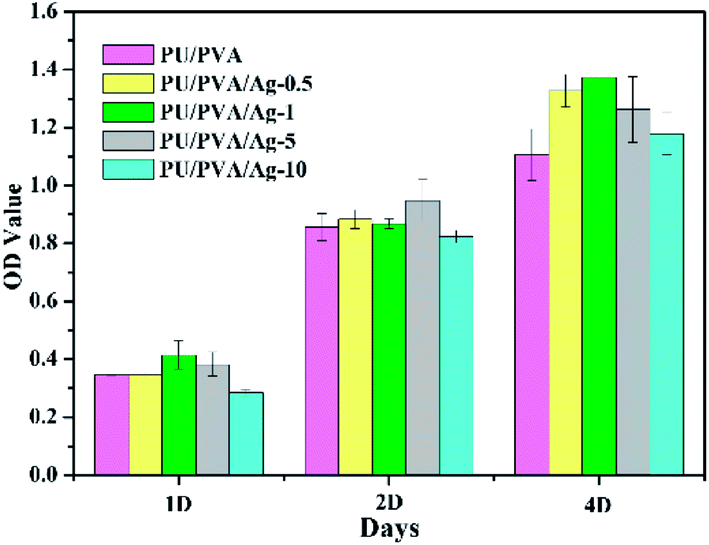 | ||
| Fig. 7 Cellular proliferation analysis of PU/PVA and PU/PVA/Ag hydrogels. | ||
Bacterial infection is a vital factor for wound healing, and thus we investigate the antimicrobial activities of the as-prepared hydrogels to estimate their promising applications as wound dressings. Fig. S1 and S2 (ESI†) show the antibacterial activities of PU/PVA, PU/PVA/Ag-0.5, PU/PVA/Ag-1, PU/PVA/Ag-5 and PU/PVA/Ag-10 hydrogels determined by the colony counts method. The results are summarized in Table 1. It is found that the PU/PVA hydrogels show no antibacterial activity, and the incorporation of silver nanoparticles in the hydrogels could induce a dose-dependent antibacterial behavior. This should be attributed to the fact that silver nanoparticles could interact with sulfur-containing proteins as well as phosphorus-containing compounds like DNA in cells, attacking the respiratory chain with cell division leading to cell death.43 For the PU/PVA/Ag-5 and PU/PVA/Ag-10 hydrogels, their disinfection rates are both higher than 90%. Based on the comprehensive investigation of the swelling ability, mechanical and tribological performance, biocompatibility, and antibacterial behaviour, PU/PVA/Ag-5 nanocomposite hydrogel is considered to be the optimum sample, showing promising applications as an artificial skin for wound healing.
| Sample | E. coli (%) | S. aureus (%) |
|---|---|---|
| PU/PVA | 0 | 0 |
| PU/PVA/Ag-0.5 | 59.84 ± 1.79 | 46.20 ± 3.78 |
| PU/PVA/Ag-1 | 85.74 ± 0.7 | 60.68 ± 6.90 |
| PU/PVA/Ag-5 | 93.60 ± 1.18 | 81.93 ± 2.13 |
| PU/PVA/Ag-10 | 97.07 ± 0.44 | 94.61 ± 0.54 |
Conclusions
A series of PU/PVA and PU/PVA/Ag hydrogels was prepared by the chemical cross-linking and in situ synthesis techniques. PU/PVA hydrogels show a uniform porous structure, good swelling ability and biocompatibility. The composition of silver nanoparticles in PU/PVA hydrogels could still maintain the abovementioned properties over a moderate range, while further improving their mechanical and tribological performance, i.e., Young's modulus, tensile strength, elongation at break and friction coefficient. In addition, the PU/PVA/Ag composite hydrogels show excellent antibacterial properties, suggesting that they could be employed for wound healing applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the CAS Pioneer Hundred Talents Program, the National Natural Science Foundation of China (81571829), and the Basic Research Projects of Higher Education Institutions in Gansu Province.Notes and references
- M. Morales-Hurtado, X. Zeng, P. Gonzalez-Rodriguez, J. E. Ten Elshof and E. van der Heide, J. Mech. Behav. Biomed. Mater., 2015, 46, 305–317 CrossRef CAS PubMed.
- I. Yannas and J. F. Burke, J. Biomed. Mater. Res., Part A, 1980, 14, 65–81 CrossRef CAS PubMed.
- C. K. Svensson, Drug Metab. Dispos., 2009, 37, 247–253 CrossRef CAS PubMed.
- J.-P. Chen, G.-Y. Chang and J.-K. Chen, Colloids Surf., A, 2008, 313, 183–188 CrossRef.
- E. Kamoun, E. Kenawy and X. Chen, J. Adv. Res., 2017, 8, 217–233 CrossRef PubMed.
- G. D. Nicodemus and S. J. Bryant, Tissue Eng., Part B, 2008, 14, 149–165 CrossRef CAS PubMed.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- L. Engel, O. Berkh, K. Adesanya, J. Shklovsky, E. Vanderleyden, P. Dubruel, Y. Shacham-Diamand and S. Krylov, Sens. Actuators, B, 2014, 191, 650–658 CrossRef CAS.
- A. Wada, S.-i. Tamaru, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5321–5330 CrossRef CAS PubMed.
- L. Wu, G. Chen and Z. Li, Small, 2017, 13, 1604070 CrossRef PubMed.
- L. Wu, C. Gao, Z. Li and G. Chen, J. Mater. Chem. C, 2017, 5, 5207–5213 RSC.
- Z. Gong, G. Zhang, X. Zeng, J. Li, G. Li, W. Huang, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2016, 8, 24030–24037 CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- Z. Hu and G. Chen, Adv. Mater., 2014, 26, 5950–5956 CrossRef CAS PubMed.
- L. Wu, Z. Hu, G. Chen and Z. Li, Soft matter, 2015, 11, 9038–9044 RSC.
- M. Kokabi, M. Sirousazar and Z. M. Hassan, Eur. Polym. J., 2007, 43, 773–781 CrossRef CAS.
- Z. Fan, B. Liu, J. Wang, S. Zhang, Q. Lin, P. Gong, L. Ma and S. Yang, Adv. Funct. Mater., 2014, 24, 3933–3943 CrossRef CAS.
- D. Staneva, D. Atanasova, E. Vasileva-Tonkova, V. Lukanova and I. Grabchev, Appl. Surf. Sci., 2015, 345, 72–80 CrossRef CAS.
- K.-D. Vorlop, A. Muscat and J. Beyersdorf, Biotechnol. Tech., 1992, 6, 483–488 CrossRef CAS.
- L. Rao, H. Zhou, T. Li, C. Li and Y. Y. Duan, Acta Biomater., 2012, 8, 2233–2242 CrossRef CAS PubMed.
- S. Bonakdar, S. H. Emami, M. A. Shokrgozar, A. Farhadi, S. A. H. Ahmadi and A. Amanzadeh, Mater. Sci. Eng., C, 2010, 30, 636–643 CrossRef CAS.
- L. Zhou, D. Liang, X. He, J. Li, H. Tan, Q. Fu and Q. Gu, Biomaterials, 2012, 33, 2734–2745 CrossRef CAS PubMed.
- H. J. Yoo and H. D. Kim, J. Biomed. Mater. Res., Part B, 2008, 85, 326–333 CrossRef PubMed.
- G. Trovati, E. A. Sanches, S. C. Neto, Y. P. Mascarenhas and G. O. Chierice, J. Appl. Polym. Sci., 2010, 115, 263–268 CrossRef CAS.
- L. Jiao, H. Xiao, Q. Wang and J. Sun, Polym. Degrad. Stab., 2013, 98, 2687–2696 CrossRef CAS.
- G. Chen, Y. Ma, X. Zheng, G. Xu, J. Liu, J. Fan, D. Shen and Z. Qi, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 654–660 CrossRef CAS.
- G. Andrade, E. Barbosa-Stancioli, A. P. Mansur, W. Vasconcelos and H. Mansur, Biomed. Mater., 2006, 1, 221 CrossRef CAS PubMed.
- N. V. Bhat, M. M. Nate, M. B. Kurup, V. A. Bambole and S. Sabharwal, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 237, 585–592 CrossRef CAS.
- A. R. Shahverdi, A. Fakhimi, H. R. Shahverdi and S. Minaian, Nanomedicine, 2007, 3, 168–171 CrossRef CAS PubMed.
- H. Kong and J. Jang, Chem. Commun., 2006, 3010–3012 RSC.
- K. Vimala, Y. M. Mohan, K. S. Sivudu, K. Varaprasad, S. Ravindra, N. N. Reddy, Y. Padma, B. Sreedhar and K. MohanaRaju, Colloids Surf., B, 2010, 76, 248–258 CrossRef CAS PubMed.
- K. Varaprasad, K. Vimala, S. Ravindra, N. N. Reddy, G. V. S. Reddy and K. M. Raju, J. Mater. Sci., 2011, 22, 1863–1872 CAS.
- T. Wang, X.-K. Zhu, X.-T. Xue and D.-Y. Wu, Carbohydr. Polym., 2012, 88, 75–83 CrossRef CAS.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- Z. Gong, G. Zhang, X. Zeng, J. Li, G. Li, W. Huang, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2016, 8, 24030–24037 CAS.
- Y. Liu, W. Zhou, Q. Zhou, K. Peng, A. Yasin and H. Yang, RSC Adv., 2017, 7, 29489–29495 RSC.
- L. Chang, Z. Zhang, C. Breidt and K. Friedrich, Wear, 2005, 258, 141–148 CrossRef CAS.
- L. Kahlman and I. M. Hutchings, Tribol. Trans., 1999, 42, 842–850 CrossRef CAS.
- A. B. Cua, K. P. Wilhelm and H. Maibach, Br. J. Dermatol., 1990, 123, 473–479 CrossRef CAS PubMed.
- F. M. Young, W. Phungtamdet and B. J. Sanderson, Toxicol. In Vitro, 2005, 19, 1051–1059 CrossRef CAS PubMed.
- Z. Fan, J. Wang, Z. Wang, H. Ran, Y. Li, L. Niu, P. Gong, B. Liu and S. Yang, Carbon, 2014, 66, 407–416 CrossRef CAS.
- J. S. Kim, E. Kuk, K. N. Yu, J.-H. Kim, S. J. Park, H. J. Lee, S. H. Kim, Y. K. Park, Y. H. Park and C.-Y. Hwang, Nanomedicine, 2007, 3, 95–101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of mechanical properties and antibacterial performance. See DOI: 10.1039/c7ra08845k |
| This journal is © The Royal Society of Chemistry 2017 |