
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fatty acid decarboxylation reaction kinetics and pathway of co-conversion with amino acid on supported iron oxide catalysts
Junjie Bian *,
Yue Wang,
Qi Zhang,
Xudong Fang,
Lijuan Feng and
Chunhu Li
*,
Yue Wang,
Qi Zhang,
Xudong Fang,
Lijuan Feng and
Chunhu Li
Key Laboratory of Marine Chemistry Theory and Technology of Ministry of Education, Ocean University of China, Qingdao 266100, Shandong, China. E-mail: junjiebian@ouc.edu.cn
First published on 6th October 2017
Abstract
Fe2O3/Al-MCM-41 nanocomposite catalysts were designed and fabricated to upgrade microalgae hydrothermal liquefaction (HTL)-derived biocrude and its model compounds (palmitic acid and glutamic acid) in the absence of hydrogen. The Fe2O3/Al-MCM-41 catalysts were prepared by encapsulating Fe2O3 particles on highly stable pure silica MCM-41 and Al-MCM-41. XRD, TEM, N2 adsorption isotherms, XPS, and UV-Vis characterizations of the catalysts revealed that 10–30 nm γ-Fe2O3 particles were well dispersed on the mesoporous support. In the range of 320–350 °C and under subcritical water, palmitic acid conversion was improved by 43–54% by the Fe2O3/Al-MCM-41 catalyst. The overall reaction rate was first order to palmitic acid for non-catalytic and catalytic conversions, and the activation energy was 336.76 kJ mol−1 for Fe2O3/Al-MCM-41. In situ DRIFTS reaction test demonstrated that the initiation temperature for the catalytic decarboxylation of palmitic acid was decreased to 195 °C, and desorption of the produced CO2 occurred at 255 °C. Based on the GC-MS results of the HTL products, the reaction pathway of the binary mixture of palmitic acid and glutamic acid was elucidated, and the chemical origins of amide, pyrrolidinone, hydroxy-butanoic acid, and pentadecane were proposed.
1. Introduction
Biomass conversion to biofuel or valuable chemicals offers an approach to sustainable, renewable energy.1–3 Microalgae is unique because of its relatively high photosynthetic efficiency, faster growth rate, and area-specific yield when compared with terrestrial plants.4,5 Thermochemical procedures are feasible for both dry (pyrolysis) and wet biomass conversion (hydrothermal liquefaction, HTL), and the latter can be carried out under mild conditions because there is no lignocellulose in the microalgae cell.6Microalgae HTL is considered an energy-efficient, environmentally friendly process, especially when (sub-) critical water is employed as a solvent and a reactant.7–9 However, the derived biocrude consists of a wide variety of compounds introduced from the decomposition of proteins, lipids, and carbohydrates or from binary mixtures of amino acids and fatty acids, amino acids and carbohydrates, or fatty acids and carbohydrates.10 Because the levels of oxygen and nitrogen contaminants are several times higher in biocrude than in traditional fossil fuels, further upgrading (deoxygenation and denitrification) the HTL-derived oil while simultaneously decreasing its acidity and increasing its stability is crucial.11
Previous researchers have exploited the ability of algae or its hydrolysis products to undergo the HTL process to obtain biocrude with lower oxygen content and higher heat value compared to the feed. Zeolites and mesoporous catalysts have been employed for algae HTL and showed good performance under relatively mild reaction conditions. Various zeolite supported non-sulfide catalysts such as noble metal(s) supported on non-sulfide zeolite catalysts such as noble metal supported on ZSM-5, HZSM-5, SAPO-11, beta-, Y-zeolite, SBA-15 and MCM-41 mesoporous materials showed considerable activity for bio-fuel conversion.12,13 MgO/MCM-41 was used in the catalytic HTL of Dunaliella tertiolecta for the production of bio-oil, and the catalyst was conducive to the improvement of conversion and bio-oil yield.14 The catalytic HTL of Dunaliella tertiolecta was performed on multifunctional SBA-15-based catalysts; high contents of furfural and its derivatives along with low contents of acids, N-containing compounds, and esters were obtained when NH2–SO3H–Co–SBA-15 and SO3H–Co–SBA-15 were used as catalysts.15
Upgrading studies have been employed to improve the quality of algae-derived oil and to eliminate oxygen and/or nitrogen containing compounds. In addition to hydrogenation and hydrotreating, the deoxygenation of oxygenated model compounds (i.e., fatty acids and esters) in the absence of hydrogen provides another efficient approach for upgrading bio-oil. The cost-intensive noble metal (Pt and Pd)-based catalysts showed remarkable performance for decarboxylation and decarbonylation. Savage et al.,16 Fu et al.,17 Strathmann et al.,18 and Kudo et al.19 recently reported the deoxygenation of bio-oil via deCOx and cracking over precious metal-based catalysts. It is necessary to develop alternative catalysts such as less expensive catalysts that have high deoxygenation ability for selective deCOx reaction. Ni nanoparticle catalysts have been investigated for the deoxygenation of fatty acids,20–22 and controlling the sizes, shapes, compositions and surface properties of these catalysts resulted in good performance for lipid conversion. Bian's group employed the optimized Fe2O3/MCM-41 catalyst for chlorella HTL and for the derived bio-oil in situ upgrading. At 342 °C, methyl palmitate conversion was 56%, and the decarboxylation selectivity for pentadecane was improved to 62% on the catalyst in sub-critical water.23 Karnjanakom et al. developed Mg-doped Al-MCM-41 for the in situ catalytic upgrading of bio-oils. Mg/Al-MCM-41 exhibited the highly selective conversion of bio-oils derived from cellulose, lignin, or sunflower stalk to high-value-added aromatic hydrocarbons via catalytic cracking, deoxygenation and aromatization.24 Crocker et al. employed a Ni–Al layered double hydroxide catalyst for the deoxygenation of purified algae oil at 300 °C and obtained diesel-like (C10–C20) hydrocarbons constituting 76 wt% of the liquid after 4 h on stream.25 Lee et al. demonstrated the deoxygenation of triolein towards paraffin catalyzed by NiO–CaO/SiO2–Al2O3, and the maximum yield of straight-chain hydrocarbons (73.3%) was achieved under the following deoxygenation condition: 7 wt% of catalyst, 340 °C, within 60 min.26
This study aims to design and fabricate supported iron oxide catalysts for in situ upgrading of model compounds those are abundant in the chlorella HTL derived oil. In this paper, the effects of the prepared Fe2O3/MCM-41 catalysts on the deoxygenation of a lipid compound (palmitic acid) are explored, and kinetic analysis is carried out. The binary interactions of fatty acids and amino acids on the iron oxide catalysts are exploited to identify the possible reaction mechanism.
2. Experiments
2.1 Catalyst preparation
A typical preparation procedure of Fe2O3/(Al-)MCM-41 catalysts is described in Fig. 1. A certain amount of FeCl3·6H2O (AR, Tianjin Basf Chemical Co., Ltd.) was dissolved in deionized water to obtain a 0.3 mol L−1 solution. Ammonia water was quickly added to adjust the pH to 11. Then, 10 mL of sodium citrate (AR, Tianjin Basf Chemical Co., Ltd.) solution was added. The obtained suspension was mixed with hexadecyltrimethyl ammonium bromide (CTAB, AR, Sinopharm Chemical Reagent Co. Ltd) solution. Then, 2 g Na2SiO3·9H2O (AR, Tianjin Basf Chemical Co. Ltd) was added slowly with stirring. The pH was adjusted to 10 to obtain mother liquid I, which was hydrothermally crystalized to obtain Fe2O3/MCM-41. Otherwise, after 4 h of aging, 2 g CTAB was added to liquid I; subsequently, 1.0 g natural clinoptilolite (Shandong Jinshan Chemical Co. Ltd) was treated with 4 mol L−1 NaOH (AR, Tianjin Basf Chemical Co., Ltd.) solution at 40 °C for 4 h to obtain colloid I, which was injected into liquid I. Concentrated sulfuric acid solution was added dropwise to adjust the pH of the mixture to 10.0, and 40 g Na2SiO3·9H2O was added. After aging for 4 h, the colloidal solution was available. The mixture was then transferred into a Teflon-lined steel autoclave and statically heated at 120 °C for 48 h. The product was dried, washed, filtered and calcined at 550 °C for 5 h with a heating rate of 10 °C min−1. Finally, the sample was designated as Fe2O3/Al-MCM-41.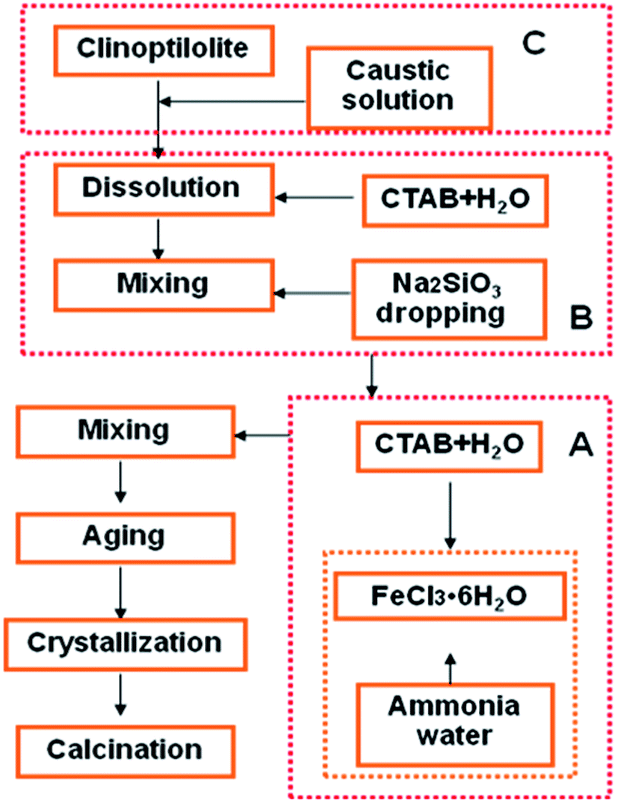 | ||
| Fig. 1 Preparation route for Fe2O3/MCM-41 (A + B) and Fe2O3/Al-MCM-41 catalysts (A + (C + B)). | ||
2.2 Catalyst characterization
Powder X-ray diffraction (XRD) analysis was carried out using a Bruker D8 ADVANCE diffractometer (wavelength = 0.154 nm, CuKα) for phase identification. Before measurements, the samples were degassed in a vacuum at 160 °C for 10 h.Transmission electron microscopy (TEM) was carried out with a JEM 2100 microscope (Japan) operated at 200 kV. The samples were ultrasonically dispersed in ethanol and laid on a Formvar-coated 200-mesh Cu grid.
UV-Vis diffuse reflectance (DR) spectra of the catalysts were collected on a Hitachi U-4100 spectrometer equipped with a diffuse reflectance attachment. An integrating sphere was coated with BaSO4 and used as a reference. The absorption intensity was expressed using the Kubelka–Munk function. The powder sample was loaded into a quartz cell, and the spectra were collected at 200–2500 nm.
X-ray photoelectron spectroscopy (XPS) measurements of the chars were carried out using a Thermo VG-Scientific, Sigma Probe spectrometer (ESCALAB250, USA) with monochromatized Al Kα radiation (1486.92 eV) in constant analyzer energy mode. The pass energy was 70 eV for survey spectra and 20 eV for high-resolution spectra.
An in situ DRIFTS reaction study of palmitic acid HTL decarboxylation was conducted using a Nicolet 6700 instrument. A high-pressure, high-temperature chamber fitted with ZnSe windows was utilized. Scans were taken at a resolution of 4 Å, and 128 scans were taken to improve the signal-to-noise ratio. Palmitic acid and glutamic acid (with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) were pre-adsorbed on the catalyst powder, and about 30 mg powder was loaded into the chamber. A steam generator consisted of a down flow tube and was equipped with an internal thermocouple.
:1 weight ratio) were pre-adsorbed on the catalyst powder, and about 30 mg powder was loaded into the chamber. A steam generator consisted of a down flow tube and was equipped with an internal thermocouple.
2.3 Catalytic activity evaluation
The HTL of chlorella and model compounds was carried out in a custom stainless-steel autoclave micro-reactor (10 mL volume) equipped with a sand-bath electric furnace and a thermocouple. In a typical test, 150 mg of reactant(s), such as palmitic acid or a mixture of palmitic acid (75 mg) and glutamic acid (75 mg), was injected into the reactor containing 10 mg catalyst and 2 mL distilled water. Liquefaction experiments performed at 320 °C, 327 °C, 335 °C, 342 °C, and 350 °C. After maintaining the temperature for 2.0 h, the autoclave was quenched rapidly to room temperature, and liquid products were collected in a comparison tube for further characterization.2.4 Product analysis
Liquid samples were dissolved in 2 mL n-heptane, and 200 mg methanol containing drops of concentrated sulfuric acid was added followed by esterification for gas chromatography (GC) analysis. Component identification was carried out on an Agilent Technologies 6890N GC equipped with a thermal conductivity detector (FID). The capillary column for GC and FID was a DB-WAX column (30 m × 0.25 mm × 0.25 μm). The carrier gas was nitrogen at a flow rate of 40 mL min−1. A volume of 1 μL was injected for each sample with a split ratio of 1/50. The injector and detector temperatures were 250° and 280 °C, respectively. GC followed the following temperature program: hold at 50 °C for 1 min and increase to 200 °C at a heating rate of 25 °C min−1. Further identification of liquid products was done on an Agilent GC-MS (7890A-5975C).Elemental analysis was carried out using a Vario EL III analyzer (CHN model), and the higher heat value was calculated from the Dulong equation.
3. Results and discussion
3.1 Physicochemical properties of the catalysts
Fig. 2 shows the XRD patterns of the prepared Fe2O3/MCM-41 and Fe2O3/Al-MCM-41 catalysts. In the large-angle range, the diffraction peaks at 31.66°, 35.56°, 43.28°, and 57.36° are attributed to the (220), (311), (400), and (511) reflections of maghemite (γ-Fe2O3; referenced to JCPDS 25-1402).27,28 Mineral clinoptilolite was employed as an aluminum source. This caused the prepared Fe2O3/Al-MCM-41 [Fig. 2(b)] to possess secondary units of natural zeolite, which was extraordinarily stable under hydrothermal and acidic conditions. Fe2O3/Al-MCM-41 retained the typical diffraction peaks of the monoclinic crystal system, with the strongest peaks appearing at 2θ values of 22.49°, 26.05°, 30.16°, and 32.02°; these peaks are coincident with the standard clinoptilolite PDF card JCPDS 33-0664 and JCPDS 39-1346.29 The good hydrothermal stability of the catalyst was attributed to the existence of clinoptilolite crystalline phase.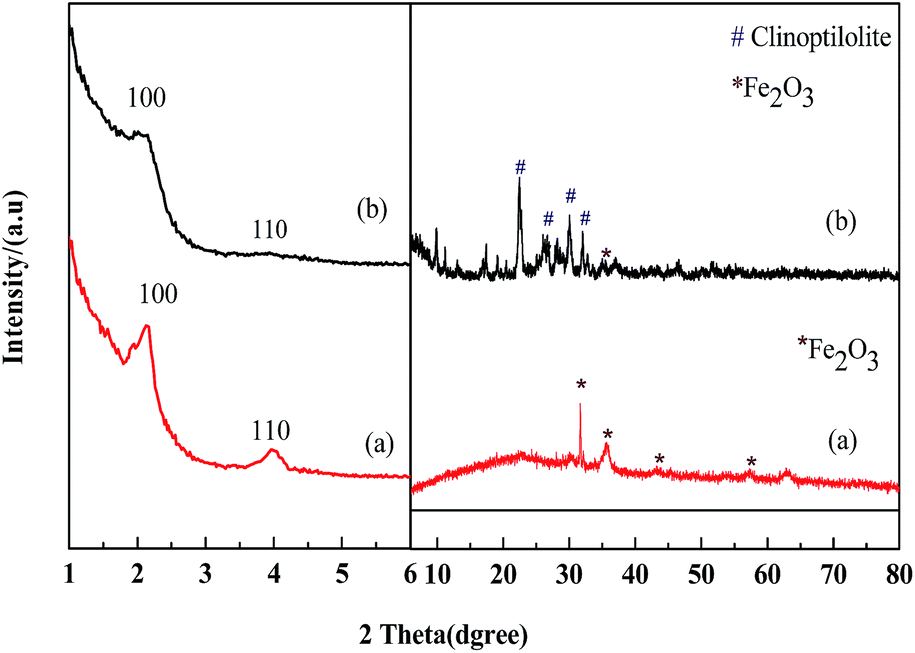 | ||
| Fig. 2 Small-angle and large-angle XRD patterns of catalysts. (a) Fe2O3/MCM-41 and (b) Fe2O3/Al-MCM-41. | ||
In the small-angle regime, the lab-synthesized pure silicon MCM-41 showed three diffraction peaks corresponding to d spacings of 3.84, 2.26 and 1.90 nm, which can be indexed to the (100), (110) and (200) reflections of the hexagonal (p6m) lattice of MCM-41.23 The Fe2O3/MCM-41 catalyst showed an obvious (100) peak, and this peak indicates structural regularity. The supported catalysts kept mesoporous structure of MCM-41 type silica. Fe2O3/MCM-41 exhibited broad diffraction peaks at 2θ values of 2.4° and 4.3° attributable to the (110) and (110) reflections, respectively. With the addition of aluminum, Fe2O3/Al-MCM-41 exhibited two weaker peaks of the hexagonal structure at 2.4° and 4.3°. The mesoporous structure was slightly affected by iron oxide incorporation, and it indicated that the Fe2O3/(Al-)MCM-41 catalysts could be well fabricated by assembling method. The prepared magnetic catalysts were chosen as HTL and decarboxylation catalysts for their reasonable distribution of active sites and good structural stability.
The main structural and textural characteristics of the prepared catalysts were determined from nitrogen adsorption–desorption isotherms. Both isotherms are type IV with hysteresis loop 25, which is characteristic of mesoporous materials. In Fig. 3(a), Fe2O3/MCM-41 shows a H2-type loop with the pore size distribution centered at 2.5 and 7.4 nm. In Fig. 3(b). Fe2O3/Al-MCM-41 shows an H1 loop and a sharp inflection in the relative pressure (p/p0) between 0.2 and 0.3, which corresponds to capillary condensation inside uniform mesopores. The sharpness of this step demonstrates the narrow pore size distribution centered at 2.6 nm. This indicates that Fe2O3/Al-MCM-41 maintained the mesoporous structure of Fe2O3/MCM-41 and enhanced the uniform hexagonal characteristics of MCM-41. Fe2O3/Al-MCM-41 provided active sites both for algae decomposition during HTL and fatty acid deoxygenation.23
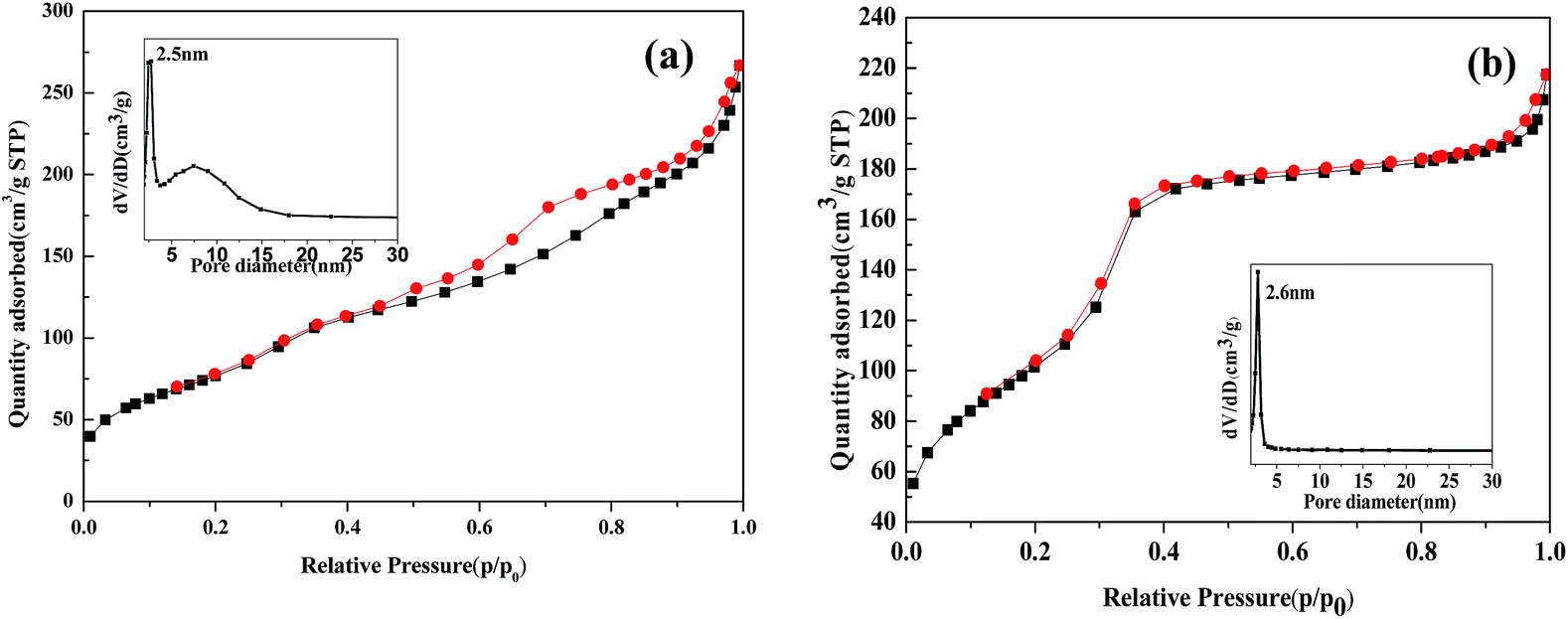 | ||
| Fig. 3 Isotherms and pore size distributions of catalysts (a) Fe2O3/MCM-41 and (b) Fe2O3/Al-MCM-41. | ||
In Fig. 4, the TEM micrographs of the prepared catalysts show an ordered hexagonal pore structure with embedded Fe2O3 nanoparticles. The TEM images of magnetic Fe2O3/(Al-)MCM-41 revealed that 10–30 nm Fe2O3 particles were uniformly distributed on the support, and in Fig. 4(c) and (d) there was no observed sintering of Fe2O3 even after 8 h time on stream of palmitic acid HTL conversion.
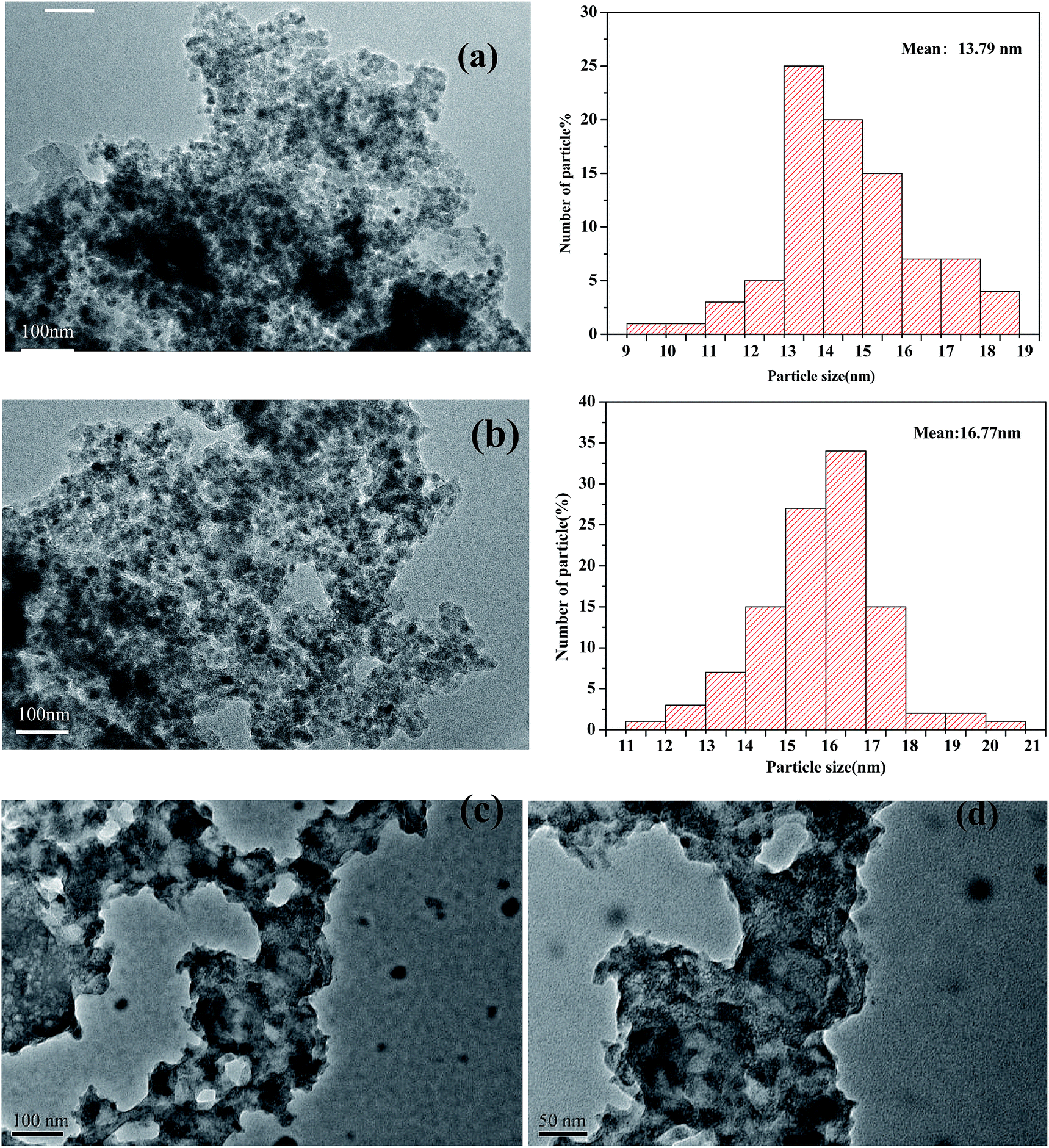 | ||
| Fig. 4 TEM images of (a) Fe2O3/MCM-41, (b) Fe2O3/Al-MCM-41, and (c), (d) Fe2O3/Al-MCM-41 catalysts after 8 h on stream. | ||
The ordered mesoporous channels of the support were recognized, and it could be deduced that the magnetic particles were located on the exterior surface of MCM-41 instead of inside the channels. The Fe2O3 maghemite cores were confirmed to have been successfully coated by the porous silica “shell” first and further enclosed with Al-MCM-41 to fabricate the supported Fe2O3/Al-MCM-41 catalyst. The consecutive coating method provided an effective approach to obtain the deoxygenation active sites, Fe2O3, that were well distributed on the decomposition sites, acidic “shell”, that made the microalgae HTL and derived biocrude in situ upgrading feasible and practical UV-Vis-NIR DR spectroscopy has been employed as a promising tool for Fe species analysis.30 The reflectance spectra of the Fe2O3/MCM-41 catalysts are shown in Fig. 4. The strong adsorption band at ∼300 nm was assigned to the band-gap transition of the maghemite core.27 The Fe2O3/MCM-41 catalyst revealed a band centered at 310 nm, while Fe2O3/Al-MCM-41 exhibited bands with maxima at 237 and 313 nm. These were attributed to Fe(III) complexes with lattice oxygen of the alumina-silicate support, and the appearance of the adsorption bands at wide wavelengths confirmed the strong interaction between iron oxide and the MCM-41 support. A weak band at about 1890 nm corresponded to the combination vibration band of water molecules.31 This band appeared in the spectra of both catalysts.
XPS is versatile surface analysis technique for compositional and chemical state analysis. The binding energies of Fe 2p1/2 and Fe 2p3/2 depend on the ionic states of Fe. The positions of the satellite peaks for the Fe 2p1/2 and Fe 2p3/2 peaks are also sensitive to the oxidation states, and these peaks have been used to qualitatively determine the ionic states of iron. In the Fe 2p narrow region of standard spectrum of Fe2O3, the binding energies of Fe 2p3/2 and Fe 2p1/2 are located at 711.8 and 725.3 eV, respectively. Fig. 5 shows the XPS spectra of the Fe2O3/(Al-)MCM-41 catalysts. There are two obvious energy peaks in the 2p area of Fe located at 712.0 and 725.5 eV for silica and 712.3 and 725.5 eV for aluminum containing the MCM-41 support. The binding energy of Fe 2p3/2 for both catalysts was slightly higher than that of the standard spectrum. This could be attributed to the strong interaction of Fe2O3 nanoclusters with the support. Similar results were also reported by Grosvenor32 and Yamashita.33 Compared with the previously reported values for silica- and alumina-supported Fe2O3, it has been confirmed that the main valence state of Fe on the catalysts was +3. Fe 2p3/2 of Fe2O3-MCM-41 has associated satellite peaks located about 8 eV higher than Fe 2p3/2. The satellite peak acquired at 719.8 eV was recognized, which is characteristic of Fe3+ in Fe2O3. Since the XRD patterns of γ-Fe2O3 and Fe3O4 were remarkably similar, the valence state of the iron oxide particles on the catalysts was further identified as Fe2O3 from the binding energy peaks in the XPS spectra.
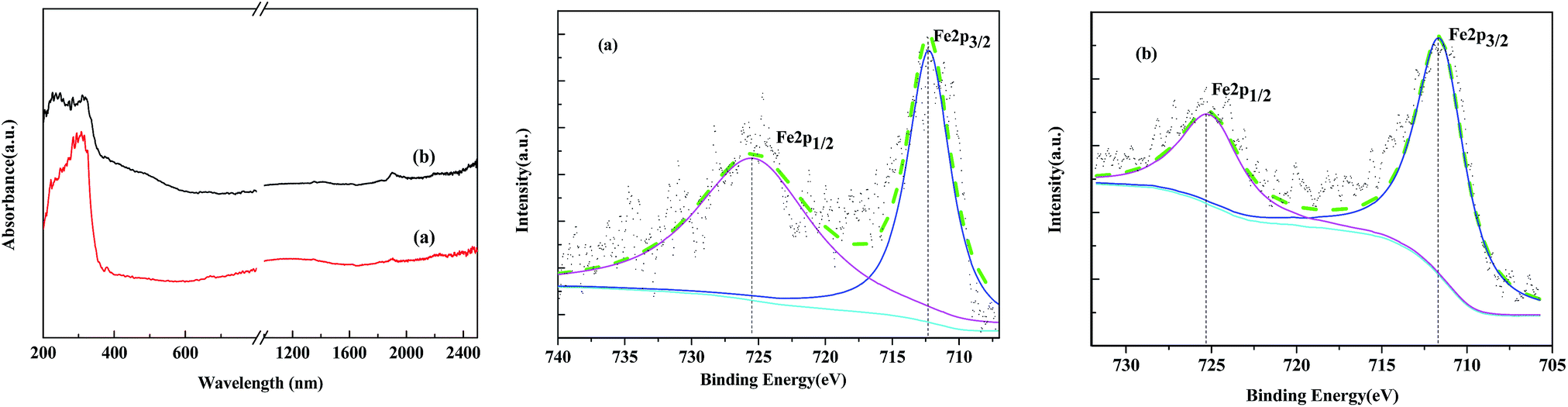 | ||
| Fig. 5 UV-Vis DR and XPS spectra of the catalysts (a) Fe2O3/MCM-41 and (b) Fe2O3/Al-MCM-41. | ||
3.2 Fe2O3/(Al-)MCM-41 for the catalytic decarboxylation of a model compound
The HTL of chlorella at 300 °C in subcritical water produced complicated organic compounds. The major components (>1.0 wt%) were ketones, esters and nitrogen compounds, as illustrated in our group's previous research.23 Palmitic acid was chosen as a model compound for catalytic upgrading.In the absence of hydrogen, the catalyst was employed for the deoxygenation of palmitic acid. As illustrated in Fig. 6, in sub-critical water and with the reaction temperature rising from 320 °C to 350 °C, the palmitic acid conversion was approximately 10.4–24.8% without catalyst addition. When the Fe2O3/MCM-41 magnetic catalyst was employed, the maximum conversion of palmitic acid was 68.1% at 342 °C, while that of Fe2O3/Al-MCM-41 reached 77.4% at 350 °C. During fatty acid deoxygenation, the desired product had one less carbon than the corresponding acid.33–35 In addition to deoxygenation reactions, a series of reactions could happen spontaneously on the Fe2O3/MCM-41 catalyst. These transformations mainly involved isomerization, cracking, hydrogenation, and cyclization reactions.36
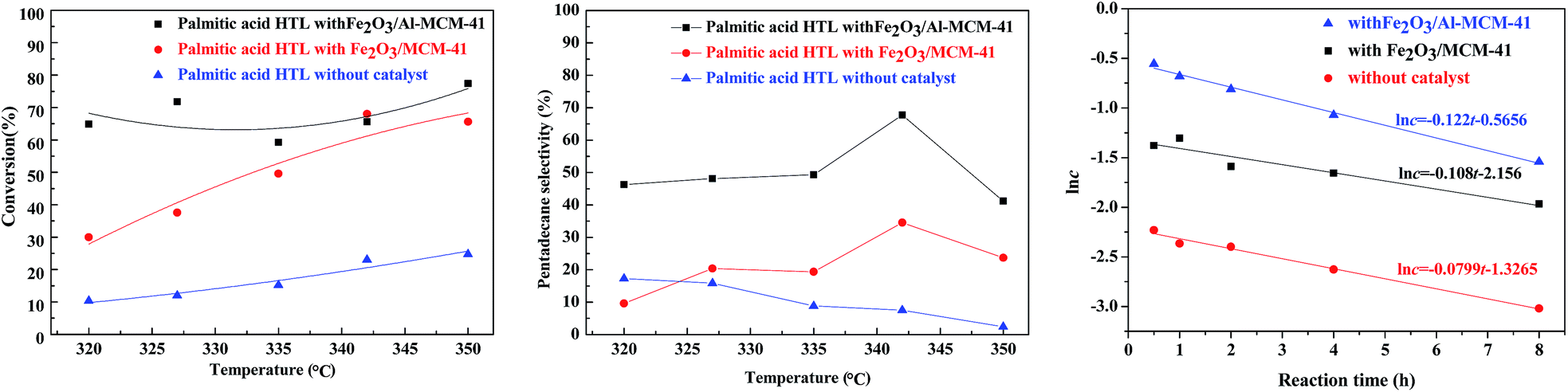 | ||
| Fig. 6 Effect of temperature on palmitic acid conversion (left) and pentadecane selectivity (middle) without and with catalysts. The correlation of palmitic acid concentration and reaction time at 342 °C (right). Reaction conditions: 150 mg feed, 10 mg catalyst, L/S = 1/75 mL mg−1. | ||
Consequently, according to the GC chromatogram for the catalytic HTL of palmitic acid with Fe2O3/MCM-41 catalyst at 342 °C in water, n-pentadecane was the predominant product obtained from the deoxygenation reactions, while other alkanes (C8–C14) and a certain amount of iso-paraffins were also formed. Judging from the selectivity to pentadecane, the selectivity was 17.3% at 320 °C without catalyst; with increasing temperature, the selectivity declined slowly to 2.4% at 350 °C. After the addition of the supporting magnetic catalyst, the selectivity for pentadecane increased to 32.3–60.7% with increasing reaction temperature from 320 °C to 342 °C. When the temperature rose to 350 °C, the selectivity was reduced. This was attributed to other prevailing side reactions such as cracking and/or isomerization on the acidic surface of the catalyst.
The existing reports regarding palmitic acid decarboxylation reveal that the overall reaction rate can be considered as first order with respect to palmitic acid.37,38 For both catalysts, the reaction rate equation was simulated on the experimental feed conversions at different reaction times. It was confirmed that the consumption rate of palmitic acid fits a first-order equation, and the apparent reaction rate constants were calculated using a batch reactor model. Fig. 6 (right) confirms the linear relationship between the natural logarithm of reactant concentration (lnc) and reaction time in the tested ranges for non-catalytic and catalytic palmitic acid conversion at 342 °C. The reaction rate constants at different temperatures were calculated, and Table 1 lists the rate constants for the non-catalytic and catalytic decarboxylation at the tested temperatures. The variation in rate constant with temperature followed the Arrhenius equation. The Arrhenius parameters were determined from the unweighted linear regression of lnk vs. 1/T. The activation energy Ea and frequency factor A were calculated and are indicated in Table 2.
| Temperature/K | k/mol−1 L h−1 | ||
|---|---|---|---|
| Without catalyst | With Fe2O3/MCM-41 | With Fe2O3/Al-MCM-41 | |
| 593.15 | 0.00775 | 0.00794 | 0.00811 |
| 600.15 | 0.0143 | 0.0182 | 0.0208 |
| 608.15 | 0.0375 | 0.0427 | 0.0566 |
| 615.15 | 0.0799 | 0.108 | 0.122 |
| 623.15 | 0.164 | 0.185 | 0.204 |
| Frequency factor A (h−1) | Activation energy Ea (kJ mol−1) | |
|---|---|---|
| Non-catalytic | 3.50 × 1022 | 320.64 |
| With Fe2O3/MCM-41 | 3.16 × 1023 | 330.66 |
| With Fe2O3/Al-MCM-41 | 1.16 × 1024 | 336.76 |
The HTL of palmitic acid on both the Fe2O3/MCM-41 catalysts was sensitive to temperature. Increasing temperature favored the non-hydrogenation decarboxylation of palmitic acid, and the apparent Ea of catalytic conversion was relatively higher (>300 kJ mol−1). The decarboxylation of palmitic acid was initiated by proton attack to produce an alkane with one less carbon. The p–π conjugation of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO led to the formation of large π bonding, and a complex was “built” between COO− and Fe3+ in the catalyst, facilitating the decarboxylation of palmitic acid. Fe3+ accelerated intermediate (carboxylate ion) formation and promoted decarboxylation.
CO led to the formation of large π bonding, and a complex was “built” between COO− and Fe3+ in the catalyst, facilitating the decarboxylation of palmitic acid. Fe3+ accelerated intermediate (carboxylate ion) formation and promoted decarboxylation.
3.3 In situ DRIFT reaction test
The reaction pathway of palmitic acid decarboxylation on Fe2O3/Al-MCM-41 catalysts was illustrated in our previous study,23 and CO2 desorption from the surface of the catalyst was identified as the rate-limiting step. Fatty acids and amino acids were abundant products of microalgae HTL or hydrolysis, and the interaction of palmitic acid and glutamic acid in the HTL process would be elucidated further. In situ DRIFTS was employed to investigate the formation and reactivity of surface species on the magnetic Fe2O3/Al-MCM-41 catalyst during the decarboxylation of palmitic acid with added glutamic acid. Scans were taken every 5 °C starting at 115 °C.As illustrated in Fig. 8, when the temperature increased to 195 °C, a band at 2349 cm−1 emerged, which was assigned to the asymmetric stretching vibration peak of OCO. The band at 1450 cm−1 weakened, which was assigned to the stretching vibration of carboxylate anion (–COO−) in L-glutamic acid.39 A band at 667 cm−1 re-appeared and corresponded to the bending peak of OCO. The desorption of weakly adsorbed CO2 on Fe2O3/Al-MCM-41 occurred at about 175 °C. The decarboxylation of glutamic acid occurred at 195 °C and led the re-appearance of the OCO peak. This means that glutamic acid decarboxylation was initiated at 195 °C.40 The shoulder peak at 1760 cm−1 could be due to the vibration of CO in carboxylic acid; this peak emerged at 215 °C and increased with increasing temperature. This indicated that glutamic acid decarboxylation occurred first, and palmitic acid then dissociated to form carboxylate ion, which was decarboxylated by proton attack to produce the alkane with one less carbon. The intramolecular dehydration of glutamic acid produced carboxyl pyrrolidinone, and the ionization of the carboxyl group occurred. This is why decarboxylation on the Fe2O3/Al-MCM-41 catalyst occurred at temperatures lower than 200 °C. The vibration peak at 1290 cm−1 was assigned to the stretching of C–N41,42 and became notable at 255 °C. It was evident that amide formed at this temperature.
As demonstrated in Fig. 7, the band at 2349 cm−1 disappeared at 255 °C, and it was deduced that glutamic acid decomposed to produce CO2, and CO2 desorption was the rate-limiting step of decarboxylation.43 Taking the activation energy of palmitic acid conversion into consideration, it was confirmed that the decarboxylation or deamination of amino acids occurred at lower temperatures than the decarboxylation or decarbonylation of fatty acids in the HTL oil. Tuning the adsorption strength of CO2 on the catalyst may improve the formation of carboxyl anion and enhance the selectivity of palmitic acid decarboxylation to alkanes.
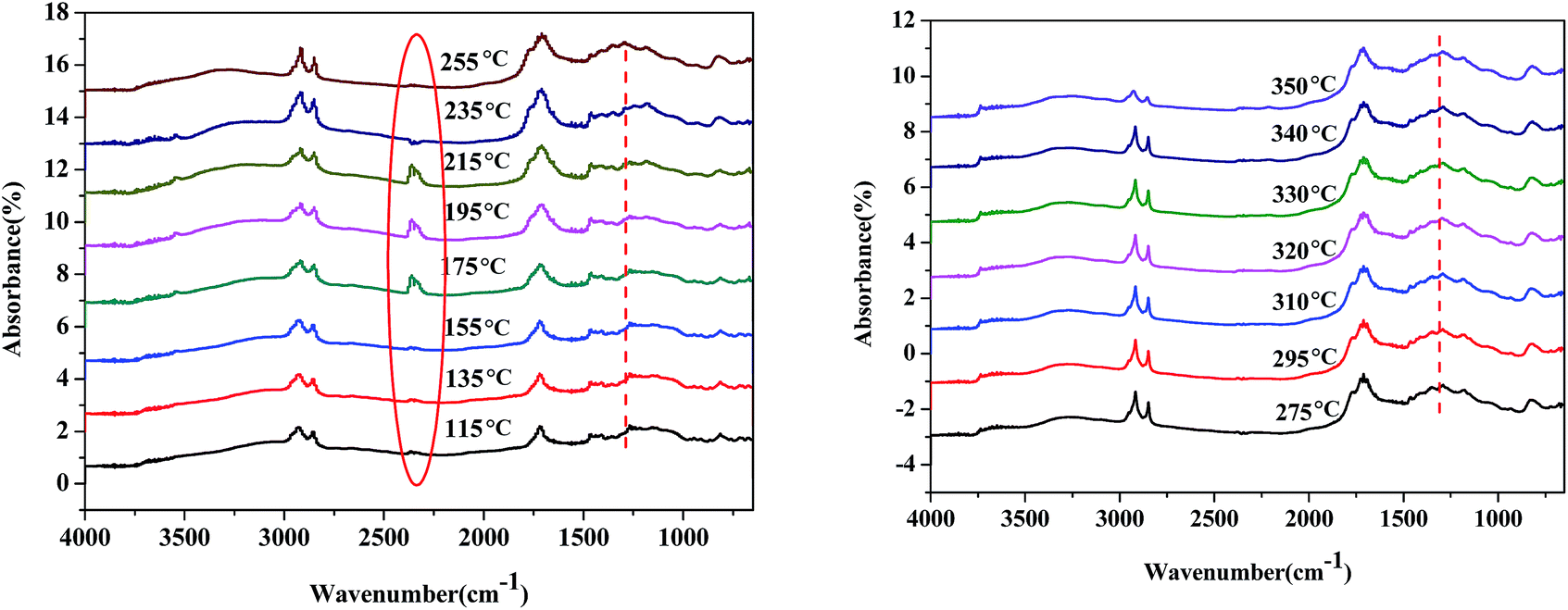 | ||
| Fig. 7 In situ DRIFT spectra of palmitic acid and glutamic acid during conversion on Fe2O3/Al-MCM-41 catalyst. | ||
3.4 GC-MS analysis
Palmitic acid and glutamic acid were subjected to co-HTL at 320 °C, and the derived CH2Cl2-soluble products were gathered for GC-MS analysis to identify the reaction pathways. As illustrated in Table 3, the main products of the binary mixture were hexadecanamide (32.00%), 4-hydroxyl-butanoic acid (24.31%), pyrrolidone (16.10%), and tetradecanoic acid (9.08%) by weight. According to their chemical origins, the products could be classified into three groups derived from glutamic acid, from palmitic acid, and from the binary interaction of amino acid and fatty acid.| Name of product | Chemical formula | wt% |
|---|---|---|
| Pentadecane | C15H32 | 4.46 |
| 4-Hydroxy-butanoic acid | C4H8O3 | 24.31 |
| 1-Propyl-2-pyrrolidinone | C7H13NO | 7.03 |
| 2-Tetradecanone | C14H28O | 1.98 |
| 2-Pyrrolidinone | C4H7NO | 9.08 |
| n-Decanoic acid | C10H20O2 | 1.92 |
| Tetradecanoic acid | C14H28O2 | 9.08 |
| Pentadecanoic acid | C15H30O2 | 2.79 |
| Valeric acid, undec-2-enyl ester | C4H7O4 | 4.53 |
| Hexadecanamide | C16H33NO | 32.00 |
The possible reaction pathways were deduced and are illustrated in Fig. 8. Amides with the general formula R–N(CxHy)n were the major products and were generated by the replacement of the hydroxyl group of palmitic acid with an amino group (route 4). Meanwhile, a certain amount of alkanes were produced via palmitic acid decomposition. Hydroxyl butanoic acid and pyrrolidone were derived from glutamic acid deamination and decarbonylation (route 3) and from intermolecular dehydration and decarboxylation (route 2). The latter, decarboxylation, is favored for biocrude upgrading.
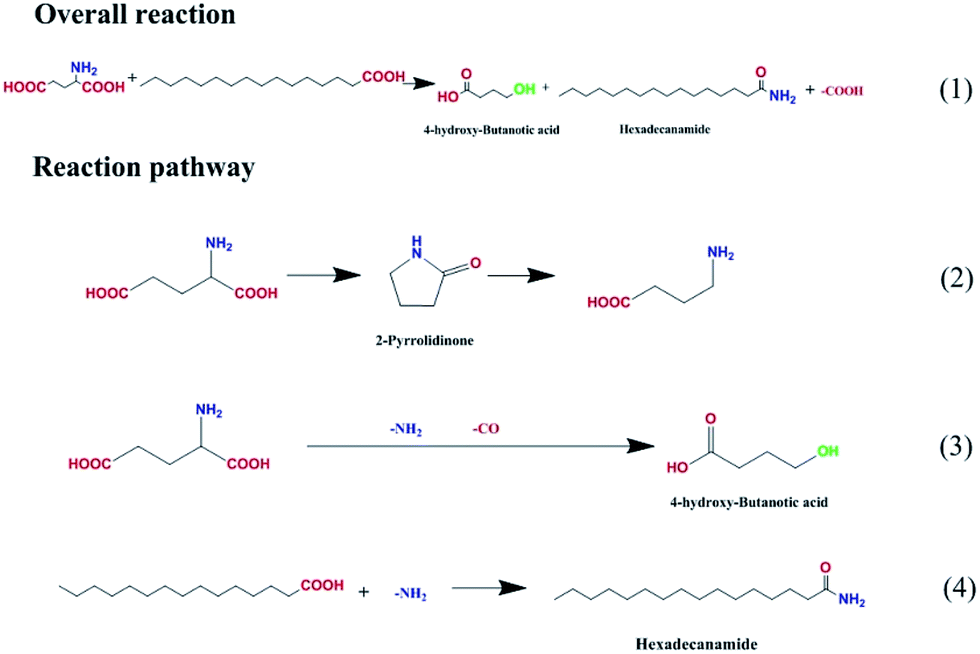 | ||
| Fig. 8 Proposed reaction pathway for the HTL of palmitic acid and glutamic acid on Fe2O3/Al-MCM-41 catalyst. | ||
For green fuel applications, the binary interaction of amino acid and fatty acid should be suppressed. We acknowledge that amide was formed at slightly higher temperature than the decarboxylation of palmitic acid, as was illustrated in the DRIFT test. Further efforts will focus on fabricating well-designed catalysts to facilitate deoxygenation and deamination.
4. Conclusions
Fe2O3/MCM-41 magnetic catalysts were designed and fabricated in a modified assembling method. An Fe2O3 “core” was prepared by precipitation, coated by porous silica, and supported on Al-MCM-41 synthesized using clinoptilolite as an aluminum source. XRD, TEM, XPS, and UV-Vis-NIR characterizations confirmed that 20–30 nm γ-Fe2O3 particles were well dispersed on the mesoporous support.The magnetic Fe2O3/MCM-41 catalysts showed good performance for chlorella HTL and upgrading of the derived biocrude. In the temperature range of 320–350 °C and under subcritical water, palmitic acid conversion was improved by 43–54% with Fe2O3/Al-MCM-41 catalyst in the absence of H2. The activation energy for the catalytic decarboxylation of palmitic acid was 336.76 kJ mol−1 on Fe2O3/Al-MCM-41.
In situ DRIFTS reaction test demonstrated that the decarboxylation initiation temperature was 195 °C, and the produced CO2 desorbed at 255 °C. A good alternative may be to “distillate” the produced CO2 from the reaction system via process intensification.
The HTL reaction pathways of the binary mixture of glutamic acid and palmitic acid were elucidated by product analysis, and the chemical origins of the main products were proposed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21376230) and The National Key Technology R&D Program of China (No. 2011BAD14B00).References
- A. L. Ahmad, N. H. M. Yasin, C. J. C. Derek and J. K. Lim, Renewable Sustainable Energy Rev., 2011, 15, 584–593 CrossRef CAS.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- O. Ellabban, H. Abu-Rub and F. Blaabjerg, Renewable Sustainable Energy Rev., 2014, 39, 748–764 CrossRef.
- D.-T. Tran, J.-S. Chang and D.-J. Lee, Appl. Energy, 2017, 185, 376–409 CrossRef CAS.
- P. Biller, A. B. Ross, S. C. Skill, A. Lea-Langton, B. Balasundaram, C. Hall, R. Riley and C. A. Llewellyn, Algal Res., 2012, 1, 70–76 CrossRef CAS.
- S. Zou, Y. Wu, M. Yang, C. Li and J. Tong, Energy Fuels, 2009, 23, 3753–3758 CrossRef CAS.
- B. Chakinala and W. Swaaij, Ind. Eng. Chem. Res., 2009, 49, 1113–1122 CrossRef.
- P. Duan, Energy Environ. Sci., 2011, 4, 1447–1456 CAS.
- M. Y. Bian and C. Promdej, Ind. Eng. Chem. Res., 2011, 50, 8492–8497 CrossRef.
- P. Duan, Ind. Eng. Chem. Res., 2010, 50, 52–61 CrossRef.
- U. Jena, K. C. Das and J. R. Kastner, Appl. Energy, 2012, 98, 368–375 CrossRef CAS.
- M. Shahinuzzaman, Z. Yaakob and Y. Ahmed, Renewable Sustainable Energy Rev., 2017, 77, 1375–1384 CrossRef CAS.
- N. S. Shamsul, S. K. Kamarudin and N. A. Rahman, Renewable Sustainable Energy Rev., 2017, 80, 538–549 CrossRef CAS.
- Y. Chen, Y. Wu, R. Ding, P. Zhang, J. Liu, M. Yang and P. Zhang, AIChE J., 2015, 61, 1118–1128 CrossRef CAS.
- Q. Lin, Y. Chen, Y. Tang, K. Wu, M. Yang, H. Hu and Y. Wu, Microporous Mesoporous Mater., 2017, 250, 120–127 CrossRef CAS.
- T. Yeh, S. Linic and P. E. Savage, ACS Sustainable Chem. Eng., 2014, 2, 2399–2406 CrossRef CAS.
- J. Fu, C. Yang, J. Wu, J. Zhuang, Z. Hou and X. Lu, Fuel, 2015, 139, 678–683 CrossRef CAS.
- D. Kim, D. R. Vardon, D. Murali, B. K. Sharma and T. J. Strathmann, ACS Sustainable Chem. Eng., 2016, 4, 1775–1784 CrossRef CAS.
- S. Idesh, S. Kudo, K. Norinaga and J.-i. Hayashi, Energy Fuels, 2013, 27, 4796–4803 CrossRef CAS.
- W. Li, Y. Gao, S. Yao, D. Ma and N. Yan, Green Chem., 2015, 17, 4198–4205 RSC.
- J. Wu, J. Shi, J. Fu, J. A. Leidl, Z. Hou and X. Lu, Sci. Rep., 2016, 6, 27820 CrossRef CAS PubMed.
- Y. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210–6224 RSC.
- J. Bian, Q. Zhang, P. Zhang, L. Feng and C. Li, Catal. Today, 2017, 293, 159–166 CrossRef.
- S. Karnjanakom, T. Suriya-umporn, A. Bayu, S. Kongparakul, C. Samart, C. Fushimi, A. Abudula and G. Guan, Energy Convers. Manage., 2017, 142, 272–285 CrossRef CAS.
- E. Santillan-Jimenez, R. Pace, S. Marques, T. Morgan, C. McKelphin, J. Mobley and M. Crocker, Fuel, 2016, 180, 668–678 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, Y. H. Taufiq-Yap, G. Abdulkrem-Alsultan, M. S. Mastuli and H. C. Ong, Energy Convers. Manage., 2017, 141, 325–338 CrossRef.
- Y. Ling, M. Long, P. Hu, Y. Chen and J. Huang, J. Hazard. Mater., 2014, 264, 195–202 CrossRef CAS PubMed.
- L. Čapek, V. Kreibich, J. Dědeček, T. Grygar, B. Wichterlová, Z. Sobalík, J. A. Martens, R. Brosius and V. Tokarová, Microporous Mesoporous Mater., 2005, 80, 279–289 CrossRef.
- J. Bian, Q. Zhang, X. Min, S. Zhang, L. Feng and C. Li, Process Saf. Environ. Prot., 2016, 101, 117–123 CrossRef CAS.
- J. Gurgul, K. Łątka, I. Hnat, J. Rynkowski and S. Dzwigaj, Microporous Mesoporous Mater., 2013, 168, 1–6 CrossRef CAS.
- G. Spoto, A. Zecchina, G. Berlier, S. Bordiga, M. G. Clerici and L. Basini, J. Mol. Catal. A: Chem., 2000, 158, 107–114 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- P. T. Huyen, L. T. H. Nam, T. Q. Vinh, C. Martínez and V. I. Parvulescu, Catal. Today, 2017 DOI:10.1016/j.cattod.2017.03.040.
- C. Miao, O. Marin-Flores, S. D. Davidson, T. Li, T. Dong, D. Gao, Y. Wang, M. Garcia-Pérez and S. Chen, Fuel, 2016, 166, 302–308 CrossRef CAS.
- H. Xin, K. Guo, D. Li, H. Yang and C. Hu, Appl. Catal., B, 2016, 187, 375–385 CrossRef CAS.
- S. V. Vassilev and C. G. Vassileva, Fuel, 2016, 181, 1–33 CrossRef CAS.
- C. Yang, R. Nie, J. Fu, Z. Hou and X. Lu, Bioresour. Technol., 2013, 146, 569–573 CrossRef CAS PubMed.
- L. Guo, M. Meng and Y. Zha, Chin. J. Catal., 2006, 27, 189–194 CrossRef CAS.
- L. Hermida, A. Z. Abdullah and A. R. Mohamed, Renewable Sustainable Energy Rev., 2015, 42, 1223–1233 CrossRef CAS.
- B. de Campos Vidal and M. L. S. Mello, Micron, 2011, 42, 283–289 CrossRef PubMed.
- O. Francioso, D. Montecchio, P. Gioacchini, L. Cavani, C. Ciavatta, O. Trubetskoj and O. Trubetskaya, Geoderma, 2009, 152, 264–268 CrossRef CAS.
- A. A. Kamnev, A. G. Shchelochkov, Y. D. Perfiliev, P. A. Tarantilis and M. G. Polissiou, J. Mol. Struct., 2001, 563, 565–572 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |