
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphorylcholine oligomer-grafted graphene oxide for tumor-targeting doxorubicin delivery†
Yu Qina,
Changyu Wanga,
Yun Jianga,
Tao Liub,
Jianyong Yangc,
Run Lin*c and
Tao Zhang *a
*a
aDepartment of Biomedical Engineering, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China. E-mail: ztnj@nju.edu.cn
bJiangsu Provincial Key Laboratory for Interventional Medical Devices, Huaiyin Institute of Technology, Huai'an 223003, China
cDepartment of Radiology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou 510080, China. E-mail: linrun5@mail.sysu.edu.cn
First published on 25th August 2017
Abstract
A novel phosphorylcholine oligomer-grafted and folate moiety-labeled graphene oxide (GO–PCn–FA) was designed, prepared, and characterized by Fourier transform infrared spectra, nuclear magnetic resonance, Raman spectra, X-ray diffraction, X-ray photoelectron spectroscopy, scanning transmission electron microscopy, transmission electron microscopy, and atomic force microscopy. GO–PCn–FA proved to be an excellent water-soluble and pH-responsive drug carrier for the targeted delivery of doxorubicin (DOX) with a drug loading content of 21%. An in vitro cytotoxicity assay and flow cytometry analysis revealed the superior biocompatibility of GO–PCn–FA compared to normal cells, while DOX-loaded GO–PCn–FA exerted efficient eradication of tumor cells, especially of those with folate receptor expression. An in vivo test showed that GO–PCn–FA was deposited mainly in the pulmonary parenchyma after intravenous administration, and no obvious adverse effect was observed. In summary, phosphorylcholine oligomer-grafted graphene oxide was developed for targeted drug delivery with optimal biocompatibility.
1. Introduction
Graphene oxide (GO) is the derivative of graphene that has great potential for application in many fields due to its unique physical and chemical properties.1,2 GO's surface functionality, ultra-high specific surface area, better biocompatibility (compared to graphene), and cost-effective features make it a potent and versatile platform for drug delivery.3–6 However, the cytotoxicity of GO, especially to certain types of normal cells, and its inadequate water solubility greatly restrict its utilization and remain the primary problems that need to be addressed.7–9Chemical modification is a critical strategy in designing and fabricating biomaterials that confers the substance's novel properties and functions, such as anti-biofouling, controlled response, altered solubility, drug encapsulation, biomarker targeting, improved biocompatibility, and other special features. Phosphorylcholine (PC) is the essential component of a biological membrane with the zwitterionic nature that offers optimized hydrophilicity, stability, biocompatibility, and, more importantly, active sites for further modification.10–13 PC-based polymers have been broadly used to improve the performance and function of biomedical materials, devices, instruments and even drug nanocarriers.14–17
The routes through which the drugs are delivered to the tumor mass can be generally divided into two categories: the passive route, which is known as the enhanced permeability and retention (EPR) effect, and the active route, which enhances the delivery by coupling targeting ligands for the specific binding of tumor biomarkers.18,19 The hypervascularized, premature, and leaky vascular wall with enlarged gaps between the endothelial cells and defective lymphatic drainage in the tumor mass account for the preferential deposition and compromised clearance leading to the passive accumulation of the drug carrier, namely the EPR effect.20,21 In addition, active targeting by conjugating moieties to specifically targeted biomarkers that are overexpressed in tumor cells to enhance delivery efficiency is another feasible and effective approach to substantially improve the drug delivery capability of the vehicle to the tumor lesions.19,22 Folic acid (folate, FA) is an important component of numerous cellular metabolic processes, particularly for DNA synthesis and repair, and rapidly proliferating cancer cells have a markedly increased requirement for folate uptake.23,24 Consequently, upregulated folate receptor (FR) expression and the key role of FA in a wide spectrum of cancers make it an ideal biomarker for targeted therapy.25–27
In previous reports, numerous modifications have been developed to improve the biocompatibility and the chemotherapeutic delivery of GO;28 this includes the use of polymeric modifiers, such as polyethylene glycol,29–31 Poloxamer,32–34 poly(amido amine),35 poly(N-isopropyl acrylamide),36 chitosan,37,38 heparin,39 and gelatin,40,41 while the employed chemotherapeutics have included doxorubicin (DOX) and derivatives,29,33,35,39 paclitaxel,42 camptothecin,36,37 and even methotrexate.41 The reported drug loading capacity in the mass percentage ranged from 5% to 380%.28 We also recently reported the synthesis of phosphorylcholine moiety-grafted GO by a different approach.43 Nevertheless, modification using PC and FA to confer GO with the improved biocompatibility and targeting capability simultaneously has yet to be investigated.
Herein, we report the development of a novel phosphorylcholine oligomer (PCn)-grafted, folic acid-coupled, and doxorubicin-loaded graphene oxide (DOX@GO–PCn–FA) for targeted tumor delivery. DOX@GO–PCn–FA was prepared through the chemical modification of GO with 2-methacryloyloxy ethyl phosphorylcholine (MPC) and 2-aminoethyl methacrylate (AMA) via the process of atom transfer radical polymerization (ATRP), the conjugation of folic acid as a targeting ligand via carbodiimide crosslinker chemistry (N-ethyl-N′-(3-(dimethyl amino)propyl) carbodiimide/N-hydroxysuccinimide (EDC/NHS) crosslinker used), and the mechanical loading of DOX through π–π stacking interactions.6,44 We studied the properties of DOX@GO–PCn–FA, including physical and chemical characteristics, DOX loading, and release profiles. We also investigated the biocompatibility on both normal and tumor cells, and evaluated the targeting specificity on tumor cells with folate receptor-positive and -negative expressions. Lastly, we performed a preliminary exploration of in vivo biocompatibility and biodistribution of GO–PCn–FA.
2. Experimental section
2.1 Materials
Natural graphite powder (>99%, with a particle size of approximately 5 μm) was purchased from Shanghai Huayuan Graphite Co., Ltd. (Shanghai, China). KMnO4 (CP), H2SO4 (98%), H2O2 (30% solution in water), 2-aminoethyl methacrylate (AMA), folic acid (FA), 1-ethyl-3-(3′-dimethylaminopropyl) carbodiimide (EDC), and N-hydroxysuccinimide (NHS) were purchased from Nanjing Reagent Co. Ltd. (Nanjing, China) and they were used as they were received. Inhibitor-free 2-methacryloyloxyenthyl phosphorylcholine (MPC, >96%) was supplied by Joy-Nature Institute of Technology (Nanjing, China). We synthesized 2-aminoethyl methacrylate (AMA) according to Narain et al.45 and verified by 1H-NMR in 2H2O. (δ = 1.88, 3.33, 4.38, 5.72, and 6.14 ppm). All of the other chemicals were purchased from Aladdin (Shanghai, China).The L929 mouse fibroblast cells, HNEPC human nasal epithelial cells, and HepG2 human hepatocellular carcinoma cells with a positive expression of folate receptor (FR), A549 human lung cancer cells with a negative expression of FR, and KB human nasopharyngeal epidermal carcinoma cells with a positive expression of FR were obtained from Zhongyuan Co., Ltd. (Beijing, China), which redistributes cell lines that initially come from ATCC (Manassas, VA, USA). Dulbecco's modified Eagle's medium (DMEM, with 4.5 g/L-glucose and L-glutamine) was purchased from Thermo Fisher Scientific (China) Co. Ltd. (Beijing, China). Fetal bovine serum (FBS) was purchased from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China). Penicillin/streptomycin (100×) was purchased from China branch of Life Technologies Co. (Shanghai, China). Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies (shanghai) Co. Ltd. (Shanghai, China). Pentobarbital sodium (>99%) was purchased from MSD China (Shanghai, China).
2.2 Preparation of GO–PCn–FA
Graphene oxide (GO) was prepared via the modified Hummers method. Briefly, 2.0 g of graphite powder was placed into 50 mL of concentrated H2SO4 (98%) in a round-bottom flask in an ice-water bath (0 °C) and 8.0 g of KMnO4 was gradually added. The mixture was continuously stirred below 5 °C for 1 h and then heated up to 35 °C for 30 min. After, 100 mL of deionized water was slowly added and the temperature was maintained at 98 °C for 30 min. Finally, 30 mL of H2O2 (30%) was added to remove the remaining KMnO4. The product was filtered out then washed with 5% HCl and deionized water until it was pH-neutral and the SO42− could not be detected by BaCl2 solution. GO was then dried under a vacuum at 60 °C overnight.Acylating chlorination and amidation processes were employed to prepare GO–MPC–AMA. Briefly, 150 mg of GO was added into 50 mL of thionyl chloride (SOCl2) and the mixture was ultrasonicated under a 40 kHz ultrasonic water bath for 3 h. Then, the remaining SOCl2 was removed by rotary evaporation. We added 200 mL of 2-aminoethanol (HO(CH2)2NH2) immediately to the flask under an ultrasonic water bath. After being stirred at room temperature for 24 h, the product (named GO–EA) was collected by centrifugal filtration and washed 3 times with anhydrous ether.
The copolymerization of MPC and AMA onto GO was processed following ATRP as shown in Fig. 1. In brief, 100 mg of GO–EA, 50 mL of tetrahydrofuran (THF), and 8.0 mL of triethylamine were added in a round-bottom flask and the mixture was continuously stirred for 30 min at room temperature. With vigorous stirring, 10 g of 2-bromo-2-methyl-propionyl bromide was added. After reacting for 30 h, 100 mL of deionized water was added and the solution was centrifuged at 8000 rpm. The precipitates (temporarily named GO–ATRP) were washed 3 times by anhydrous ether and dried in a vacuum oven at 40 °C. We dissolved 40 mg of GO–ATRP in 10 mL of methanol and 0.9 g of MPC and 0.1 g of AMA were added under a N2 atmosphere. After stirring for 30 min, 20 mg of CuBr and 50 mg of 2,2′-bipyridine were introduced and reacted for another 24 h at room temperature. The final product (temporarily named GO–MPC–AMA) was collected by centrifugal filtration and washed 3 times with anhydrous ether.
 | ||
| Fig. 1 Synthetic route of GO–PCn–FA. | ||
FA was then conjugated onto GO–MPC–AMA via EDC/NHS chemistry between the NH2 groups in GO–MPC–AMA and the carboxy (–COOH) groups in the FA molecules. Briefly, EDC and NHS were added to the GO–MPC–AMA suspension (in DMF) and the mixture was ultrasonicated for 1 h. Then, 5% FA was added and stirred for 24 h. The unreacted reagents were separated out by dialyzing against deionized water for 48 h. The final product (GO–PCn–FA) was dried in a vacuum oven at 30 °C overnight.
2.3 Characterizations
Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker AR×500 NMR spectrometer (Bruker, Germany) using 2H2O as the solvent. Fourier transform infrared spectra (FTIR) were recorded on a PE GX spectrometer (Perkin-Elmer, USA) at room temperature on KBr pellets with sample concentrations of ∼1% from 4000 to 400 cm−1 and a resolution of 4 cm−1. The Raman spectra were measured using a confocal Raman micro spectrometer (Renishaw InVia, Derbyshire, England) with an excitation wavelength of 514 nm. X-ray diffraction (XRD) measurements were performed on a Rigaku ULTIMA-3 setup with a Mar 345 image plate as the detector, the Cu Kα as the source (wavelength of 0.1542 nm), the recorded region of 2θ = 5° to 40°, and a scanning speed of 2° min−1. X-ray photoelectron spectroscopy (XPS) of GO–PCn–FA and GO were recorded on a VG Scientific ESCA Lab MK-II spectrometer (West Sussex, England) equipped with a monochromatic Mg-Kα X-ray source. XPS Peak software (v 4.1) was used to analyze and deconvolute the XPS peaks, and the peak deconvolutions were performed using Gaussian components after a Shirley background subtraction. Transmission electron microscope (TEM) imaging was performed using a Titan3 G2 (FEI, OR, USA) with a field emission gun (XFEG) operating at an accelerating voltage of 80 kV. Scanning transmission electron microscope (STEM) imaging was acquired by high-angle annular dark-field detector (HAADF) with a camera length of 115 mm. A tapping mode atomic force microscope (AFM) (Cypher, Asylun Research, CA, USA) was utilized to observe the micromorphology of samples.2.4 Loading and releasing profiles of DOX with GO–PCn–FA
DOX was loaded onto GO–PCn–FA or GO via π–π stacking interactions by mechanically mixing 0.5 mg mL−1 of DOX with 1.0 mg mL−1 of GO–PCn–FA or GO in pure water at different volume ratios overnight. Then, the DOX-loaded GO–PCn–FA composites (DOX@GO–PCn–FA) were filtered out from the mixture with a 0.22 μm membrane, washed 3 times, vacuum dried, and stored at 4 °C.The drug loading content was defined as the mass percentage of DOX presented in DOX@GO–PCn–FA composites, and the drug loading efficiency was expressed as the proportion of the added DOX that was encapsulated in DOX@GO–PCn–FA. The drug loading content and loading efficiency were determined using a Waters 2695 high-performance liquid chromatography (HPLC) system (Milford, MA, USA) equipped with a 4.6250 mm Discovery HS F5 HPLC column (Supelco, Bellefonte, PA, USA) and a UV detector (Waters 2487). The determinations were proceeded by a mobile phase composed of CH3OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (0.1% TFA):CH3CN (0.1% TFA) = 17:54:29 (volume ratio) at a flow rate of 1.0 mL min−1 at 25 °C with the sample injection volume of 10 μL. In order to analyze the release profile of DOX, 10 mg of DOX@GO–PCn–FA was placed into each dialysis bag and dialyzed in 10 mL of PBS as medium (0.1 mol L−1 with pH 7.2 and 5.5, respectively) at 37 ± 1 °C. At each scheduled time point, 1 mL of PBS was taken out for HPLC analysis and an additional 1 mL of medium was replaced to keep the volume.
:H2O (0.1% TFA):CH3CN (0.1% TFA) = 17:54:29 (volume ratio) at a flow rate of 1.0 mL min−1 at 25 °C with the sample injection volume of 10 μL. In order to analyze the release profile of DOX, 10 mg of DOX@GO–PCn–FA was placed into each dialysis bag and dialyzed in 10 mL of PBS as medium (0.1 mol L−1 with pH 7.2 and 5.5, respectively) at 37 ± 1 °C. At each scheduled time point, 1 mL of PBS was taken out for HPLC analysis and an additional 1 mL of medium was replaced to keep the volume.
2.5 In vitro cell cytotoxicity analysis
L929, HNEPC, HepG2, KB, and A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% FBS, 100 μg mL−1 of streptomycin, and 100 μg mL−1 of penicillin in culture flasks, and they were incubated at 37 °C in a humidified incubator (Heracell model 150i, Thermo Scientific, USA) containing 5% CO2 and 95% air.The cytotoxicity experiments of the samples were performed using a CCK-8 assay and they were repeated at least in sextuplicate. Briefly, 1 × 104 viable cells were placed in each well of 96-well plates and cultured for 24 h prior to the addition of GO, GO–PCn–FA, free DOX, and DOX@GO–PCn–FA of different concentrations (25, 50, 75, 100, and 150 μg mL−1 for GO and GO–Pn–FA; 1, 2.5, 5, 7.5 and 10 μg mL−1 for free DOX and DOX@GO–PCn–FA), respectively. After being cultured for 24 h or 48 h at 37 °C, 20 μL of CCK-8 (5 mg mL−1 in culture medium) was added and the cells were incubated for 1 h at 37 °C. The absorbance of the solution at 450 nm was recorded with a Rayto RT-6000 microplate reader (Rayto Life and Analytical Sciences Co., Ltd., Shenzhen, China). The mean absorbance of the untreated cells was used as the reference value to determine 100% cellular viability.
2.6 Flow cytometry analysis
Flow cytometry analysis was carried out to ascertain and compare the percentages of apoptotic cells between the groups subjected to different treatments. HepG2 human hepatocellular carcinoma cells were placed in 6-well plates (1.5 × 105 cells per well) and cultured overnight, allowing the cells to attach. The cells were then treated with GO (30 μg mL−1), GO–PCn–FA (30 μg mL−1), DOX@GO–PCn–FA (30 μg mL−1), and free DOX (5 μg mL−1) at 37 °C, respectively. After 8 h of incubation, the HepG2 cells were detached and collected by centrifugation at 1000 rpm for 3 min, and they were then resuspended in 100 μL of 1× loading buffer. We added 5 μL of annexin-V into the suspension, which was kept in a dark place for 15 min before 5 μL of propidium iodide (PI) was added. After 5 min of incubation on ice, each sample was analyzed using a flow cytometer (BD FACSAria II, BD Biosciences, CA, USA), and FlowJo Workplace (V.x.0.7, FlowJo LLC, Ashland, OR, USA) was used to analyze the data.2.7 In vivo biodistribution study of GO–PCn–FA
The animal test was approved by the Animal Care Committee of Sun Yat-sen University (Guangzhou, China), and the experiments were performed in accordance with the National Institutes of Health's guidelines for the use of experimental animals. Male adult specific pathogen-free (SPF) SD rats weighing between 250 and 300 g were obtained from the Laboratory Animal Center of Sun Yat-sen University.Rats were anesthetized with pentobarbital sodium (30 mg kg−1 via intraperitoneal injection). GO–PCn–FA was dispersed in 0.9% saline at a concentration of 5.0 mg mL−1 and the suspension was ultrasonicated for 5 min using a Branson digital sonifier (Emerson, Danbury, CT, USA) S-250D with a 1/8-in tapered microtip (200 W; 20 kHz; amplitude: 40%; 2 s/2 s) in an ice-cold bath. Immediately after homogenization, the GO–PCn–FA suspension was intravenously injected through the tail vein at the dosage of 20 mg kg−1. The rats were killed 24 h after administration and the major organs, including the brain, lungs, heart, liver, spleen, kidneys, intestines, and muscles were collected. Samples were subjected to hematoxylin–eosin (HE) staining and scrutinized with a Leica (Leica Microsystems Inc., IL, USA) DM 2500 microscope with a Leica DFC425 C camera.
3. Results and discussions
3.1 Characterizations
The 13C-NMR spectrum of GO–PCn–FA reveals the characteristic peaks of the methyl (30.18 ppm), the methylene (61.22 ppm and 66.85 ppm), and the methyne (126.83 ppm) in the phosphorylcholine oligomer chains, while the peak at 215.34 ppm is very likely the resonance of carbon atoms that belong to the phenyl structure of folic acid (Fig. 2A). In the 1H-NMR spectrum of GO–PCn–FA, the distinct peak at 4.78 ppm is the resonance of H2O when the peak at 3.14, 4.22, 3.58, and 1.86 ppm should be the resonance of the hydrogen atoms in the methyl group, which comes from the phosphorylcholine oligomer structure (Fig. 2A). These results proved the successful grafting of the phosphorylcholine oligomer and the conjugation of folic acid onto the GO sheets since the pure GO is superparamagnetic without these characteristic peaks. | ||
| Fig. 2 (A) 13C-NMR and 1H-NMR spectra of GO–PCn–FA; (B) FTIR spectra of graphite, free DOX, GO, GO–PCn–FA, and DOX@GO–PCn–FA; and (C) Raman spectrum and (D) XRD patterns of graphite, GO, and GO–PCn–FA. | ||
Fig. 2B shows the FTIR spectra of graphite, free DOX, GO, GO–PCn–FA, and DOX@GO–PCn–FA. On the GO spectrum, the peaks at 1730 and 1630 cm−1 correspond to the stretching vibration of carboxyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the deformation of the hydroxyl (–OH) groups in water that present in GO, respectively. The peak centers at 1388 cm−1, which is attributed to the deformation vibration of the C–OH bond, while an intense band at 1051 cm−1 is due to the stretching vibration of the C–O bonds in GO.46 For GO–PCn–FA, the double peaks at 3034 and 2956 cm−1 are associated with the transformation of methylene carbon. The peak at 1489 cm−1 is related to P–O–alkyl and the absorbance at 964 cm−1 can be assigned to the tertiary amine group. In addition, the peak at 1605 cm−1 is due to the deformation vibration N–H amide-II of the amine group in FA-conjugated GO–PCn, which confirms the existence of the chemical bonds between FA and GO–PCn. On the FTIR spectrum of DOX@GO–PCn–FA, the stretching vibration band of C–O–C (∼1085 cm−1) and C–O–H (∼1285 cm−1) are detected. The broad peak between 3600 cm−1 and 3000 cm−1 represents the hydroxyl (OH) stretching and NH-stretching vibration bands.
O) and the deformation of the hydroxyl (–OH) groups in water that present in GO, respectively. The peak centers at 1388 cm−1, which is attributed to the deformation vibration of the C–OH bond, while an intense band at 1051 cm−1 is due to the stretching vibration of the C–O bonds in GO.46 For GO–PCn–FA, the double peaks at 3034 and 2956 cm−1 are associated with the transformation of methylene carbon. The peak at 1489 cm−1 is related to P–O–alkyl and the absorbance at 964 cm−1 can be assigned to the tertiary amine group. In addition, the peak at 1605 cm−1 is due to the deformation vibration N–H amide-II of the amine group in FA-conjugated GO–PCn, which confirms the existence of the chemical bonds between FA and GO–PCn. On the FTIR spectrum of DOX@GO–PCn–FA, the stretching vibration band of C–O–C (∼1085 cm−1) and C–O–H (∼1285 cm−1) are detected. The broad peak between 3600 cm−1 and 3000 cm−1 represents the hydroxyl (OH) stretching and NH-stretching vibration bands.
Raman spectroscopy was carried out to investigate the carbon structures of pristine graphite, GO, and GO–PCn–FA. As shown in Fig. 2C, pristine graphite has a very weak D band at 1350 cm−1, while both GO and GO–PCn–FA have an increased D band. Furthermore, the 2D band of pristine graphite at 2700 cm−1 decreases after modification and a new peak (called the D + G peak) in 2940 cm−1 appears. This result indicates that the sample becomes amorphous after oxidation and grafting, and the sample is a mixture of a single-layer and multilayer structure.47
In the XRD investigation, the graphite presented a strong diffraction peak at 26.2° and GO showed at 10.1°, while GO–PCn–FA did not show an evident diffraction peak in this range (Fig. 2D). This was because the lamellar spacing increased from 0.34 nm for pristine graphite to 0.81 nm for GO. The formation of oxygen-containing functional groups on the carbon basal planes led to the large lamellar spacing of GO.43,48 After conjugating with phosphorylcholine chains and FA, the lamellar spacing of GO–PCn–FA became even greater, and the 001 diffraction peak decreased sharply and shifted to an angle that is too low to be observed in the scanning range.
The XPS results reveal that only the carbon and oxygen elements can be observed in the spectrum of GO, while the additional nitrogen and phosphorus elements emerge in the spectrum of GO–PCn–FA, indicating the successful introduction of phosphorylcholine oligomer chains (Fig. 3A and B). On the C 1s intensive scan of GO–PCn–FA, the CO bond at 288.3 eV, the C–C bond at 285.9 eV, and the C–N bond at 284.3 eV are observed, which confirms the linkage between GO and phosphorylcholine (Fig. 3C). On the O 1s intensive scan of GO–PCn–FA, the peak at 529.4 eV is caused by the O–P bond, which cannot be observed in the O 1s XPS spectrum of GO (Fig. 3D). In addition, the peak at 401.9 eV in the N 1s intensive scan (Fig. 3E) as well as the peak at 132.4 eV in the P 2p intensive scan (Fig. 3F) demonstrate the presence of phosphorylcholine and FA moieties in GO–PCn–FA. Therefore, the aforementioned results confirm the successful preparation of GO–PCn–FA.
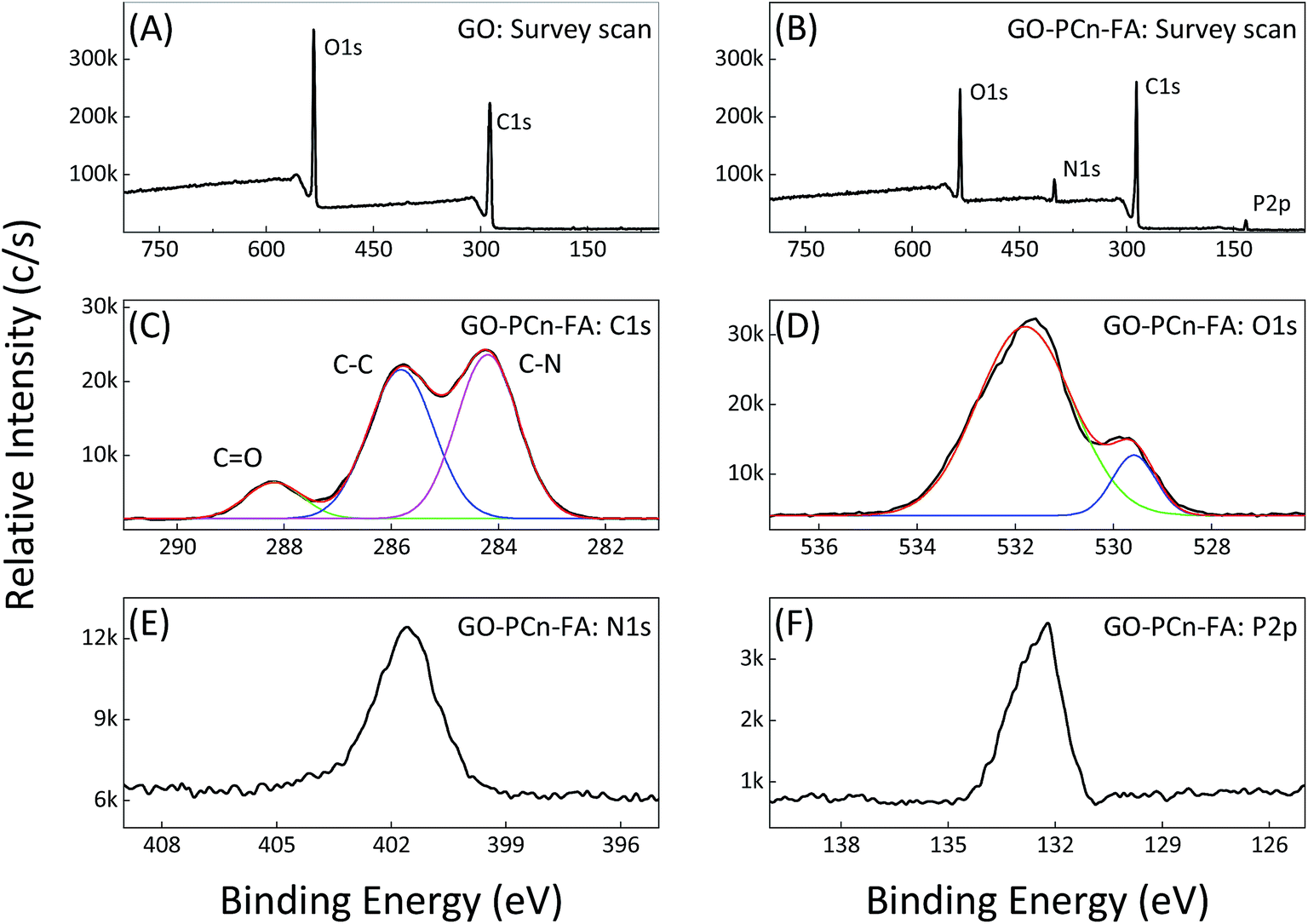 | ||
| Fig. 3 XPS spectra of GO and GO–PCn–FA. (A) Survey scan of pristine GO; (B) survey scan of GO–PCn–FA; and (C) C 1s, (D) O 1s, (E) N 1s, and (F) P 2p intensive scan of GO–PCn–FA, respectively. | ||
3.2 Micromorphological investigation of GO and GO–PCn–FA
The STEM and TEM studies showed that both GO and GO–PCn–FA displayed a paper-like structure with the area of a few to dozens of square microns (Fig. 4A–D), but no distinct differences on morphology between GO and GO–PCn–FA were observed at this contrast resolution. The morphology of as-prepared GO and GO–PCn–FA was also characterized by AFM (Fig. 4E and F). Furthermore, both GO and GO–PCn–FA presented with a sheet-like shape with the thickness of about 1.2 nm, which was in accord with data reported in other literature,49 confirming single- to multiple-layer structures of GO and GO–PCn–FA. As compared to GO, GO–PCn–FA sheets were smaller and more fragmented, with the average lateral dimension of 2.0 μm.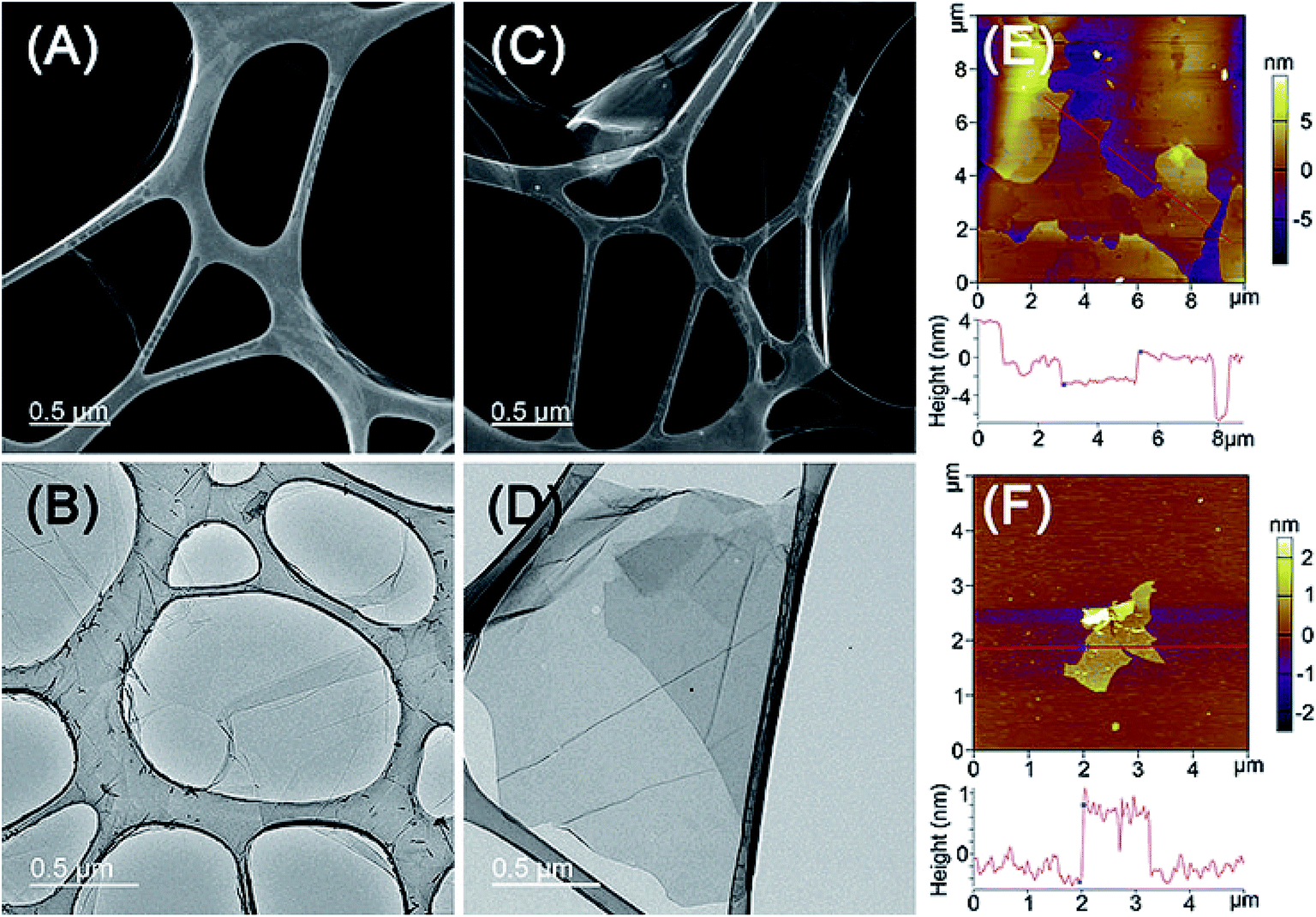 | ||
| Fig. 4 Micromorphology of GO and GO–PCn–FA. (A) STEM and (B) TEM images of GO; (C) STEM and (D) TEM images of GO–PCn–FA; and tapping mode AFM images on the mica surface of (E) GO and (F) GO–PCn–FA sheets. | ||
3.3 Loading and releasing of DOX with GO–PCn–FA
Loading of DOX onto GO–PCn–FA or GO is postulated to occur through the non-covalent interaction between the graphene aromatic layer and the aromatic structure in DOX molecules known as the π–π stacking effect.6,50 The loading efficiency as well as the loading content of DOX onto GO–PCn–FA or GO were determined by HPLC as shown in Fig. 5A. For GO–PCn–FA, with the increase of the mass ratio of DOX to GO–PCn–FA, the loading efficiency declined, while the loading content increased (Fig. 5A). The balance was achieved at the ratio of 1.5, in which the DOX loading efficiency reached up to about 18% and the corresponding drug loading content was 21% (0.27 mg of DOX on per mg of GO–PCn–FA), and the continuing increase of the ratio did not promote the DOX loading contents (Fig. 5A). Thus, in the following preparation of DOX@GO–PCn–FA, the mass ratio of DOX to GO–PCn–FA was kept at 1.5, and the DOX concentration in DOX@GO–PCn–FA was determined as 21%. As a control, on the pristine GO, the DOX loading contents kept increasing with the increase of the mass ratios of DOX to GO and reached around 63.5% (1.74 mg of DOX on per mg of pristine GO) at a mass ratio of 2. The huge difference of drug loading contents between GO and GO–PCn–FA should be attributed to the grafting of hydrophilic phosphorylcholine oligomers. The grafted moieties covered up a part of the GO sheet surface thus decreased the space for DOX loading. Moreover, the hydrogen bonding between the carboxy group on pristine GO sheets and amino group in DOX molecule can also strengthen the interactions of GO sheets and DOX, thus increased the DOX loading contents. However, the drug loading content, which is determined largely by intrinsic properties, such as the size of GO used in this study, can be promoted to a higher level using a distinct batch of GO. As the exfoliated graphite was employed, the DOX loading content of GO–PCn–FA, prepared by an identical approach, reached above 45% (ESI†).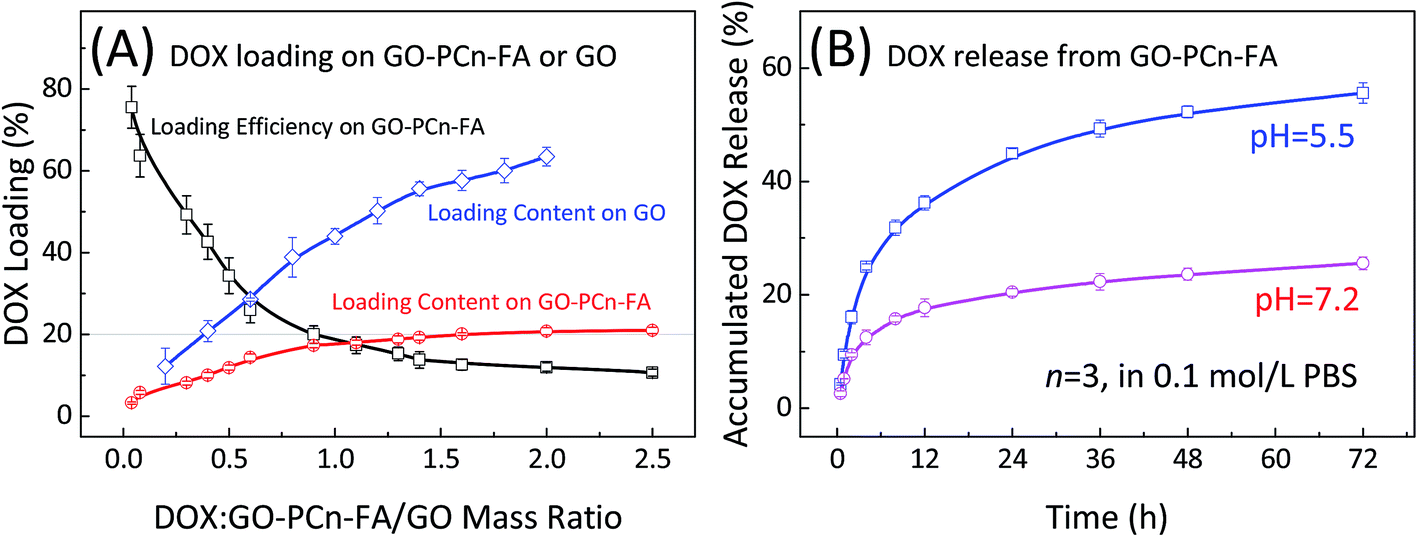 | ||
| Fig. 5 (A) Loading efficiency and loading content of DOX onto GO–PCn–FA or GO; and (B) the release behavior of DOX from DOX@GO–PCn–FA in PBS (0.1 mol L−1, 37 °C, pH 5.5 and 7.2, respectively). | ||
The release profiles of DOX from DOX@GO–PCn–FA were measured at pH 5.5 and pH 7.2, respectively. As shown in Fig. 5B, a slow release feature was observed at pH 7.2, in which less than 20% of DOX was off-loaded from DOX@GO–PCn–FA within 24 h, whereas rapid release occurred in an acidic environment (pH 5.5) in which around 45% of DOX was released in 24 h. This pH-dependent DOX release behavior might stem from increased hydrophilicity via protonation of the NH2 group of DOX in an acidic condition, which substantially weakened the π–π interaction between DOX and the GO layers.51 Given that both the extra- and intracellular microenvironments of the tumors tissues are acidic, the pH-dependent drug release behavior is beneficial for treating tumors.
3.4 In vitro cytotoxicity assay of DOX, GO, and derivatives
The cytotoxicity of GO and GO–PCn–FA were investigated by CCK-8 assay. The L929, HNEPC, and HepG2 cells were selected as the model cell lines. As shown in the first row of Fig. 6, the viability of the L929, HNEPC, and HepG2 cells remained above 75% without a significant difference after 24 h of incubation as the dosage was increased to 100 μg mL−1, which implied good biocompatibility of GO and GO–PCn–FA to the cells under these circumstances. After being exposed to GO and GO–PCn–FA for 48 h, cell viabilities remained over 60% (second row of Fig. 6). It is noteworthy that GO–PCn–FA exerted a considerably lower cytotoxic effect on the HNEPC cells as compared to GO at high dosages over 100 μg mL−1, suggesting that the surface modification of phosphorylcholine oligomer improved biocompatibility.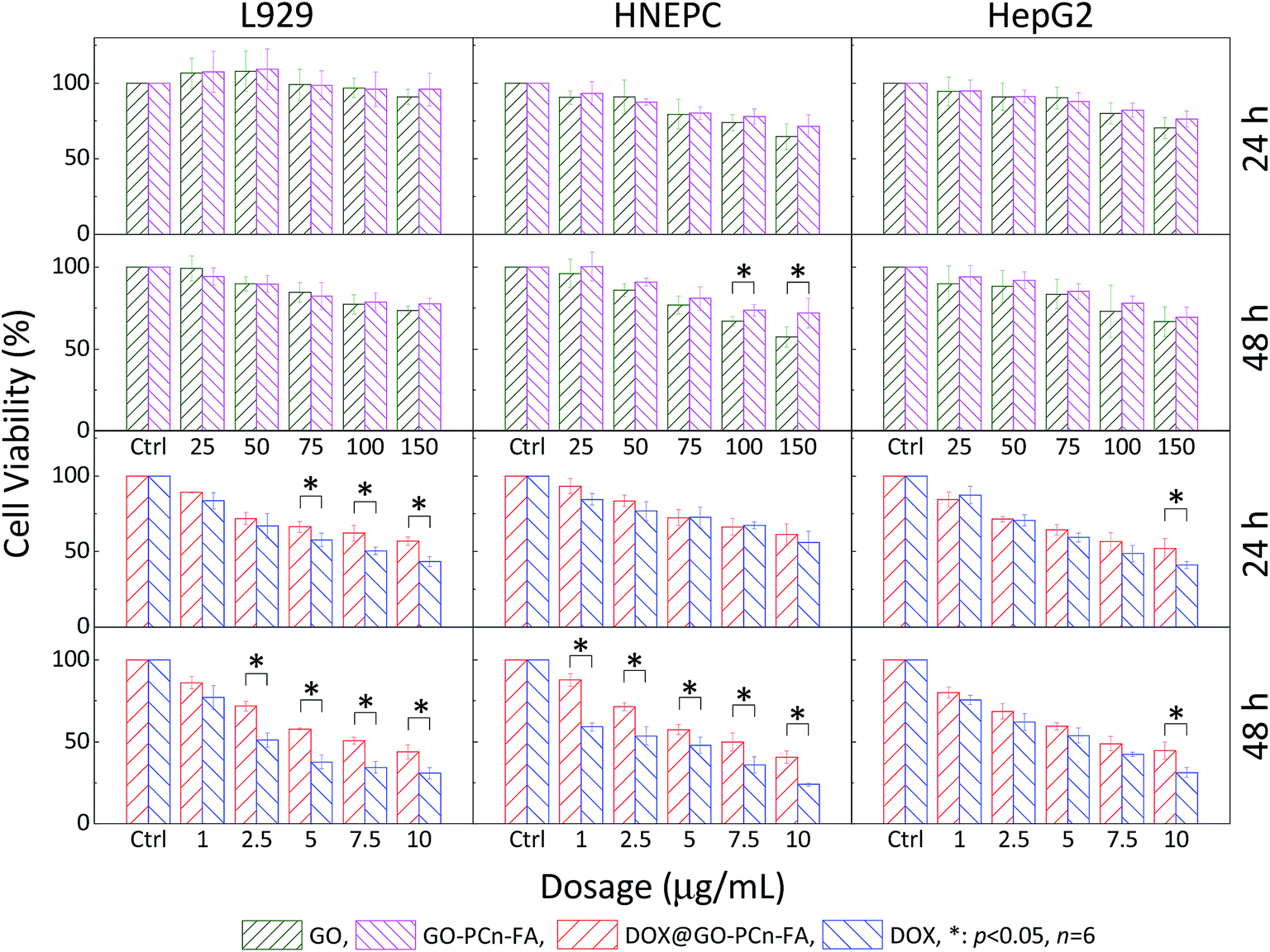 | ||
| Fig. 6 Cytotoxicity assays of GO, GO–PCn–FA, DOX@GO–PCn–FA, and free DOX against L929, HNEPC, and HepG2 cells, respectively (*p < 0.05). | ||
Evaluations comparing the cytotoxic effects of free DOX and DOX@GO–PCn–FA on normal cells as well as tumor cells were also performed (third and fourth row of Fig. 6). For the normal cells, DOX@GO–PCn–FA had an apparently lower detrimental effect on cell viability in comparison with free DOX, which was mainly attributable to the postponed uptake of DOX@GO–PCn–FA, which was substantially limited by steric hindrance because the micron-sized vehicle and the cells were of the same order of magnitude in size. Moreover, the slow release feature of DOX from the vehicle (approximate 45% of DOX was released in 24 h) geared down the translocation of DOX into the cell nucleus and resulted to the postponed cytotoxicity.
For the HepG2 hepatocellular carcinoma (HCC) cells, both free DOX and DOX@GO–PCn–FA exhibited remarkable inhibition against the tumor cells in a dosage- and duration-dependent manner. The cell viability of HepG2 of both groups was reduced to about 50% at the dosage of 7.5 μg mL−1, and it kept decreasing as the dosage increased and the duration was prolonged. The difference in antitumor effects between the groups became statistically significant only when the dosage was raised up to 10 μg mL−1 of DOX, which was also due to the slow release and limited uptake characteristics of DOX@GO–PCn–FA as described above.
More importantly, considering the inherent and remarkable chemoresistance of the HCC cells52,53 as well as the normal cells, especially those dividing rapidly (e.g., fibroblast cells, epithelial cells, and mucosal cells), which are extremely susceptible to chemotherapeutic agents,54,55 chemotherapy agents are supposed to cause much more pronounced damage to normal cells as compared to HCC cells. However, our data revealed that DOX@GO–PCn–FA exerted a comparable cytotoxic effect on both the normal and HCC cells without a significant difference. The possible and reasonable explanation is that PC modification reduced the toxicity to normal cells and, on the other hand, the targeting ligand (FA) maintained the cytotoxicity against the HepG2 HCC cells with folate receptor expression,56,57 leading to the targeted cytotoxic effect of DOX@GO–PCn–FA on tumor cells. Succinctly put, DOX@GO–PCn–FA simultaneously presented optimized biocompatibility to normal cells and preferential suppression in the viability of tumor cells.
Cytotoxicity was further investigated by the flow cytometry analysis of HepG2 cells incubated for 8 h with GO, GO–PCn–FA, DOX@GO–PCn–FA, and free DOX, respectively (Fig. 7). There was little difference in cell viabilities, with no statistically significant difference between the control group and the groups subjected to GO or GO–PCn–FA at 8 h post treatment (Fig. 7A–C), suggesting the optimized biocompatibility of GO and GO–PCn–FA in the experimental setting. On the other hand, after incubation for 8 h with DOX@GO–PCn–FA, 45.8% of the cells remained viable, while most of the damaged cells were at the stage of late apoptosis or necrosis (Fig. 7D). Moreover, around 32% of cells treated with free DOX survived when the majority of the injured cells were at the early apoptotic stage (Fig. 7E). These outcomes suggested that DOX@GO–PCn–FA was more prone to induce necrosis while free DOX tended to trigger apoptosis. The distinct effects of DOX@GO–PCn–FA and DOX on cells could be ascribed at least partly to the slow release behavior of DOX from DOX@GO–PCn–FA. Another speculated mechanism might be that after the slow internalization of the micron-sized DOX@GO–PCn–FA, the intracellular release of DOX was enabled, which considerably promoted and accelerated the process of apoptosis and necrosis of the cells.
 | ||
| Fig. 7 Flow cytometry analysis of the cytotoxicity of materials to HepG2 cells. (A) Control group; cells incubated for 8 h with (B) GO at 30 μg mL−1, (C) GO–PCn–FA at 30 μg mL−1, (D) DOX@GO–PCn–FA at 30 μg mL−1, and (E) free DOX at 5 μg mL−1. | ||
3.5 Specific targeting effect of folate modification
In addition to the enhanced permeability and retention (EPR) effect, active targeting is another feasible and effective strategy to improve drug delivery capability of the vehicle to the tumor lesions. The conjugation of ligands confers the drug carrier with the ability to identify and bind to the targeting cells, tissues, or organs with high sensitivity and specificity. As is shown in Fig. 8, the coupling of folic acid (FA) provided DOX@GO–PCn–FA with a formidable and enhanced eradication effect over DOX@GO–PCn against KB tumor cells with folate receptor expression. For example, at 50 μg mL−1, the cell viabilities after treatment were 27.4% for DOX@GO–PCn–FA and 44.3% for DOX@GO–PCn, respectively (Fig. 8B). With respect to folate receptor-negative A549 cells, DOX@GO–PCn–FA and DOX@GO–PCn introduced a parallel cytotoxic effect without a significant difference (Fig. 8A). These results verified the targeting specificity of DOX@GO–PCn–FA towards folate receptor-expressing cells, representing the active targeting approach, which is frequently used to prepare biomarker-oriented drug delivery systems. Furthermore, as for the specificity of linked ligands, GO–PCn–FA has the great potential to be a versatile platform to target a wide spectrum of biomarkers.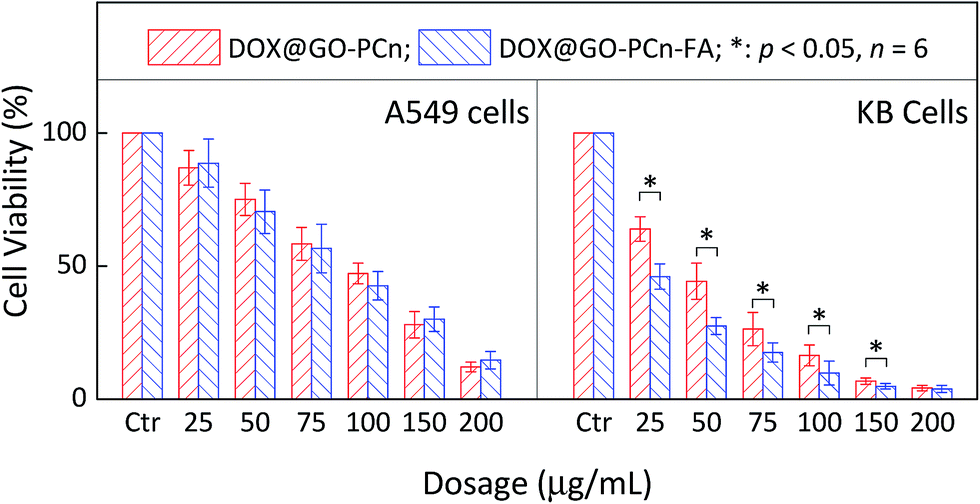 | ||
| Fig. 8 Cell viability of A549 and KB cells treated with different dosages of DOX@GO–PCn and DOX@GO–PCn–FA for 24 h. | ||
3.6 Preliminary exploration of in vivo biodistribution and biocompatibility of GO–PCn–FA
In order to evaluate in vivo biodistribution and biocompatibility, GO–PCn–FA suspension (5 mg mL−1 in saline) was intravenously injected into SD rats through the tail vein at the dosage of 20 mg kg−1. The rats were killed 24 h after administration and the major organs, including the lung, brain, heart, liver, spleen, kidneys, intestines, and muscles were collected and sectioned into thin slices for hematoxylin–eosin (HE) staining. As shown in Fig. 9, GO–PCn–FA was predominantly deposited in pulmonary parenchyma (indicated by the blue arrows in Fig. 9A), whereas no accumulation of GO–PCn–FA was detected in other organs. Furthermore, no obvious pulmonary toxicity, such as pulmonary edema induced by the as-prepared GO observed in earlier reports58, was evidenced in this study. Our data demonstrated that the surface modification of the phosphorylcholine oligomer offered GO improved biocompatibility. In addition, GO–PCn–FA has great application potential in the management of respiratory diseases as it presented the propensity for accumulating in pulmonary parenchyma. More studies are warranted to fully evaluate the applications and the long-term toxicology profiles of GO–PCn–FA.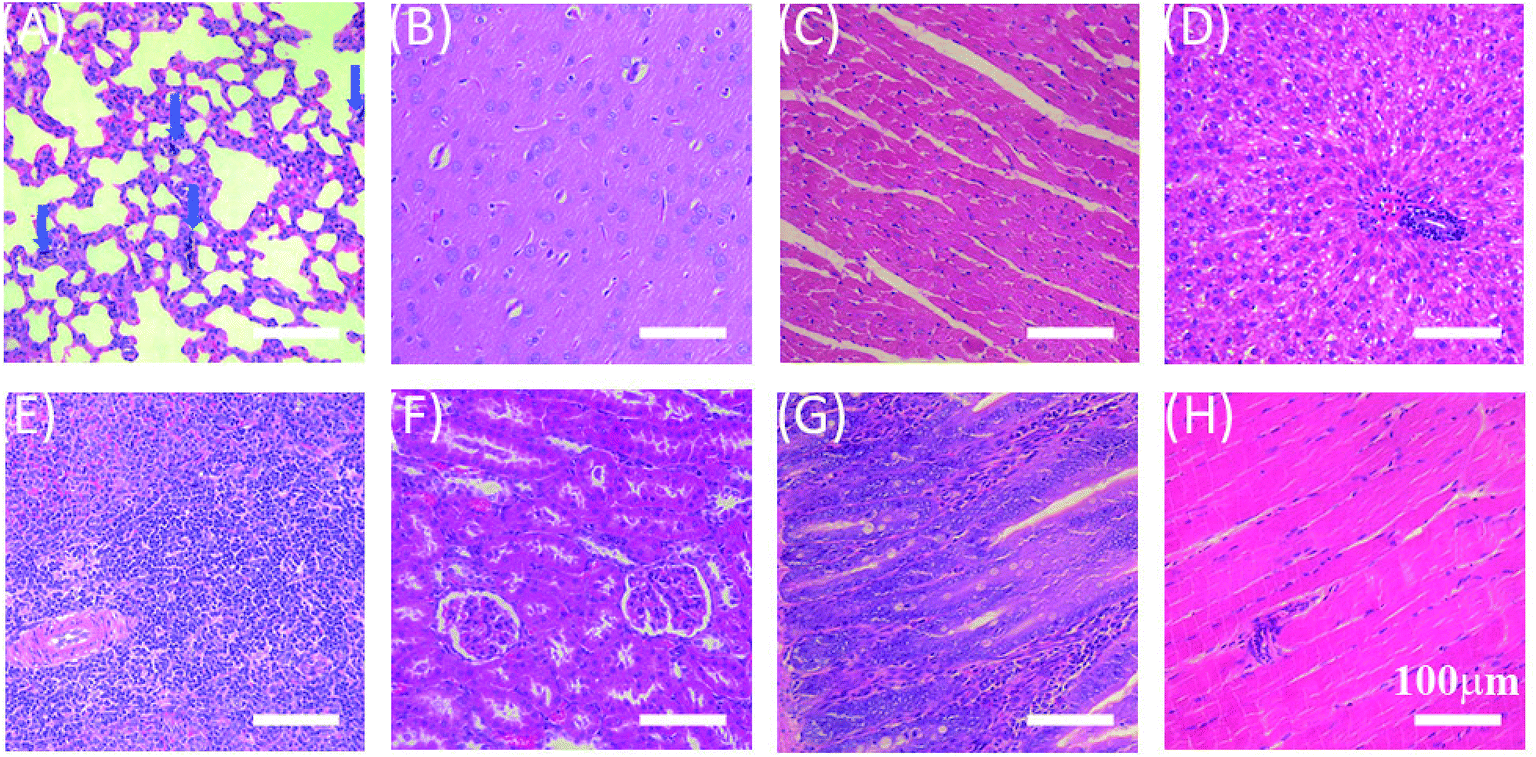 | ||
| Fig. 9 HE-stained histological sections of the organs and tissues. (A) Lung, (B) brain, (C) heart, (D) liver, (E) spleen, (F) kidney, (G) intestines, and (H) muscle. | ||
4. Conclusions
In conclusion, we developed an efficient approach to prepare phosphorylcholine oligomer-grafted, folic acid-labeled, doxorubicin-loaded GO (DOX@GO–PCn–FA) for targeted drug delivery. GO–PCn–FA presented superior biocompatibility to normal cells as compared to GO, while DOX@GO–PCn–FA presented preferred cytotoxicity over DOX@GO–PCn against tumor cells with folate receptor expression, indicating the specific targeting effect of DOX@GO–PCn–FA. An in vivo biodistribution and biocompatibility study suggested that GO–PCn–FA was predominantly deposited in pulmonary parenchyma without a significant adverse effect after intravenous administration. Our work demonstrated the great potential of phosphorylcholine oligomer-grafted graphene oxide as a versatile platform for biomarker-directed delivery with optimal biocompatibility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Yu Liu (Laboratoire Mécanique des Sols, Structures et Matériaux, Université Paris-Saclay, France) is acknowledged for his kind help in the micromorphology investigation with STEM and TEM. The authors acknowledge the support of Natural Science Foundation of China (No. 81671792), the support of National Key R&D program of China (2016YFC0104100), the Foundation of Jiangsu Provincial Key Laboratory for Interventional Medical Devices (No. JR1504). The works are also a part of the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC
.
- Y. W. Chen, Y. L. Su, S. H. Hu and S. Y. Chen, Adv. Drug Delivery Rev., 2016, 105, 190–204 CrossRef CAS PubMed
- T. Ren, L. Li, X. Cai, H. Dong, S. Liu and Y. Li, Polym. Chem., 2012, 3, 2561–2569 RSC
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC
- R. Cheng, R. Zou, S. Ou, R. Guo, R. Yan, H. Shi, S. Yu, X. Li, Y. Bu, M. Lin, Y. Liu and L. Dai, Polym. Chem., 2015, 6, 2401–2406 RSC
- G. Shim, M. G. Kim, J. Y. Park and Y. K. Oh, Adv. Drug Delivery Rev., 2016, 105, 205–227 CrossRef CAS PubMed
- X. Zhang, W. Hu, J. Li, L. Tao and Y. Wei, Toxicol. Res., 2012, 1, 62–68 RSC
- Y. Ma, H. Shen, X. Tu and Z. Zhang, Nanomedicine, 2014, 9, 1565–1580 CrossRef CAS PubMed
- W. Zhang, Y. Sun, Z. Lou, L. Song, Y. Wu, N. Gu and Y. Zhang, Colloids Surf., B, 2017, 151, 215–223 CrossRef CAS PubMed
- A. L. Lewis, Z. L. Cumming, H. H. Goreish, L. C. Kirkwood, L. A. Tolhurst and P. W. Stratford, Biomaterials, 2001, 22, 99–111 CrossRef CAS PubMed
- H. Chen, L. Yuan, W. Song, Z. Wu and D. Li, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS
- T. Blin, A. Kakinen, E. H. Pilkington, A. Ivask, F. Ding, J. F. Quinn, M. R. Whittaker, P. C. Ke and T. P. Davis, Polym. Chem., 2016, 7, 1931–1944 RSC
- S. Li, Y. Cai, J. Cao, M. Cai, Y. Chen and X. Luo, Polym. Chem., 2017, 8, 2472–2483 RSC
- G. Xie, C. Ma, X. Zhang, H. Liu, X. Guo, L. Yang, Y. Li, K. Wang and Y. Wei, Colloids Surf., B, 2017, 157, 166–173 CrossRef CAS PubMed
- B. Wang, T. Blin, A. Kakinen, X. Ge, E. H. Pilkington, J. F. Quinn, M. R. Whittaker, T. P. Davis, P. C. Ke and F. Ding, Polym. Chem., 2016, 7, 6875–6879 RSC
- Q. Jin, Y. Chen, Y. Wang and J. Ji, Colloids Surf., B, 2014, 124, 80–86 CrossRef CAS PubMed
- R. Matsuno and K. Ishihara, Nano Today, 2011, 6, 61–74 CrossRef CAS
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, J. Controlled Release, 2012, 161, 164–174 CrossRef CAS PubMed
- S. D. Weitman, R. H. Lark, L. R. Coney, D. W. Fort, V. Frasca, V. R. Zurawski Jr and B. A. Kamen, Canc. Res., 1992, 52, 3396–3401 CAS
- N. Gonen and Y. G. Assaraf, Drug Resist. Updates, 2012, 15, 183–210 CrossRef CAS PubMed
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2012, 64, 342–352 CrossRef
- J. A. Ledermann, S. Canevari and T. Thigpen, Ann. Oncol., 2015, 26, 2034–2043 CrossRef CAS PubMed
- B. Wu, P. Yu, C. Cui, M. Wu, Y. Zhang, L. Liu, C. X. Wang, R. X. Zhuo and S. W. Huang, Biomater. Sci., 2015, 3, 655–664 RSC
- G. Shim, M.-G. Kim, J. Y. Park and Y.-K. Oh, Adv. Drug Delivery Rev., 2016, 105, 205–227 CrossRef CAS PubMed
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed
- H. Shen, M. Liu, H. He, L. Zhang, J. Huang, Y. Chong, J. Dai and Z. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 6317–6323 CAS
- W. Miao, G. Shim, S. Lee, S. Lee, Y. S. Choe and Y.-K. Oh, Biomaterials, 2013, 34, 3402–3410 CrossRef CAS PubMed
- M. Kakran, N. G. Sahoo, H. Bao, Y. Pan and L. Li, Curr. Med. Chem., 2011, 18, 4503–4512 CrossRef CAS PubMed
- H. Hu, J. Yu, Y. Li, J. Zhao and H. Dong, J. Biomed. Mater. Res., Part A, 2012, 100, 141–148 CrossRef PubMed
- A. Sahu, W. I. Choi, J. H. Lee and G. Tae, Biomaterials, 2013, 34, 6239–6248 CrossRef CAS PubMed
- A. Siriviriyanun, M. Popova, T. Imae, L. V. Kiew, C. Y. Looi, W. F. Wong, H. B. Lee and L. Y. Chung, Chem. Eng. J., 2015, 281, 771–781 CrossRef CAS
- Y. Pan, H. Bao, N. G. Sahoo, T. Wu and L. Li, Adv. Funct. Mater., 2011, 21, 2754–2763 CrossRef CAS
- H. Bao, Y. Pan, Y. Ping, N. G. Sahoo, T. Wu, L. Li, J. Li and L. H. Gan, Small, 2011, 7, 1569–1578 CrossRef CAS PubMed
- C. Wang, S. Ravi, U. S. Garapati, M. Das, M. Howell, J. Mallela, S. Alwarappan, S. S. Mohapatra and S. Mohapatra, J. Mater. Chem. B, 2013, 1, 4396–4405 RSC
- G. Shim, J.-Y. Kim, J. Han, S. W. Chung, S. Lee, Y. Byun and Y.-K. Oh, J. Controlled Release, 2014, 189, 80–89 CrossRef CAS PubMed
- K. Liu, J.-J. Zhang, F.-F. Cheng, T.-T. Zheng, C. Wang and J.-J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC
- J. An, Y. Gou, C. Yang, F. Hu and C. Wang, Mater. Sci. Eng., C, 2013, 33, 2827–2837 CrossRef CAS PubMed
- A. Angelopoulou, E. Voulgari, E. K. Diamanti, D. Gournis and K. Avgoustakis, Eur. J. Pharm. Biopharm., 2015, 93, 18–26 CrossRef CAS PubMed
- Y. Liu, Y. Zhang, T. Zhang, Y. Jiang and X. Liu, Carbon, 2014, 71, 166–175 CrossRef CAS
- C. He, Z. Q. Shi, C. Cheng, H. Q. Lu, M. Zhou, S. D. Sun and C. S. Zhao, Biomater. Sci., 2016, 4, 1431–1440 RSC
- R. Narain and S. P. Armes, Biomacromolecules, 2003, 4, 1746–1758 CrossRef CAS PubMed
- G. I. Titelman, V. Gelman, S. Bron, R. L. Khalfin, Y. Cohen and H. Bianco-Peled, Carbon, 2005, 43, 641–649 CrossRef CAS
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed
- Y. Matsuo, K. Tahara and Y. Sugie, Carbon, 1997, 35, 113–120 CrossRef CAS
- T. Mitra, P. J. Manna, S. T. K. Raja, A. Gnanamani and P. P. Kundu, RSC Adv., 2015, 5, 98653–98665 RSC
- L. G. Xu, L. Cheng, C. Wang, R. Peng and Z. Liu, Polym. Chem., 2014, 5, 1573–1580 RSC
- X. Yang, Y. Wang, X. Huang, Y. Ma, Y. Huang, R. Yang, H. Duan and Y. Chen, J. Mater. Chem., 2011, 21, 3448–3454 RSC
- X. Chenivesse, D. Franco and C. Brechot, J. Hepatol., 1993, 18, 168–172 CrossRef CAS PubMed
- U. Asghar and T. Meyer, J. Hepatol., 2012, 56, 686–695 CrossRef PubMed
- R. W. Johnstone, A. A. Ruefli and S. W. Lowe, Cell, 2002, 108, 153–164 CrossRef CAS PubMed
- R. S. Kerbel and B. A. Kamen, Nat. Rev. Cancer, 2004, 4, 423–436 CrossRef CAS PubMed
- Z. Luo, X. Ding, Y. Hu, S. Wu, Y. Xiang, Y. Zeng, B. Zhang, H. Yan, H. Zhang, L. Zhu, J. Liu, J. Li, K. Cai and Y. Zhao, ACS Nano, 2013, 7, 10271–10284 CrossRef CAS PubMed
- S. Kugel Desmoulin, L. Wang, E. Hales, L. Polin, K. White, J. Kushner, M. Stout, Z. Hou, C. Cherian, A. Gangjee and L. H. Matherly, Mol. Pharmacol., 2011, 80, 1096–1107 CrossRef PubMed
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, C. Fan and Q. Huang, Carbon, 2011, 49, 986–995 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08287h |
| This journal is © The Royal Society of Chemistry 2017 |