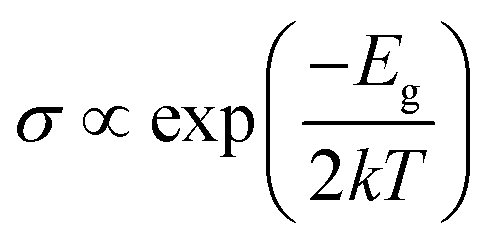DOI:
10.1039/C7RA08579F
(Paper)
RSC Adv., 2017,
7, 43521-43530
Adsorption sensitivity of graphane decorated with B, N, S, and Al towards HCN: a first-principles study
Received
3rd August 2017
, Accepted 5th September 2017
First published on 8th September 2017
Abstract
The geometric structure, adsorption energy, electronic structure, and magnetic properties of hydrogenated graphene (graphane) with the adsorption of a HCN molecule were investigated by first-principles calculations. Compared with graphane, the adsorption of HCN on H-vacancy defected graphane (VHG) exhibited higher stability, which implied that the H-vacancy improved the sensitivity of graphane. However, the small adsorption energies and large bond distance indicated that the weak adsorption of a HCN molecule on the graphane and VHG substrates was due to physisorption. By introducing dopants (B, N, S, and Al), the activity of graphane was significantly improved. The adsorption of HCN changed to chemisorption on the graphane with dopants. Meanwhile, the opening of band gaps by HCN adsorption can be used as an electronic signal to detect HCN gas. Interestingly, the spin polarized density of states (PDOS) results suggested that the adsorption of HCN on VHG and S-doped VHG exhibited magnetic character and half-metallicity behavior. These results could provide useful information to design gas sensors for HCN or spintronic devices based on graphane.
1. Introduction
The controlling of air pollution is now regarded as an important area and the monitoring of toxic gases by sensors is a great technological challenge.1–3 Hydrogen cyanide (HCN) vapor is extremely virulent to human beings and animals, because it inhibits the consumption of oxygen by body tissues.4 A small concentration will result in damage to the health of human beings,5 and thus it is important to explore effective methods to detect HCN gas. A promising gas sensor usually exhibits some advantages such as fast response time, miniature size, high sensitivity, and low cost.2,6,7 Carbon-based materials like graphene present promising potential in the application of gas sensors due to their large surface to volume ratio and relatively high reactivity. Graphene, a two-dimensional sp2 connected carbon monolayer, has attracted great interest since it was first experimentally synthesized in 2004.8–11 Recently, graphene used as gas sensors to detect many molecules has been explored.12–16 However, the results indicated that the pure graphene exhibits relative low sensitivity to the gas molecules and the zero-gap restrict its application in some important area like gas sensors. Therefore, the introducing of defects and dopants has been successfully attempted to improve the interaction between the graphene and absorbents. In this regard, graphane is a promising compound, which was first experimentally prepared by attaching hydrogen atoms on the graphene in 2009.17
In graphane, the full hydrogenation of graphene leads the sp2 hybridization of C–C bond in graphene changed to sp3 hybridization in graphane. Graphane has been studied in recent past due to its unique electronic and magnetic properties.18–22 Although the highly mechanical strength and wide band-gap of graphane add to its excellence in applications, the bonds (C–C and C–H bonds) formed in graphane cause weak activity to adsorbents. However, the sensitivity of graphane can be significantly improved by introducing of H-vacancy, which also induced magnetism in graphane.23–28 Berashevich et al. investigated the influence of adsorbates on the electronic and magnetic properties of graphane, and they found that H vacancies significantly improved the chemical reactivity of graphane to environment.23 Islam et al. investigated the sensing aptitude of graphane by crafting with H-vacancy and dopants, and the results suggested that interaction of graphane towards CH4 gas molecules was enhanced by introducing H-defect and dopants.29 Therefore, it is worth investigating the sensitivity to gas molecules of graphane with defects and dopants.
In this work, we systematically investigated the geometry, adsorption stability, electronic and magnetic properties of HCN adsorption on graphane. The modified graphane substrates considered here were pure, H-defected, and doped graphane, and the dopants were B, N, S and Al. We hope our study provides useful information to the application based on graphane for gas sensing.
2. Computational methods and model
All the DFT calculations were performed by the Dmol3 code.30 We used the generalized gradient approximation (GGA) for exchange-correlation functional, as described by Perdew–Burke–Ernzerhof (PBE).31 Basiuk et al. reported that PBE functional along with Grimme dispersion correction can be suggested as the most suitable for theoretical studies of the interaction between molecules and graphene.32 Therefore, the all results were obtained with Grimme-corrected PBE. We selected DFT semicore pseudopotential (DSSP) to replace core electrons as a single effective potential.33 A double numerical plus polarization (DNP) was employed as the basis set. The DNP basis set corresponds to a double-ζ quality basis set with a p-type polarization functions to hydrogen and d-type polarization functions added to heavier atoms, which was comparable with the Gaussian 6-31G (d, p) basis set and exhibited a better accuracy.34
We applied 5 × 5 × 1 supercell with periodic boundary condition on the x and y axes to model the infinite graphene sheet. The vacuum space of 20 Å was set in the direction normal to the sheets to avoid the interactions between periodic images. A 15 × 15 × 1 mesh of k-point and the global orbital cutoff of 5.0 Å were set in the spin-unrestricted calculations. All atoms are allowed to relax. Convergence in energy, force, and displacement were set at 2 × 10−5 Ha, 0.004 Ha Å−1, and 0.005 Å, respectively.
We analyzed the adsorption of HCN on hydrogenated graphene (HG). The adsorption energy was obtained from the expression
| Eads = E(HCN/HG) − E(HG) − E(HCN) |
where
E(HCN/HG) is the total energy of HCN molecule adsorbed on hydrogenated graphene,
E(HG) is the total energy of the HG, and
E(HCN) is the total energy of a free HCN molecule, respectively.
3. Results and discussion
3.1 Structures of HG, VHG, BVHG, NVHG, SVHG, and AlVHG
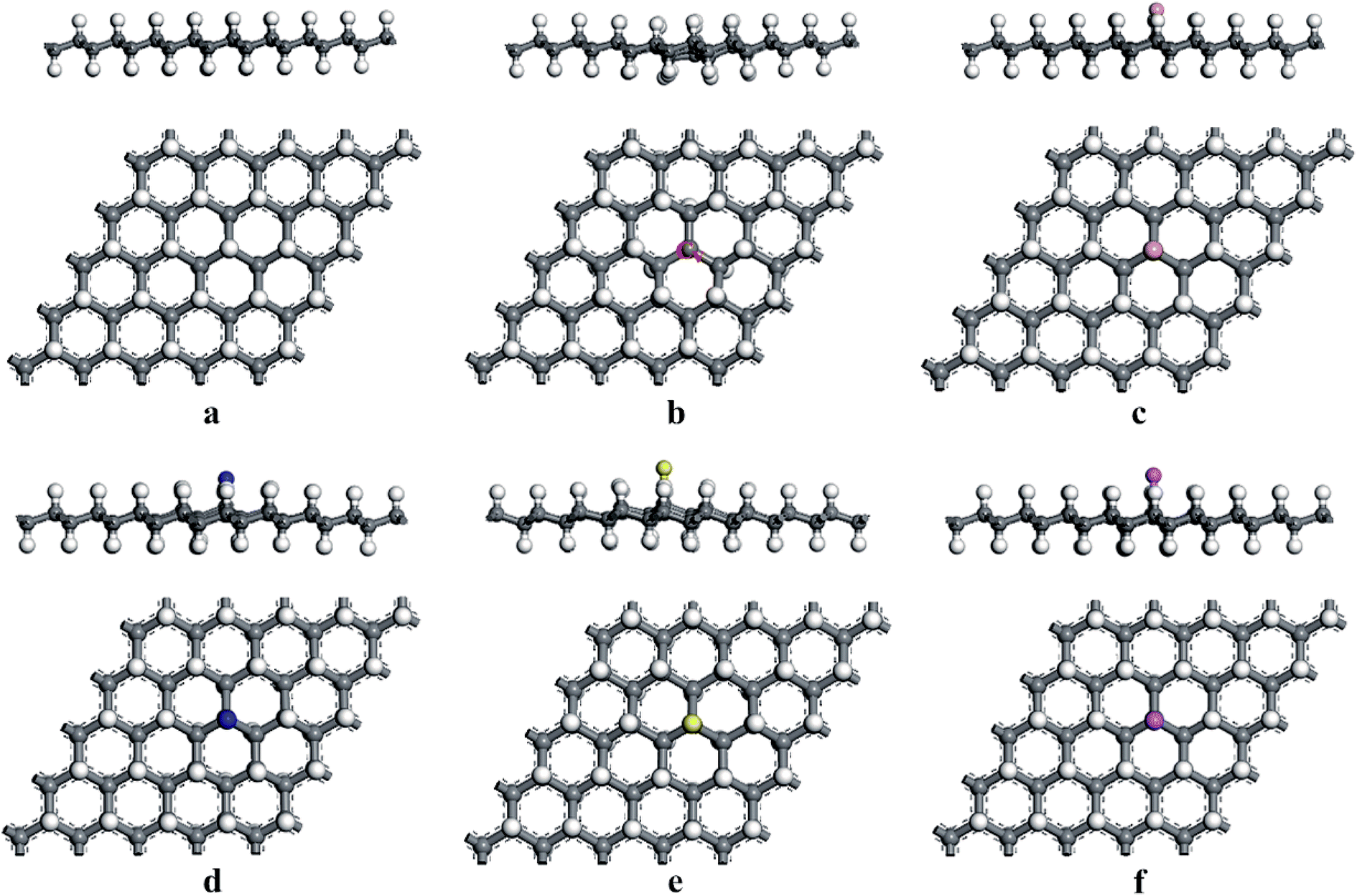
At first, we considered the optimized geometries of the graphane (HG) and H-vacancy defected graphane (VHG), and the relaxed structures were shown in Fig. 1. In the HG, the bond lengths of C–C and C–H were 1.496 Å and 1.109 Å, which was in accord with previous report.35 The H-vacancy was created by removing an H atom from the perfect hydrogenated graphene. Experimentally, the appearance of H-vacancy is very common during the synthetic process, while the graphene is exposed in the hydrogen plasma. After fully relaxation, the Cv atom, which lost a H atom, exhibited unpaired localized electron due to the appearance of H-vacancy. As shown in Fig. 1b, the Cv was labeled. The calculated results indicated that the bond length of C–C around the Cv atom decreased to 1.469 Å, which was consistent with previous literatures.25,29
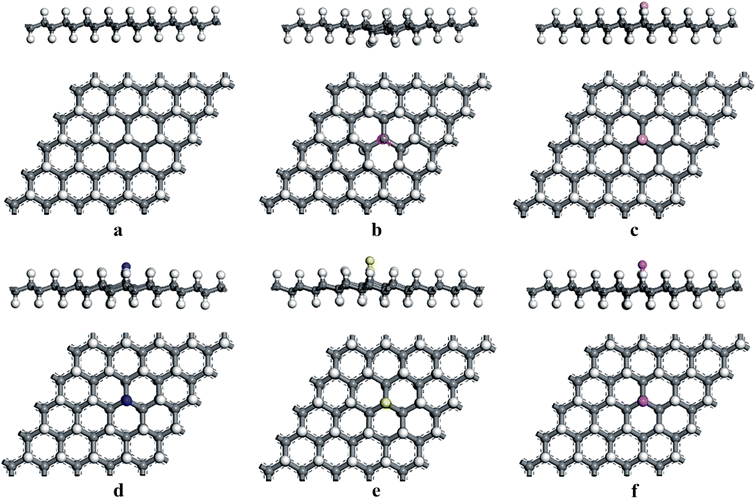 |
| | Fig. 1 The optimized structures of (a) HG, (b) VHG, (c) BVHG, (d) NVHG, (e) SVHG, and (f) AlVHG. | |
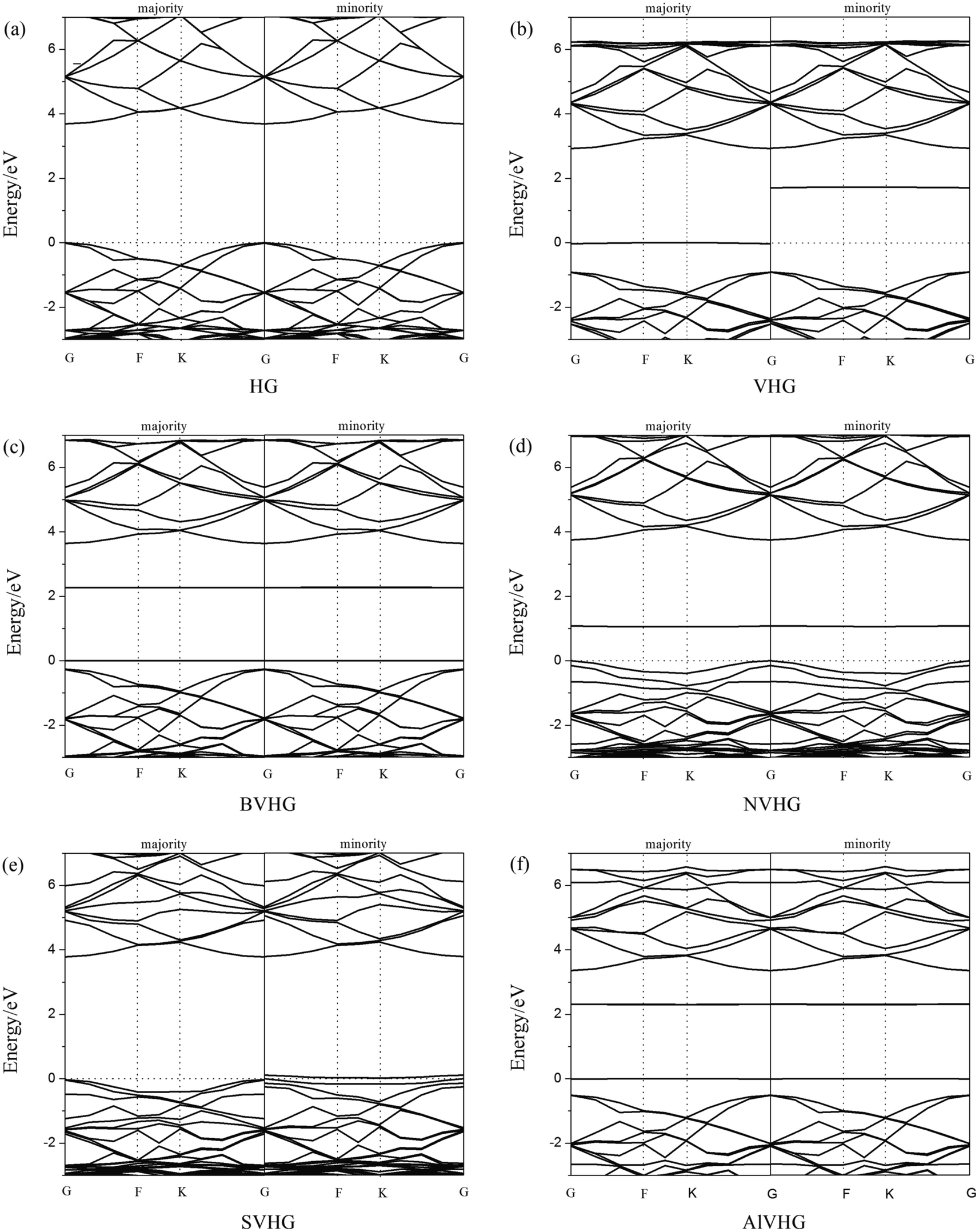
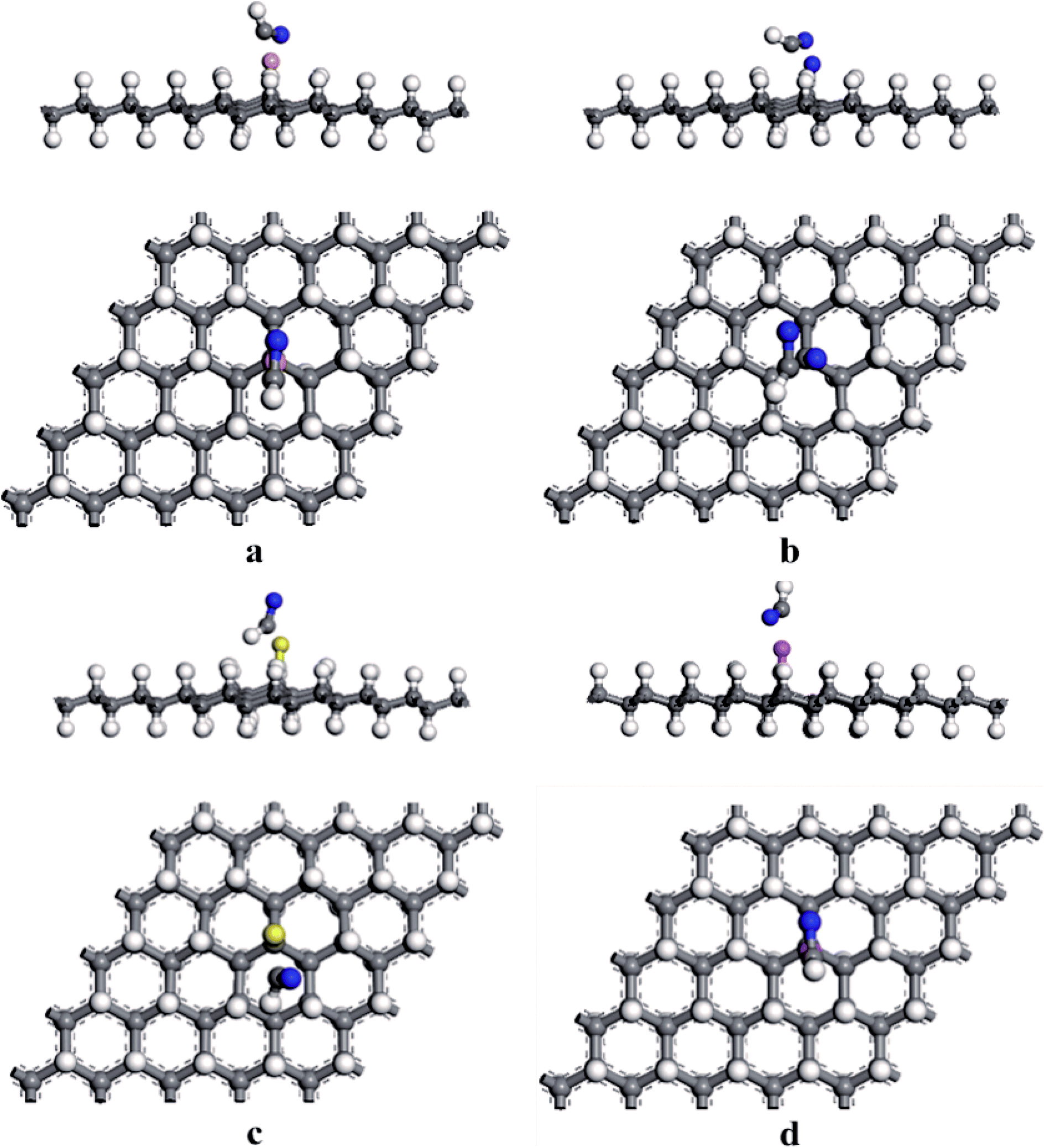
According to previous results, the appearance of H-vacancy increased the activity of graphane, and the Cv site exhibited high sensitivity towards foreign atoms or molecules. Thus, we considered the introducing of dopants to VHG, and the dopants included B, N, S, and Al atoms. The optimized doped configurations were performed in Fig. 1c–f. Furthermore, the calculated results suggested that the binding energies of B, N, S, and Al were 2.80 eV, 2.23 eV, 2.55 eV and 3.37 eV, respectively. The calculated Eb were larger than their cohesive energies, which enhanced the stability of the geometry. To explore the influence of H-vacancy and dopants on the graphane, we performed the band structures of the configurations in Fig. 2, and the spin-up (majority) and spin-down (minority) bands were separated in each case. For HG (Fig. 1a), both band structures were symmetrical, and the band gap was 3.686 eV, which was in agreement with the results of Leenaerts et al.36 and Islam et al.29 In the case of VHG, the majority and minority bands were mismatched and the band gap decreased to 1.704 eV, which indicated that the structure exhibited net magnetic moments. The appearance of H-vacancy released the p electron on the C atom of the graphane and formed a localized state possessing the 0.995 μB, which induced nonzero magnetization and was consistent with the result of Berashevich et al.23 Furthermore, the spin-up bands showed metallic and the spin-down bands exhibited semiconductivity generating a half-metallic character in the band structure of VHG. After the introducing of dopants, the BVHG, NVHG, and AlVHG were nonmagnetic, which were resulted from the unpaired electron of dopants interacted with the localized electron of Cv atom. Moreover, the band gaps for the BVHG, NVHG, and AlVHG were 2.68 eV, 1.056 eV, and 2.296 eV, respectively. In the case of SVHG, the Fermi level located within the conduction bands in the spin-down bands, while the spin-up bands maintained a band gap of 2.78 eV. Namely, a half-metallic behavior was observed. All in all, the impurity bands appeared around the Fermi level in the doped systems, and then the activity of graphane substrate was expected to be improved to interact with adsorbents.
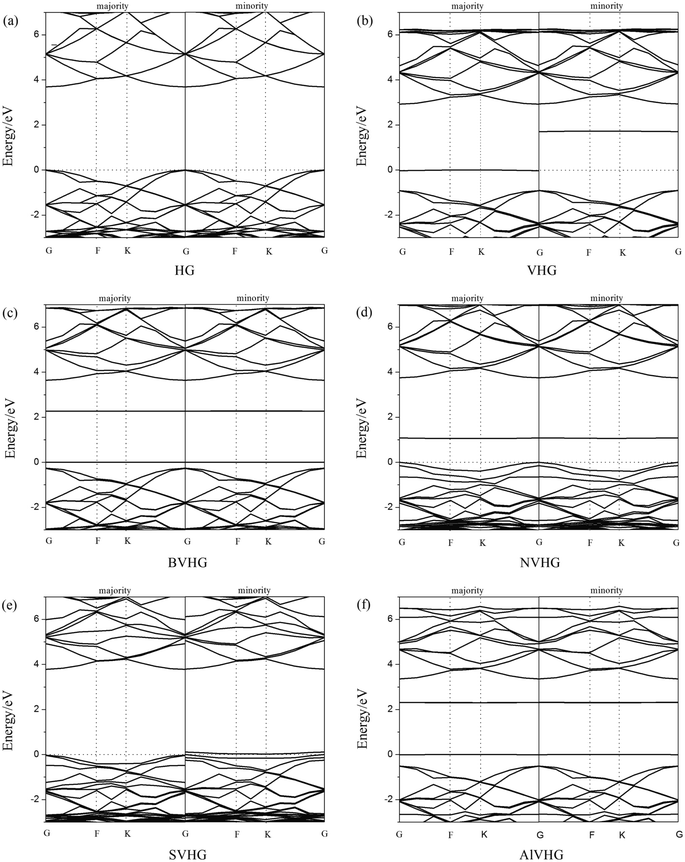 |
| | Fig. 2 The band structures of (a) HG, (b) VHG, (c) BVHG, (d) NVHG, (e) SVHG, and (f) AlVHG. | |
3.2 Adsorption of HCN on HG and VHG
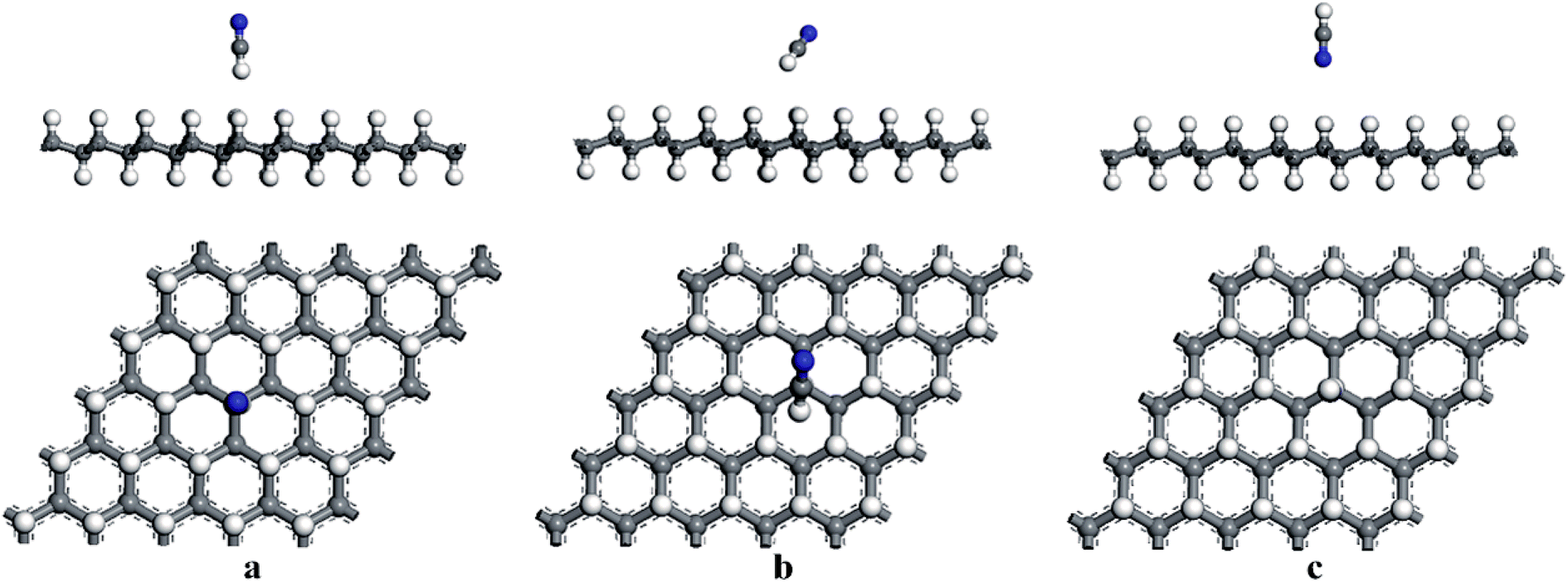
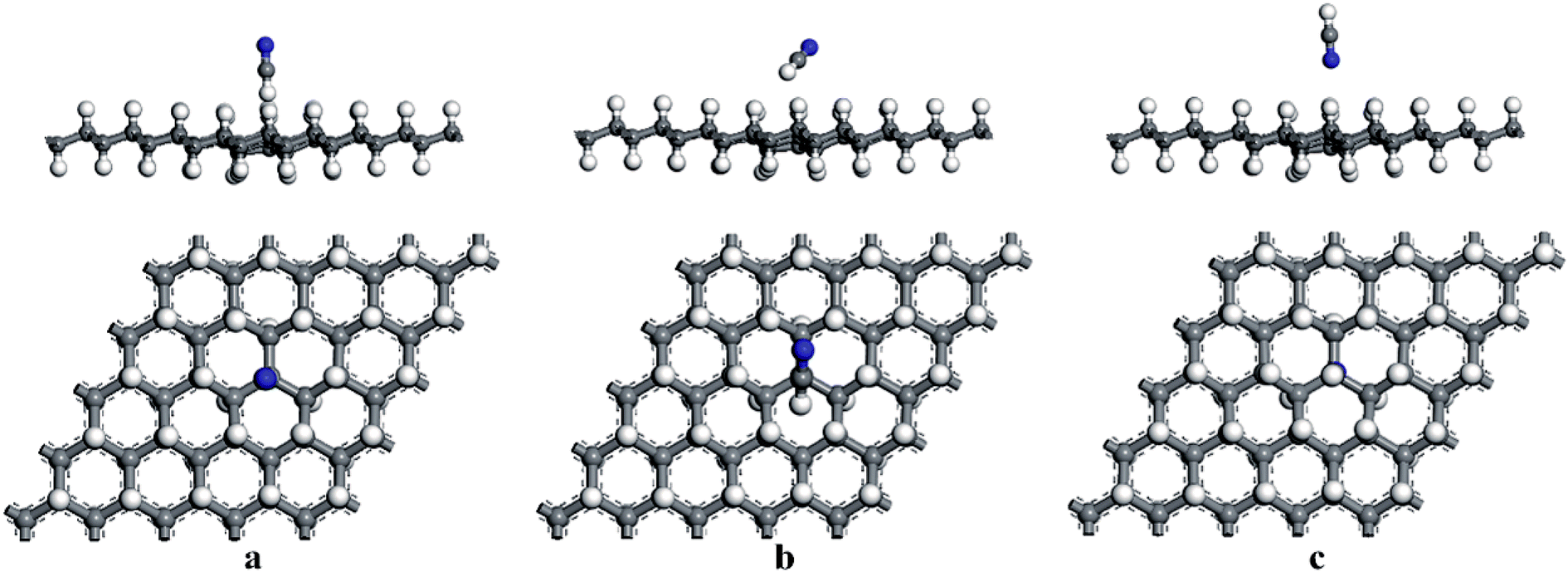
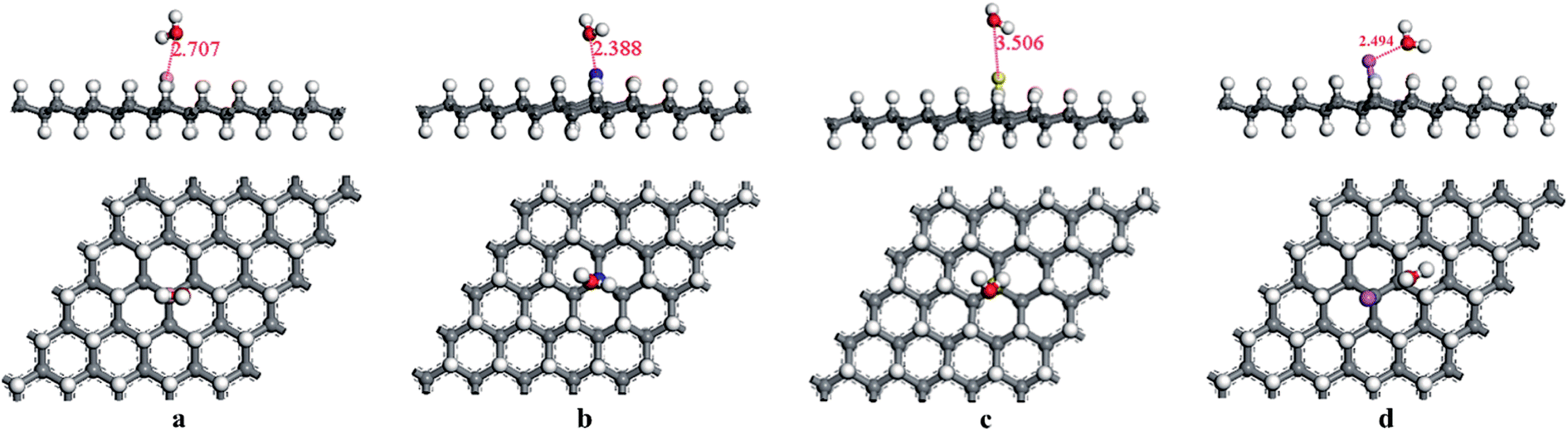
First, the adsorption of HCN molecule on the surface of HG and VHG were investigated. In order to explore the most stable adsorption structure, the initial adsorption structures for HCN molecule were considered as follows: the H atom close to graphane with the HCN molecule perpendicular to the surface (H site), the C atom close to graphane with the HCN molecule parallel to the surface (C site), and the N atom close to graphane with the HCN molecule perpendicular to the surface (N site). The optimized adsorption systems of H–HCN/HG, C–HCN/HG, and N–HCN/HG were performed in Fig. 3. Meanwhile, for the “C site” in VHG, the C atom of HCN molecule was close to the Cv atom of VHG. After optimization, the adsorption structures of HCN on VHG were also showed in Fig. 4. Furthermore, the calculated bond lengths of HCN molecule, adsorption distance between the HCN and the graphane, and the adsorption energy were summarized in Table 1. In the cases of HCN/HG, the adsorption energies were −0.019 eV, −0.021 eV, and −0.011 eV, respectively. Compared with HCN/HG, the adsorption of HCN molecule on VHG exhibited higher stability. The results indicated that the C–HCN/VHG was the most stable system, of which the Eads was −0.088 eV. The larger adsorption energies indicated that the appearance of H-vacancy in graphane improved the activity of graphane substrate. For the two adsorption substrates, the “C site” systems exhibited most stability than the other two adsorption structures. However, the relative large adsorption distance and small adsorption energies showed that the interaction between the HCN molecule and the HG and VHG surface were physisorption.
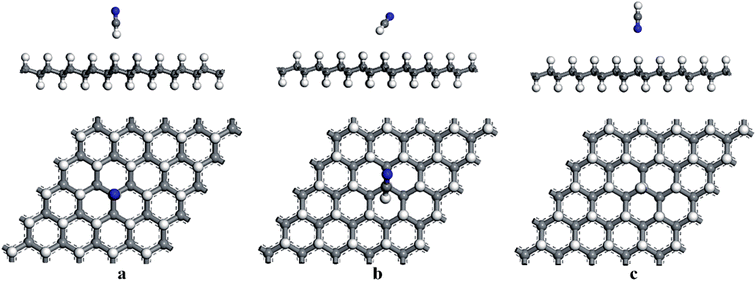 |
| | Fig. 3 The optimized structures of (a) H–HCN/HG, (b) C–HCN/HG, and (c) N–HCN/HG. | |
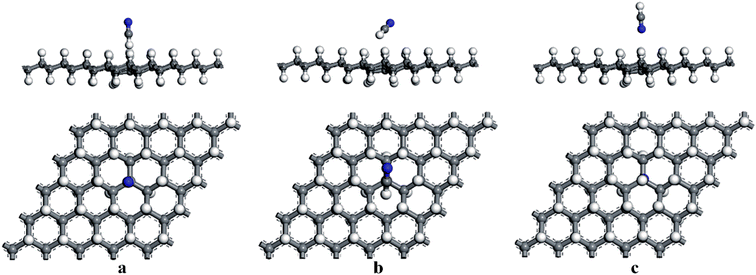 |
| | Fig. 4 The optimized structures of (a) H–HCN/VHG, (b) C–HCN/VHG, and (c) N–HCN/VHG. | |
Table 1 Summary of the results for the HCN molecule adsorbed on pure graphane (HG) and H-vacancy defected graphane (VHG). The properties listed are as follows: the distance between the H and C atom of HCN molecule (d1), the distance between the N and C atom of HCN molecule (d2), the distance between the molecule HCN and graphane surface (dM–G), the adsorption energy (Eads)
| System |
Site |
d1 (Å) |
d2 (Å) |
dM–G (Å) |
Eads (eV) |
| HCN/HG |
H |
1.078 |
1.163 |
3.239 |
−0.019 |
| C |
1.077 |
1.163 |
3.207 |
−0.021 |
| N |
1.076 |
1.162 |
3.686 |
−0.011 |
| HCN/VHG |
H |
1.087 |
1.163 |
3.583 |
−0.048 |
| C |
1.077 |
1.162 |
2.568 |
−0.088 |
| N |
1.076 |
1.162 |
3.794 |
−0.027 |
3.3 Adsorption of HCN on BVHG, NVHG, SVHG, and AlVHG
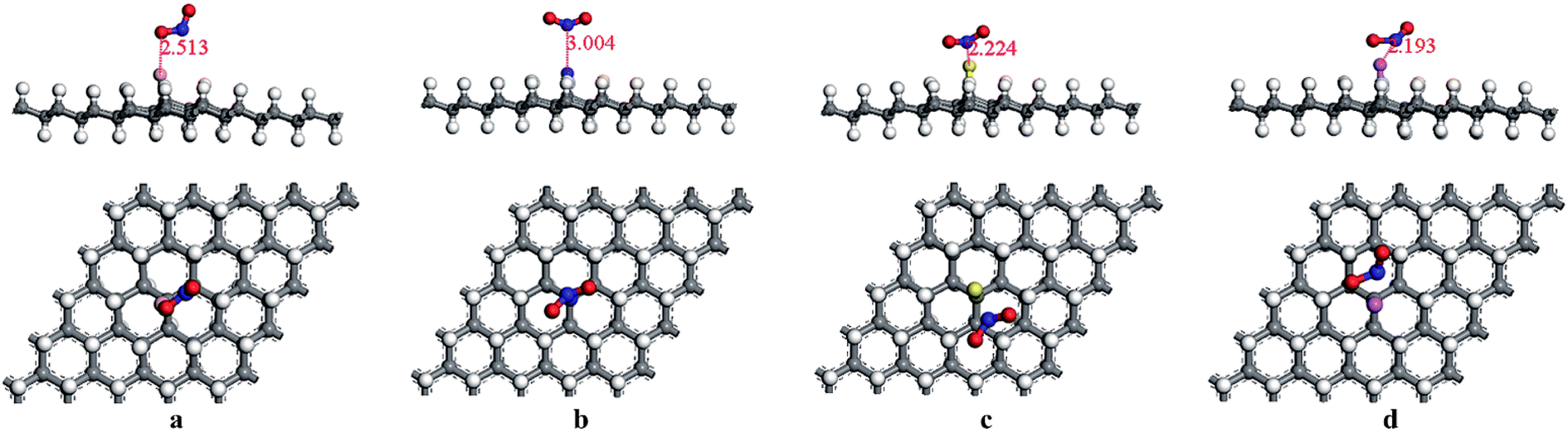
According to the discussion in 3.2, for the adsorption of HCN on HG and VHG, the initial adsorption system for “C site” was the most energetically favorable system. Thus, we set the HCN molecule parallel to the doped graphane (BVHG, NVHG, SVHG, and AlVHG) with the C atom close to the dopants in the adsorption configurations. The optimized configurations of HCN/BVHG, HCN/NVHG, HCN/SVHG, and HCN/AlVHG were shown in Fig. 5. Furthermore, the corresponding parameters detailedly describing the adsorption systems were summarized in Table 2. As shown in Fig. 5, the HCN molecule did not maintain the linear bond structure after interacting with the doped graphane. The bond lengths of C atom of HCN molecule and the dopants were 1.406 Å, 1.381 Å, 1.796 Å, and 1.867 Å for the adsorption complexes, respectively. As performed in Table 2, the HCN/NVHG exhibited the largest Eads with the value of −1.144 eV. Meanwhile, the adsorption energies varied from −0.115 eV to −1.144 eV, which were all larger than that of HG and VHG. The results suggested that the introducing of dopants (B, N, S, and Al) improved the sensitivity of graphane to HCN gas molecules. Moreover, the charge transfer of HCN molecule was analyzed by Hirshfeld method which seems to be more reliable and yield chemically meaningful charges in comparison with Mulliken method.37,38 Take the case of HCN/NVHG as example, the N-dopant accepted 0.359 e and the HCN molecule lost 0.125 e, which indicated that some electrons transferred from HCN molecule and graphane substrate to the N-dopant, illustrating that the dopants can facilitate the interaction between the HCN molecule and the graphane substrate.
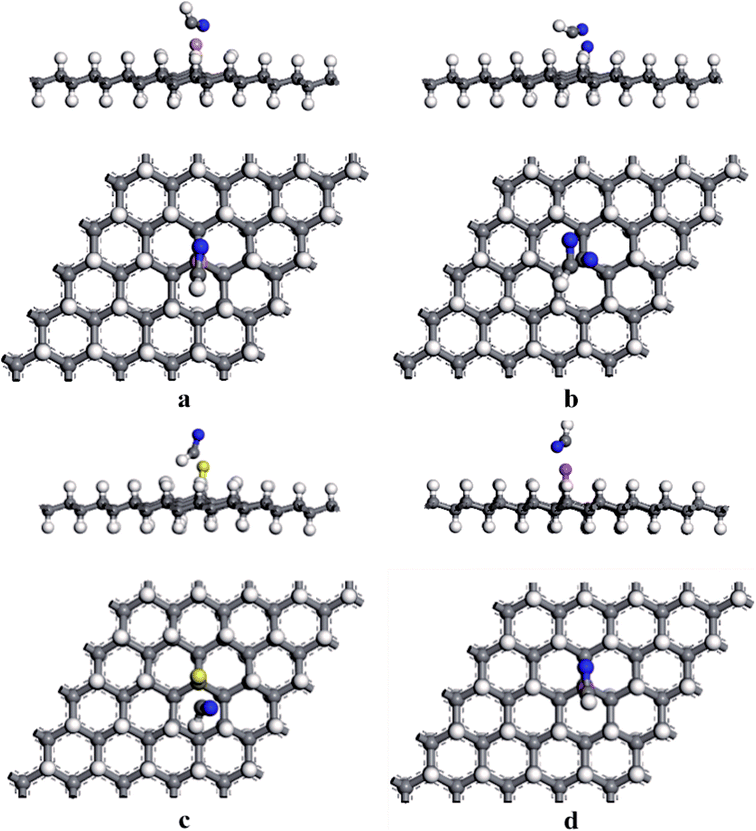 |
| | Fig. 5 The optimized adsorption structures of (a) HCN/BVHG, (b) HCN/NVHG, (c) HCN/SVHG, and (d) HCN/AlVHG. | |
Table 2 Summary of the results for the HCN molecule adsorbed on VHG with dopants (B, N, S, Al). The properties listed are as follows: the distance between the H and C atom of HCN molecule (d1), the distance between the N and C atom of HCN molecule (d2), the bond length of Cv atom and the dopant (d3), the distance between the molecule C atom of HCN molecule and dopant (d4), the adsorption energy (Eads), the band gap of adsorption system (Eg), and the charge transfer of the HCN molecule (Q)
| System |
d1 (Å) |
d2 (Å) |
d3 (Å) |
d4 (Å) |
Eads (eV) |
Eg (eV) |
Q (e) |
| HCN/BVHG |
1.095 |
1.315 |
1.575 |
1.406 |
−1.040 |
2.400 |
−0.121 |
| HCN/NVHG |
1.093 |
1.258 |
1.496 |
1.381 |
−1.144 |
2.426 |
0.125 |
| HCN/SVHG |
1.101 |
1.254 |
1.896 |
1.796 |
−0.115 |
0.821 |
−0.036 |
| HCN/AlVHG |
1.090 |
1.300 |
2.015 |
1.867 |
−0.412 |
2.406 |
−0.394 |
As shown in Table 2, we calculated the band gaps of the adsorption systems. The adsorption of HCN induced a band open to the configurations. In case of NVHG, the band gap changed from 1.056 eV to 2.426 eV by the interaction with the HCN molecule. For other cases, the band gaps of BVHG (2.268 eV), SVHG (0.020 eV), and AlVHG (2.296 eV) increased to 2.400 eV, 0.821 eV, and 2.406 eV, respectively. It is well known the change of Eg induces the electric conductivity change of graphane substrate according to the following equation:39
where
σ is the electric conductivity of the configurations,
k is the Boltzmann's constant, and
T is the thermodynamic temperature. Therefore, the change of conductance of graphane substrate can be seen as signal to detect HCN gas molecules.
Moreover, the adsorptions of H2O and NO2 molecules on the modified graphane substrates were investigated, aiming to explore the selectivity of the structures used as sensors to detect the HCN gas. The most stable final adsorption configurations were performed in Fig. 6 and 7. The adsorption energies of the H2O molecules on BVHG, NVHG, SVHG, and AlVHG were −0.012 eV, −0.030 eV, −0.007 eV, and −0.025 eV, respectively. The interaction distances were labeled in Fig. 6. According to the calculated results, the small adsorption energies and large bond distances indicated that the adsorption were physisorptions. Furthermore, the modified substrates were slightly changed by the adsorption of H2O molecule. Compared with the case of HCN, the presence of H2O molecules cannot interfere in the detection of HCN.
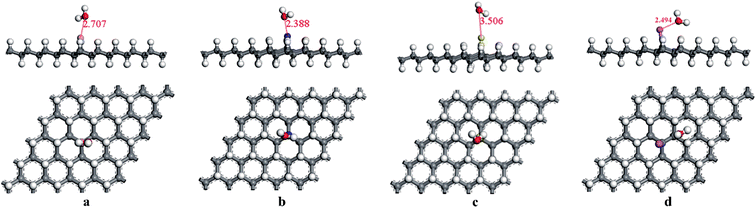 |
| | Fig. 6 The optimized adsorption structures of (a) H2O/BVHG, (b) H2O/NVHG, (c) H2O/SVHG, and (d) H2O/AlVHG. | |
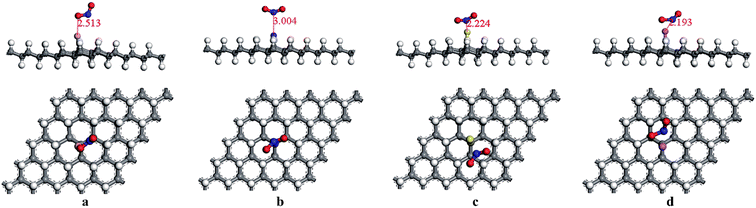 |
| | Fig. 7 The optimized adsorption structures of (a) NO2/BVHG, (b) NO2/NVHG, (c) NO2/SVHG, and (d) NO2/AlVHG. | |
The most stable structure of the NO2 adsorption systems was NO2/AlVHG, and the Eads was −0.105 eV and the band gap changed by 0.076 eV. The interaction was somewhat strong, though the change of band gap was smaller than the case of HCN. Therefore, we think that the presence of NO2 gas molecule may interfere in detecting of the HCN gas.
3.4 Electronic properties
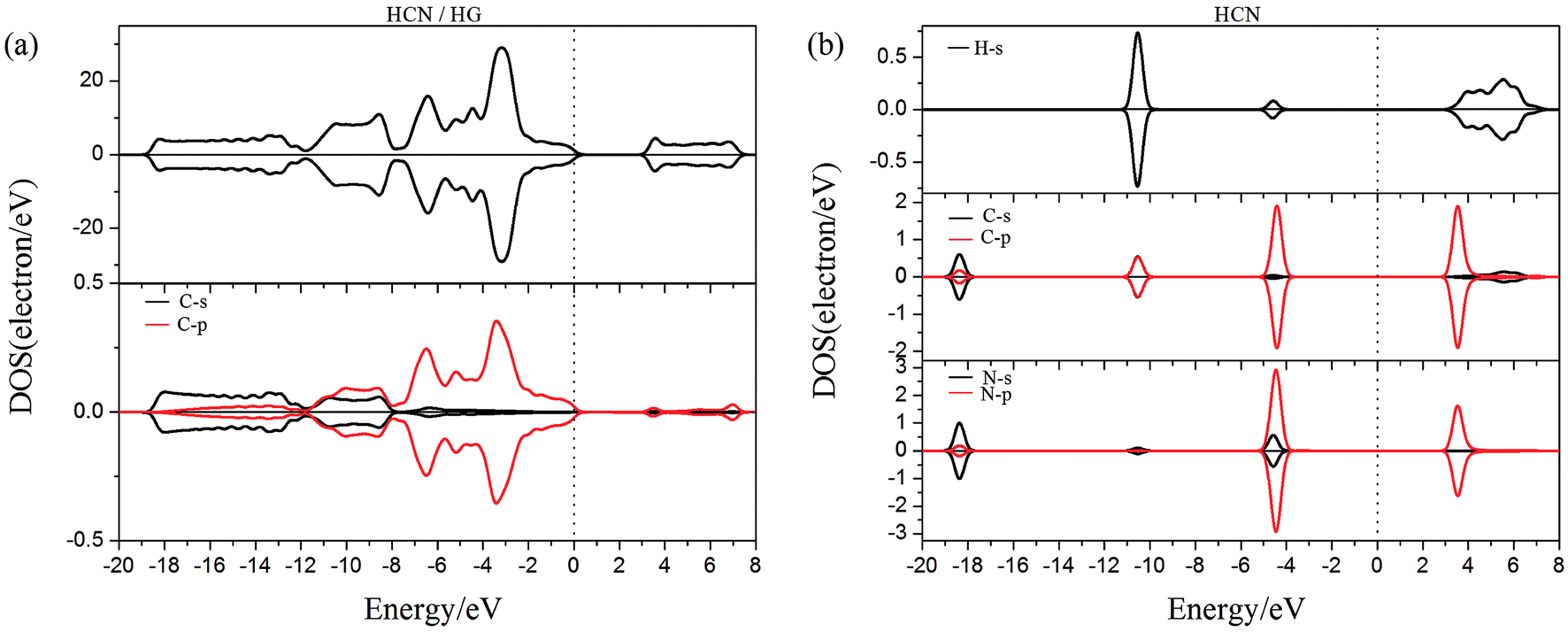
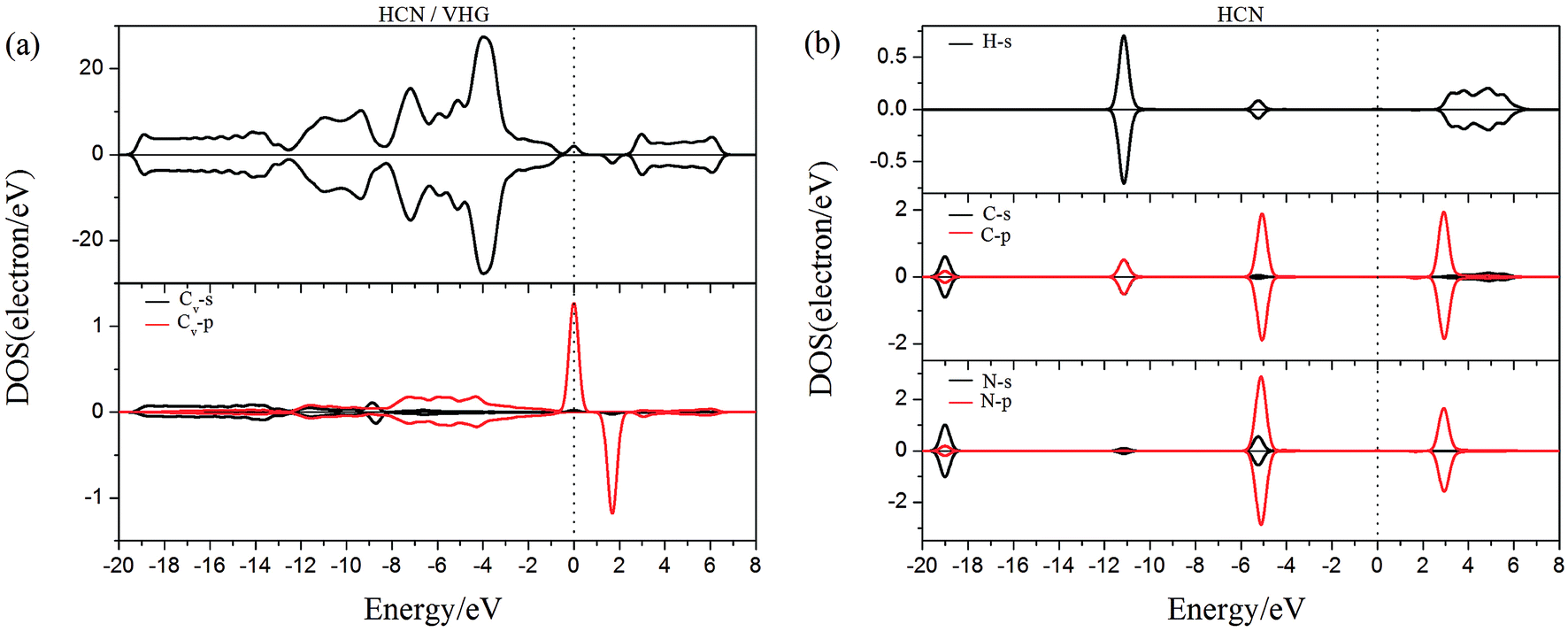
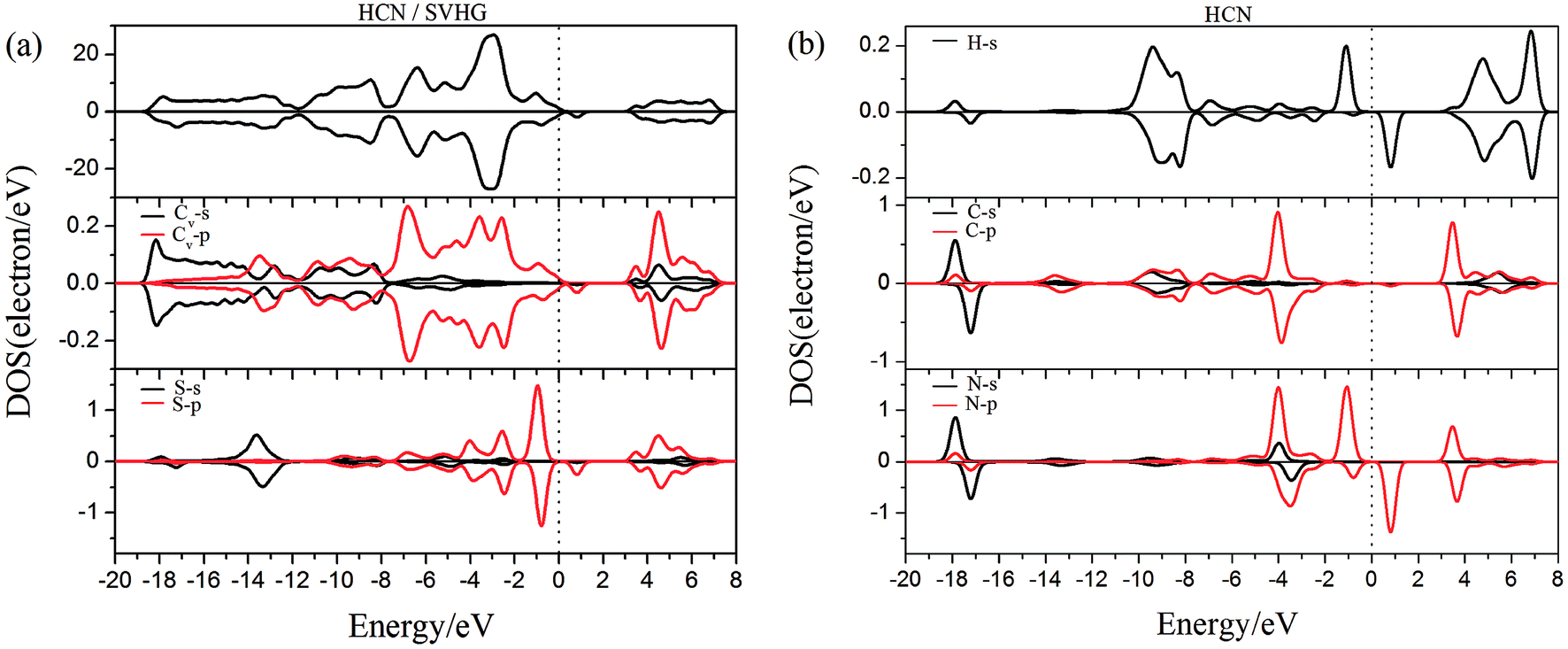
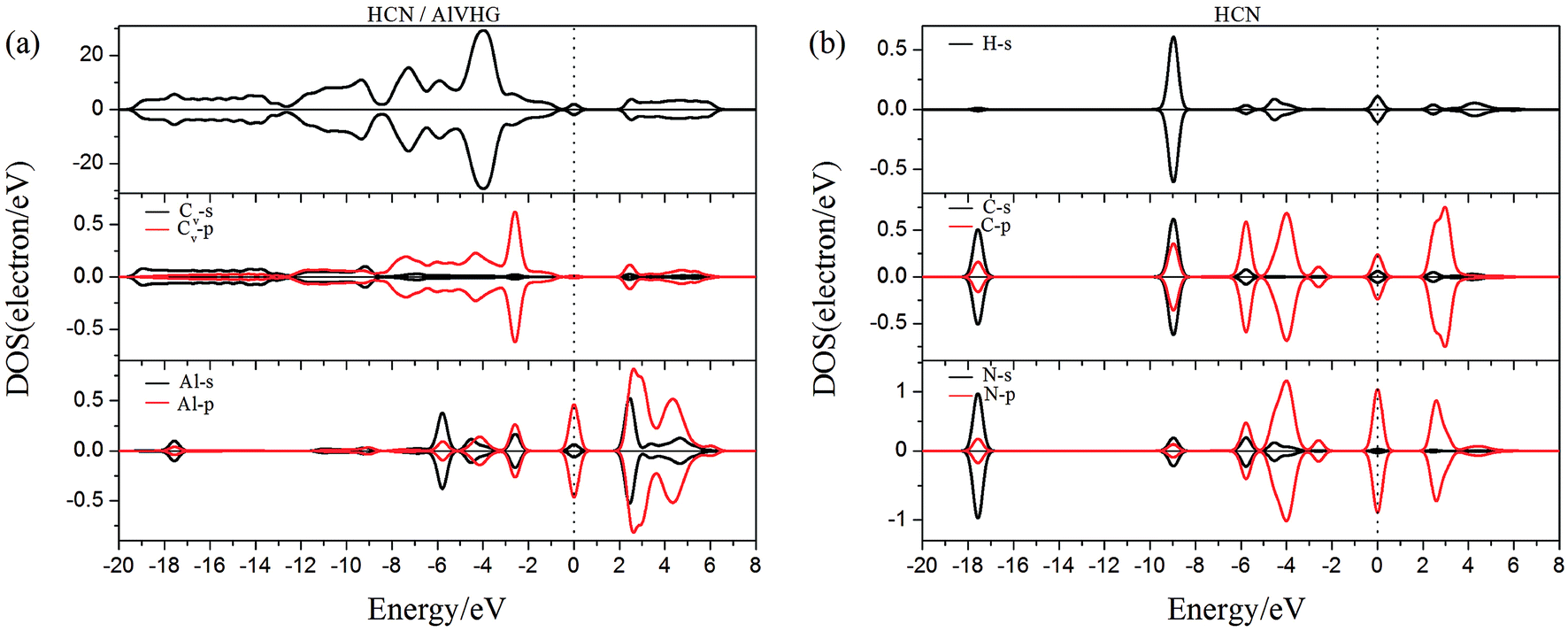
To gain more insight into the effect of the H-vacancy and dopants on the adsorption of HCN molecule on the graphane, we investigated the change of electronic structures by performing the total density of states (TDOS) and spin polarized density of states (PDOS) of the adsorption complexes in Fig. 8–13. As shown in Fig. 8 and 9, the DOS showed that the HCN/HG exhibited nonmagnetic, while the HCN/VHG exhibited magnetic characters. The PDOS of Cv atom in Fig. 9a showed that the H-vacancy induced the localized electron of Cv showed a obvious half-metallic behavior and complete spin-polarized states around the Fermi level. The PDOS plots was consistent with the band structures of VHG performed in Fig. 2b, which implied that the appearance of H-vacancy produced significant change in the electronic and magnetic properties of HG. Furthermore, a few overlaps between the HCN molecule and the HG and VHG substrates, which indicated that the interaction were very weak.
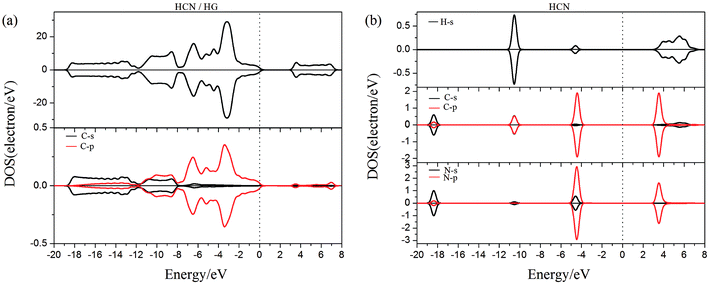 |
| | Fig. 8 The PDOS of configurations of (a) HCN/HG and (b) HCN molecule. | |
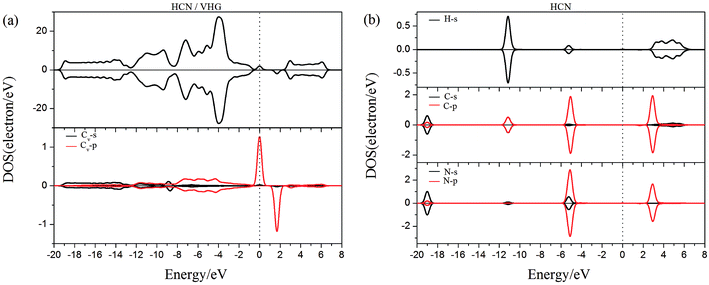 |
| | Fig. 9 The PDOS of configurations of (a) HCN/VHG and (b) HCN molecule. | |
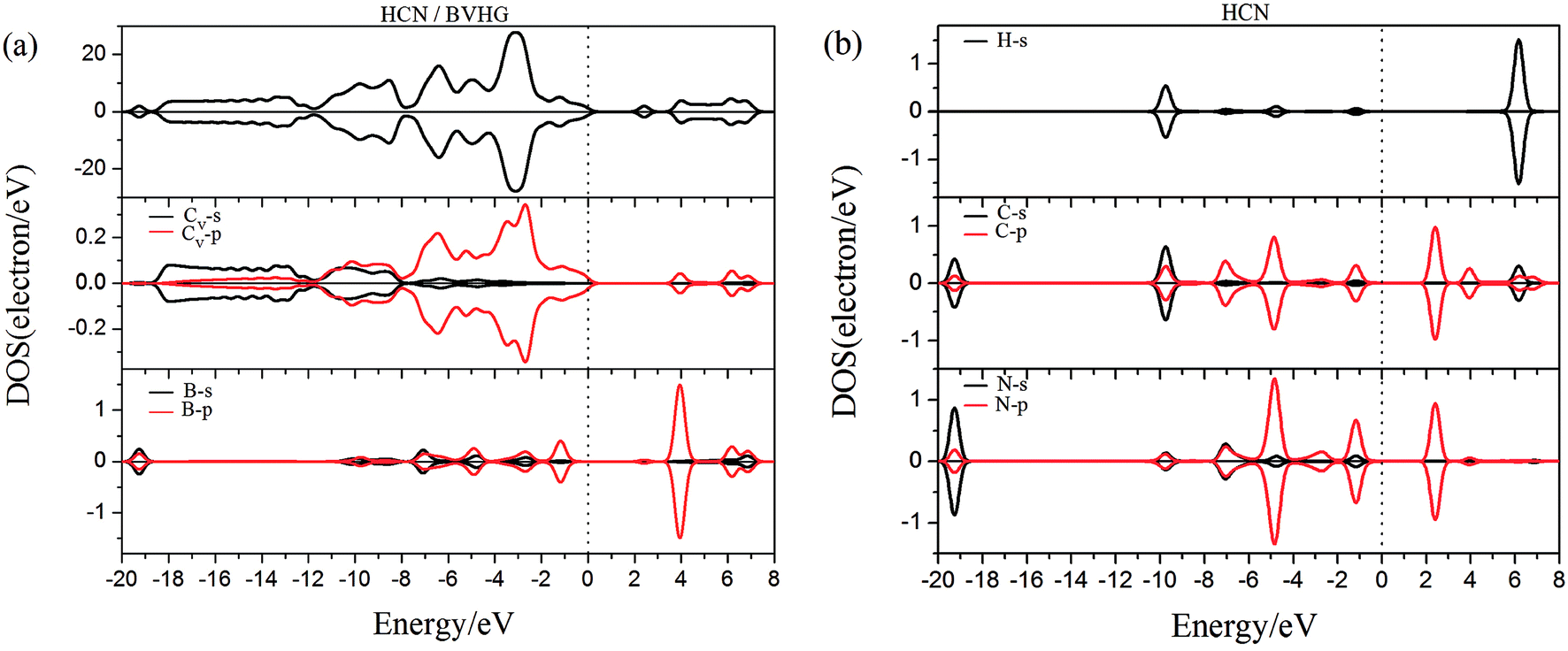
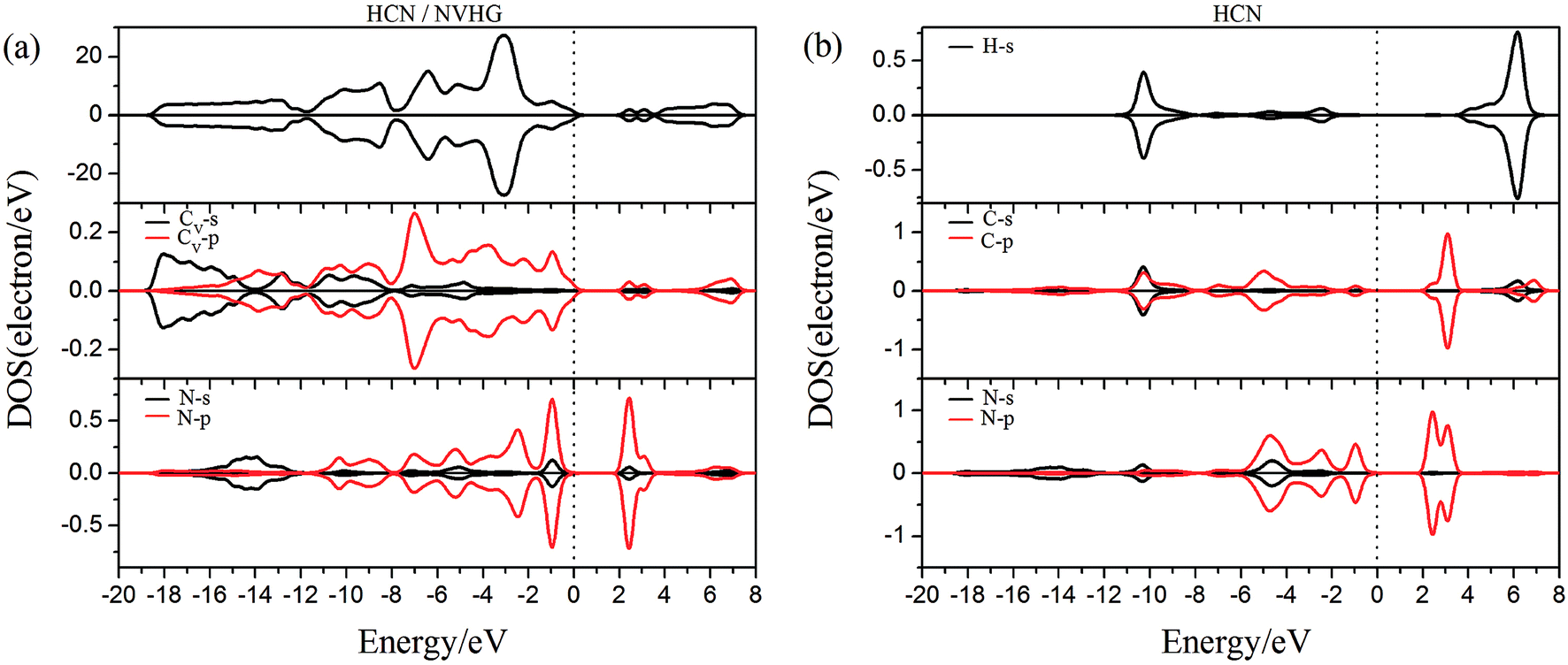
As performed in Fig. 10a–13a, a large area of hybridization between the dopants (B, N, S, Al) and the Cv atom was observed below and above the Fermi level. It was well agreement with the large binding energies of the VHG for the dopants and chemical bonds were formed between the dopants and Cv atom. Furthermore, the HCN/BVHG, HCN/NVHG, and HCN/AlVHG exhibited nonmagnetic according to the symmetry of spin channels, while the HCN/SVHG exhibited net magnetic moment. Compared with the structures without absorbing HCN molecules, the results suggested that the adsorption of HCN hardly influence the magnetic character of the VHG with dopants. In the case of HCN/NVHG, the most stable adsorption complex, the obvious hybridization between the N-2p orbital of substrate and C-2p orbital of HCN molecule below the Fermi level indicated that their strong interaction, which was responsible for the large adsorption energy of HCN molecule. As shown in Fig. 12, the spin-up and spin-down channels of HCN molecule became asymmetric and then the HCN/SVHG configuration exhibited magnetic property with the net moment of 0.998 μB. Furthermore, the spin-down channel exhibited nearly zero-gap semiconductivity, while the spin-up channel showed non-zero DOS at the Fermi level. The result indicated that the HCN/SVHG system maintained the property of half-metallicity. Based on the above discussions, the HCN molecule on the doped HG exhibited higher adsorption stability than that of HG and VHG. The PDOS plots indicated that the large hybridization between the HCN molecule and dopants, which meant that the strong adsorption of HCN on the doped substrates.
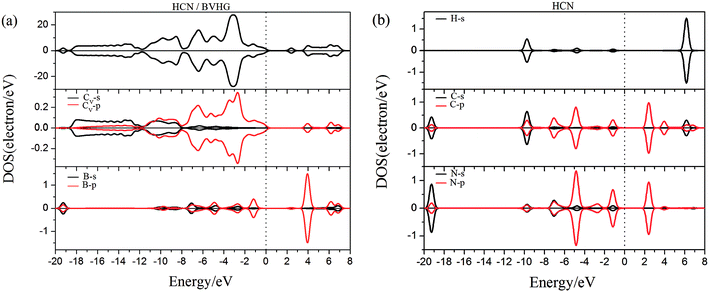 |
| | Fig. 10 The PDOS of configurations of (a) HCN/BVHG and (b) HCN molecule. | |
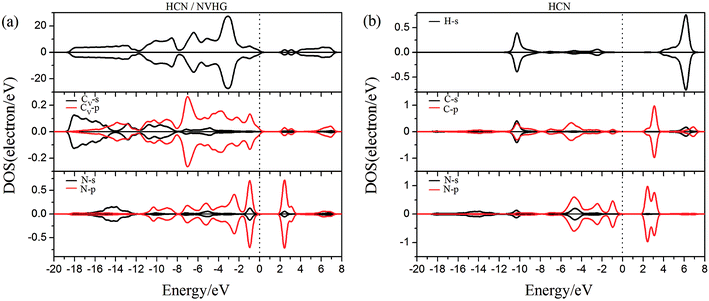 |
| | Fig. 11 The PDOS of configurations of (a) HCN/NVHG and (b) HCN molecule. | |
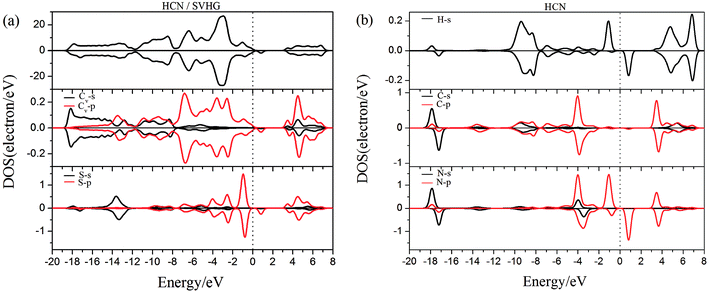 |
| | Fig. 12 The PDOS of configurations of (a) HCN/SVHG and (b) HCN molecule. | |
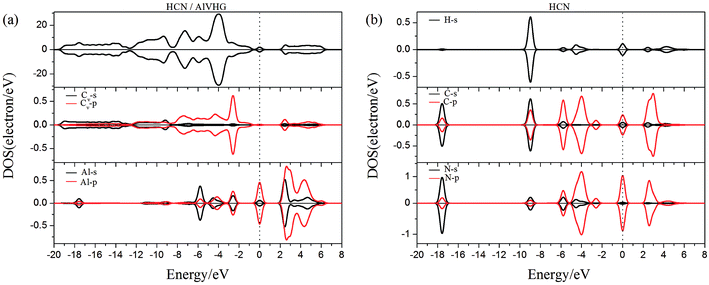 |
| | Fig. 13 The PDOS of configurations of (a) HCN/AlVHG and (b) HCN molecule. | |
4. Conclusions
Based on the first-principles calculations, we investigated the stable structures of hydrogenated graphene (graphane) doped with B, N, S, and Al, which used as potential gas sensing device to detect HCN molecules. The results indicated that the appearance of H-vacancy improved the activity of graphane and induced a half-metallicity behavior. However, the weak interaction between the HCN molecule and the HG and VHG substrates illustrated the adsorption was physisorption. After introducing the dopants, the chemical bonds formed between the dopants and the Cv atom. Moreover, the PDOS results suggested that the impurity peaks appeared around the Fermi level, which indicated that the dopants improved the sensitivity to adsorbents. The large adsorption energies and small bond distance indicated that the adsorption of HCN on doped HG substrates transferred into chemisorption. Furthermore, the adsorption of HCN induced a band gap open on the BVHG, NVHG, SVHG, and AlVHG systems, which could change the conductance of configurations. Therefore, the controlling of electronic and magnetic properties of graphane by the adsorption of HCN molecules is expected to provide meaningful approach for designing HCN gas sensing device.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (NSFC, Grant No. 11604080, 11404096, U1404609, 11304080, 11547153) and the Innovation Team of Henan University of Science and Technology (No. 2015XTD001).
References
- Y. Qin, X. Sun, X. Li and M. Hu, Sens. Actuators, B, 2012, 162, 244–250 CrossRef CAS
 .
. - S. Capone, A. Forleo, L. Francioso, R. Rella, P. Siciliano, J. Spadavecchia, D. S. Presicce and A. M. Taurino, ChemInform, 2004, 35, 1335–1348 CrossRef .
- F. Hossein-Babaei and A. Amini, Sens. Actuators, B, 2012, 166, 419–425 CrossRef .
- R. Wang, D. Zhang, Y. Liu and C. Liu, Nanotechnology, 2009, 20, 505704 CrossRef PubMed .
- A. Zabardasti, A. Kakanejadifard and M. Ghasemian, Struct. Chem., 2012, 23, 1155–1161 CrossRef CAS .
- N. Barsan, D. Koziej and U. Weimar, Sens. Actuators, B, 2007, 121, 18–35 CrossRef CAS .
- A. Kolmakov and M. Moskovits, Annu. Rev. Mater. Res., 2004, 35, 151–180 CrossRef .
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed .
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed .
- M. Sun and Y. Peng, Appl. Surf. Sci., 2014, 307, 158–164 CrossRef CAS .
- Z. Dai and Y. Zhao, Appl. Surf. Sci., 2014, 305, 382–385 CrossRef CAS .
- Y. Tang, W. Chen, C. Li, L. Pan, X. Dai and D. Ma, Appl. Surf. Sci., 2015, 342, 191–199 CrossRef CAS .
- J. W. Gu, C. Liang, X. Zhao, B. Gan, H. Qiu, Y. Guo, X. Yang, Q. Zhang and D. Y. Wang, Compos. Sci. Technol., 2017, 139, 83–89 CrossRef CAS .
- X. Zhang, L. Yu, Y. Gui and W. Hu, Appl. Surf. Sci., 2016, 367, 259–269 CrossRef CAS .
- A. S. Rad and E. Abedini, Appl. Surf. Sci., 2016, 360, 1041–1046 CrossRef CAS .
- B. S. Li, P. W. Yong and K. D. Hai, Appl. Surf. Sci., 2015, 329, 330–336 CrossRef .
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson and A. K. Geim, Science, 2009, 323, 610–613 CrossRef CAS PubMed .
- H. Şahin, C. Ataca and S. Ciraci, Appl. Phys. Lett., 2009, 95, 222510 CrossRef .
- T. Hussain, B. Pathak, M. Ramzan, T. A. Maark and R. Ahuja, Appl. Phys. Lett., 2012, 100, 302–304 CrossRef .
- S. Haldar, D. G. Kanhere and B. Sanyal, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155426 CrossRef .
- T. Hussain, A. D. Sarkar and R. Ahuja, Appl. Phys. Lett., 2012, 101, 103907 CrossRef .
- C. Zhou, S. Chen, J. Lou, J. Wang, Q. Yang, C. Liu, D. Huang and T. Zhu, Nanoscale Res. Lett., 2014, 9, 26 CrossRef PubMed .
- J. Berashevich and T. Chakraborty, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 876–877 CrossRef .
- B. Julia and C. Tapash, Nanotechnology, 2010, 21, 355201 CrossRef PubMed .
- R. E. Mapasha, M. P. Molepo and N. Chetty, Phys. E, 2016, 79, 52–58 CrossRef CAS .
- C. K. Yang, Carbon, 2010, 48, 3901–3905 CrossRef CAS .
- T. Hussain, P. Panigrahi and R. Ahuja, Phys. Chem. Chem. Phys., 2014, 16, 8100–8105 RSC .
- H. Tanveer, P. Puspamitra and A. Rajeev, Nanotechnology, 2014, 25, 325501 CrossRef PubMed .
- M. S. Islam, T. Hussain, G. S. Rao, P. Panigrahi and R. Ahuja, Sens. Actuators, B, 2016, 228, 317–321 CrossRef CAS .
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed .
- V. A. Basiuk and L. V. Henaoholguín, J. Comput. Theor. Nanosci., 2014, 11, 1609–1615 CrossRef CAS .
- D. J. Chadi, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef .
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef .
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef .
- O. Leenaerts, H. Peelaers, A. D. Hernández-Nieves, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195436 CrossRef .
- C. Fonseca Guerra, J. W. Handgraaf, E. J. Baerends and F. M. Bikelhaupt, J. Comput. Chem., 2004, 25, 189 CrossRef PubMed .
- A. E. Clark, J. L. Sonnenberg, P. J. Hay and R. L. Martin, Chem. Phys., 2004, 121, 2563–2570 CAS .
- S. S. Li, Semiconductor physical electronics, Springer, USA, 2nd edn, 2006 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac,
Weiwei Jua,
Xiangying Sua,
Yongliang Yonga,
Xiaohong Lia,
Zhibing Fub and
Chaoyang Wangb
*ac,
Weiwei Jua,
Xiangying Sua,
Yongliang Yonga,
Xiaohong Lia,
Zhibing Fub and
Chaoyang Wangb