
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphology control and enhancement of 1.5 μm emission in Ca2+/Ce3+ codoped NaGdF4:Yb3+, Er3+ submicrorods
Jiangyun Dai a,
Chao Yanga,
Hong Zhanga,
Hua Zhanga,
Guoying Feng*a and
Shouhuan Zhou*ab
a,
Chao Yanga,
Hong Zhanga,
Hua Zhanga,
Guoying Feng*a and
Shouhuan Zhou*ab
aInstitute of Laser and Micro/Nano Engineering, College of Electronics and Information Engineering, Sichuan University, Chengdu 610064, China. E-mail: guoing_feng@scu.edu.cn
bNorth China Research Institute of Electro-Optics, Beijing 100015, China. E-mail: zhoush@scu.edu.cn
First published on 13th October 2017
Abstract
The successful synthesis of rare earth doped nano/microcrystals with the desired morphology, uniform monodispersity, and intense infrared emission is of fundamental significance, and would facilitate their application in infrared emission-related miniaturization and integration in optoelectronics. However, simultaneously controlling the morphology and enhancing the infrared emission through simple and effective methods has not been well explored. In this study, via Ca2+ doping with an optimized concentration of 25 mol% in the hydrothermal solution, irregular NaGdF4:Yb/Er submicrocrystals converted into highly uniform submicrorods. Meanwhile, an obvious enhancement of visible upconversion (UC) and near-infrared (NIR) downconversion (DC) emission were obtained, probably due to the improved morphology and lowering the local crystal field symmetry around the rare earth ions induced by Ca2+ doping. Furthermore, Ce3+ ions with an optimized concentration of 2 mol% were codoped to intentionally tailor the branching ratio through efficient energy transfer from Er3+ to Ce3+. As a result, the ∼1530 nm emission intensity of the NaGdF4: 25Ca/2Ce/20Yb/2Er submicrorods increased by about 6.4 times in comparison with the NaGdF4:20Yb/2Er counterpart.
1. Introduction
In the past decades, rare earth doped materials, which are able to exhibit infrared DC emission in addition to their visible UC emission of lanthanide ions, have attracted considerable attention due to their favorable merits, such as large anti-Stokes shifts, long excited-state lifetimes, narrow emission bandwidths and high photostability.1–4 Based on these inherent advantages, rare earth doped materials play an important role in fabricating the active optical components in optoelectronic applications like laser materials, flat-panel displays, and optical sensors.5–9 Notably, rare earth doped nano/microcrystals are of great importance for expanding the applications of miniaturized and integrated optoelectronics.10,11 Among the rare earth ions, Er3+ is especially attractive because of its NIR emission wavelength of approximately 1530 nm, which lies in the low-loss telecommunication window in optical communication.12,13 However, the low intra-4f-configurational absorption cross section of Er3+ commonly leads to a weak emission intensity upon inefficient pumping.14 Fortunately, Yb3+ ions possess a higher absorption cross section and its 2F5/2 energy level is resonant with the 4I11/2 energy level of Er3+. Thus, introduction of Yb3+ as an ideal sensitizer can realize efficient NIR emission of Er3+ upon excitation at 980 nm.It is well established that the optical, electrical and magnetic properties of nano/microcrystals are depended on not only their composition, but also their geometrical factors including phase, morphology and size. Especially, when the rare earth doped materials are synthesized in the form of the one-dimensional nano/microstructures, they would stimulate great attention due to their marked shape-specific and quantum-confinement effects.15,16 Although several methods have been used to synthesize the one-dimensional nano/microcrystals with a well defined morphology, finding a novel and facile strategy is still imperative.15 Furthermore, to realize their practical applications, simultaneously obtaining an efficient infrared optical signal is also desired. Hence, many efforts have been devoted to optimizing the performance of rare earth doped materials by adopting novel strategies, such as dye sensitizing,17 core–shell structure,18,19 and host lattice manipulation.20 Impressively, host lattice manipulation via impurity doping, which has the ability to modify the electron charge density and crystal symmetry of rare earth ions, could simultaneously enhance the emission intensity and control the well-defined morphology in a facile and straightforward way.21,22 Although many previous works have adopted this strategy to enhance the visible or NIR UC emission,23–25 enhancing the luminescence in the NIR DC emission range has rarely been studied.
For Er3+ doped materials, NIR emission at 1530 nm is always accompanied by the visible UC emission upon excitation at 980 nm. However, the visible emission is undesired in the field of infrared application (e.g., optical amplifiers), which would reduce the intensity of NIR emission (∼1530 nm) of Er3+ ions. One of the approaches to circumvent these obstacles is to improve the low branching ratio for the NIR emission. Recent reports show doping with Ce3+ ions can significantly enhance the 4I11/2 → 4I13/2 transition probability of Er3+.26,27 The possible mechanism is the non-radiative phonon-assisted energy transfer 2F5/2(Ce3+) + 4I11/2(Er3+) → 2F7/2(Ce3+) + 4I13/2(Er3+) which facilitates the population of 4I13/2 of Er3+ and decreases UC emission. Therefore, based on the forementioned theories, codoping Ca2+ and Ce3+ in the crystals would synergistically enhance NIR emission.
In this work, via doping impurity Ca2+ ions into the host lattices by substituting Gd3+ ions, we demonstrated the irregular NaGdF4:Yb/Er submicrocrystals converted into well-defined submicrorods, and the visible UC and NIR DC emission were enhanced through a facile and effective hydrothermal method. Additionally, benefiting from the highly efficient energy transfer from Er3+ to Ce3+ by co-doping Ce3+, the NIR DC was further enhanced by about 6.4 times in comparison with that of the NaGdF4:Yb/Er counterpart.
2. Experimental
2.1 Materials and reagents
Ethylenediamine tetraacetic acid (EDTA > 99.4%) was supplied by Sigma-Aldrich. Gd(NO3)3·6H2O (99.99%), Er(NO3)3·5H2O (99.9%), Yb(NO3)3·5H2O (99.9%), sodium hydroxide (NaOH > 96.0%), ammonium fluoride (NH4F > 98.0%), and highly concentrated nitric acid were purchased from Aladdin Chemical Reagent Co. All chemicals were of analytical grade and were used directly as received without further purification, deionized water was used throughout.2.2 Synthesis of 20% Yb3+, 2% Er3+:NaGdF4 submicrocrystals
The rare-earth doped NaGdF4 (1 mmol) submicrocrystals were synthesized by a hydrothermal method. Firstly, 12.5 ml of aqueous solution containing 0.4 g EDTA and 1.05 ml of NaOH aqueous solution (5.0 M) were mixed under stirring until the solution becomes even. Subsequently, 5 ml of Ln(NO3)3 (Ln = Gd, Yb, Er) aqueous solutions (0.2 M), 8 ml of NH4F (2.0 M) aqueous solutions and 7 ml dilute nitric acid (1 M) were added to the mixture. After agitating at room temperature for 90 min, the resultant solution was poured into a 50 ml Teflon-lined autoclave and heated subsequently to 190 °C for 48 h. Finally, the autoclave was cooled to room temperature naturally. The products deposited at the bottom of the vessel were collected by centrifugation, washed with deionized water and ethanol several times, and then dried at 60 °C for 24 h in air. The procedure for the synthesis of the Ca2+ doped, Ce3+ doped and Ca2+/Ce3+ codoped 20% Yb3+, 2% Er3+:NaGdF4 submicrocrystals were similar to the case of the 20% Yb3+, 2% Er3+:NaGdF4 submicrocrystals.2.3 Characterization
The X-ray powder diffraction (XRD) patterns were carried out with a powder diffractometer (Japan Rigaku D/max-rB) with Cu Kα radiation (λ = 0.154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 178 nm). The morphology was studied using a field-emission scanning electron microscope (FESEM) operated at an acceleration voltage of 5 kV (HitachiSU-8220). The NIR luminescence spectroscopy was recorded by a monochromator (Princeton Instrument SP2750) equipped with an InGaAs infrared detector (ACTON), the UC luminescence spectroscopy were measured by a single mode fiber with an Ocean Optics USB4000 spectrometer (optical resolution ∼0.2 nm) using a continuous 980 nm laser from an semiconduct laser as the excitation source, and the area of the samples that were exposed to the laser radiation was about 1.5 mm2. The decay time was measured by a digital phosphor oscilloscope (Tektronix DPO 4032) using the 980 nm laser from an OPO (an optical parametric oscillation, INNOLAS) as the excitation source. All the measurements were implemented at room temperature (RT).
178 nm). The morphology was studied using a field-emission scanning electron microscope (FESEM) operated at an acceleration voltage of 5 kV (HitachiSU-8220). The NIR luminescence spectroscopy was recorded by a monochromator (Princeton Instrument SP2750) equipped with an InGaAs infrared detector (ACTON), the UC luminescence spectroscopy were measured by a single mode fiber with an Ocean Optics USB4000 spectrometer (optical resolution ∼0.2 nm) using a continuous 980 nm laser from an semiconduct laser as the excitation source, and the area of the samples that were exposed to the laser radiation was about 1.5 mm2. The decay time was measured by a digital phosphor oscilloscope (Tektronix DPO 4032) using the 980 nm laser from an OPO (an optical parametric oscillation, INNOLAS) as the excitation source. All the measurements were implemented at room temperature (RT).
3. Results and discussion
3.1 Morphology and crystal structure
It should be pointed out that rare earth doped fluorides with high quality and anisotropic morphology could be synthesized in an appropriately surplus F−/Ln3+ molar ratio.28–31 In this view, the F−/Ln3+ molar ratio was set to be 16. A series of 20% Yb/2% Er:NaGdF4 crystals doped with different Ca2+ and Ce3+ concentration were synthesized and the corresponding XRD patterns are shown in Fig. 1. XRD patterns (Fig. 1a) suggest that the samples of different Ca2+ doping concentration are identical with JCPDS Card (no. 27-0699), showing that the Ca2+ are completely embedded into the crystal lattice of NaGdF4 by replacing the sites of Gd3+ ions, without finding obvious shifting of diffraction peaks due to the F− vacancies and ionic radius difference between Gd3+ and Ca2+.24 Fig. 1b shows the X-ray diffraction patterns of 20% Yb/2% Er:NaGdF4 crystals doped with different Ce3+ concentration. No additional peaks are observed, indicating that the doping Ce3+ ions have a negligible effect on the phase structure and Ce3+ ions have substituted the Gd3+ sites in the NaGdF4 host. Notably, the X-ray diffraction patterns of 20% Yb/2% Er:NaGdF4 crystals codoped with 25% Ca2+ and 2% Ce3+ are also given in Fig. 1b, which can be still indexed to the β-NaGdF4 phase. The schematic diagram of β-NaGdF4 crystal structure and coordination environment of Na+/Gd3+ ions are shown in Fig. 1c. There are three types of cationic sites in the unit cell of β-NaGdF4: a six-fold coordinated one occupied by Na+ and vacancies, a nine-fold coordinated position occupied by Gd3+, and another nine-fold coordinated site occupied randomly by Na+ and Gd3+. The two different Gd3+ sites are coordinated in the form of tricapped trigonal prisms.32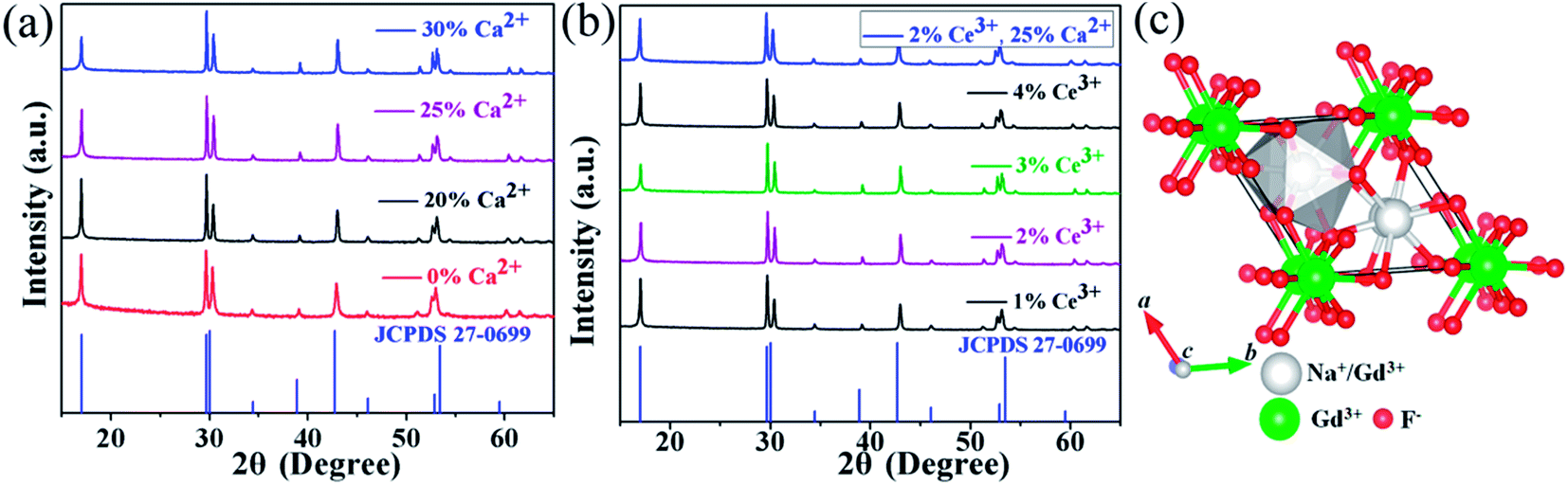 | ||
| Fig. 1 XRD patterns of the NaGdF4:xCa/yCe/20Yb/2Er samples, (a) x = 0%, 20%, 25%, 30%, y = 0; (b) x = 0, y = 1%, 2%, 3%, 4%. (c) Schematic illustration of β-NaGdF4 structure and coordination environment of the Na+/Gd3+. | ||
FESEM were then carried out to study the morphology and size of NaGdF4:20% Yb/2% Er samples doped with different Ca2+ and Ce3+ doping concentrations. Fig. 2a presents the FESEM image of the NaGdF4:20% Yb/2% Er submicrocrystals without Ca2+ doping and shows irregular microrods. Notably, many cracks are observed on the surface, which could be ascribed to the microrods deriving from decreasing the core of spherical aggregates and extruding between the growing microrods.33 Interestingly, once doped 20% Ca2+ in the NaGdF4:20% Yb/2% Er sample, the crystals (Fig. 2b) exhibit uniform hexagonal submicrorods, although with a wide size distribution. Further increasing the doping content of Ca2+ results in a more even size distribution. Furthermore, when the crystal size is denoted as “length × radius”, the mean size of NaGdF4:xCa/20Yb/2Er (x = 0%, 20%, 25%, 30%) crystals are measured to be 0.38 × 0.96, 0.24 × 1.26, 0.21 × 1.34 and 0.33 × 1.82 μm, respectively. Without the help of Ca2+ doping, the morphology of NaGdF4:xCe/20Yb/2Er (x = 1%, 2%, 3%, 4%) crystals still inherit irregular submicrorods as that of NaGdF4:20Yb/2Er, the typical FESEM image of NaGdF4:2Ce/20Yb/2Er crystal is shown in Fig. 2e. With codoping of 25% Ca2+ and 2% Ce3+, the sample keep highly uniform submicrorods (Fig. 2f).
 | ||
| Fig. 2 FESEM images of NaGdF4:20% Yb/2% Er submicrocrystals doped with (a) 0% Ca2+, (b) 20% Ca2+, (c) 25% Ca2+, (d) 30% Ca2+, (e) 2% Ce3+, (f) 2% Ce3+, 25% Ca2+. | ||
Based on the results stated above, the effect induced by Ca2+ doping on the morphology evolution of NaGdF4 submicrocrystals is proposed, as schematically shown in Fig. 3. First of all, it is essential to have a full understanding of the crystal growth of NaGdF4 submicrocrystals without Ca2+ doping. As is well known, hexagonal NaGdF4 has a very close crystalline structure with hexagonal NaYF4, so they may undergo a same crystal growth kinetic process in the similar hydrothermal condition.31 The irregular NaGdF4 submicror-ods with cracked surface were obtained due to two main factors. One is that final submicrorods originate from decreasing the core of spherical aggregates and extruding between the growing submicrods.33 And another one is that the fast growth rate of the top/bottom planes would hinder the continuing growth of six energetically equivalent prismatic side planes.29,34 Once upon doping Ca2+ in the reaction solution, extra F− ions are remaining in system due to charge compensation and a transient electric dipole with the positive pole pointing outward is also formed on the grain surface.24,35,36 Both of them accelerate the diffusion of F− ions in the system under the control of monomer diffusion to the grain surface by attractive interaction,36,37 finally facilitating the anisotropic growth of NaGdF4 submicrorods. Hence, along with increasing Ca2+ doping content, the surface is becoming more smooth and the size distribution more even. It should be noting that the sharp ends of NaGdF4 crystals tend to be more flat ends, which is indirect evidence of F− concentration increasing only induced by Ca2+ doping.38
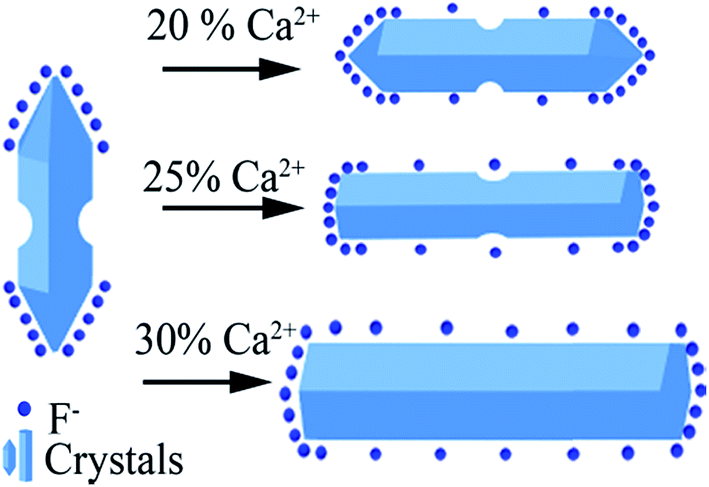 | ||
| Fig. 3 Schematic illustration of possible mechanism of Ca2+ doping and its effect on morphology evolution of NaGdF4 submicrocrystals. | ||
3.2 Optical properties
Fig. 4a shows the visible UC emission spectra of the NaGdF4:20% Yb/2% Er submicrocrystals doped with different Ca2+ concentrations, which are recorded under 980 nm laser excitation with the pump power of 150 mW. It is observed that there are three UC bands, centred at 519, 537, and 652 nm, which are ascribed to 2H11/2 → 4I15/2 (519 nm), 4S3/2 → 4I15/2 (537 nm), 4F9/2 → 4I15/2 (652 nm), respectively. Meanwhile, an obvious enhancement of the UC emission is observed upon 20% Ca2+ doping. The enhancement reaches the most at 25% Ca2+ doping, then declining with further increasing the Ca2+ concentration to 30%. Furthermore, the integrated visible UC emission intensity of crystals doped with 25% Ca2+ is about 5.1 times higher than that of the crystals without Ca2+ doping. To investigate the effect of Ca2+ doping on the UC emission, we first determined the number of photons required for the red and green luminescence intensity, which obey the following relationship with the pumping power:39| Iup = Pn | (1) |
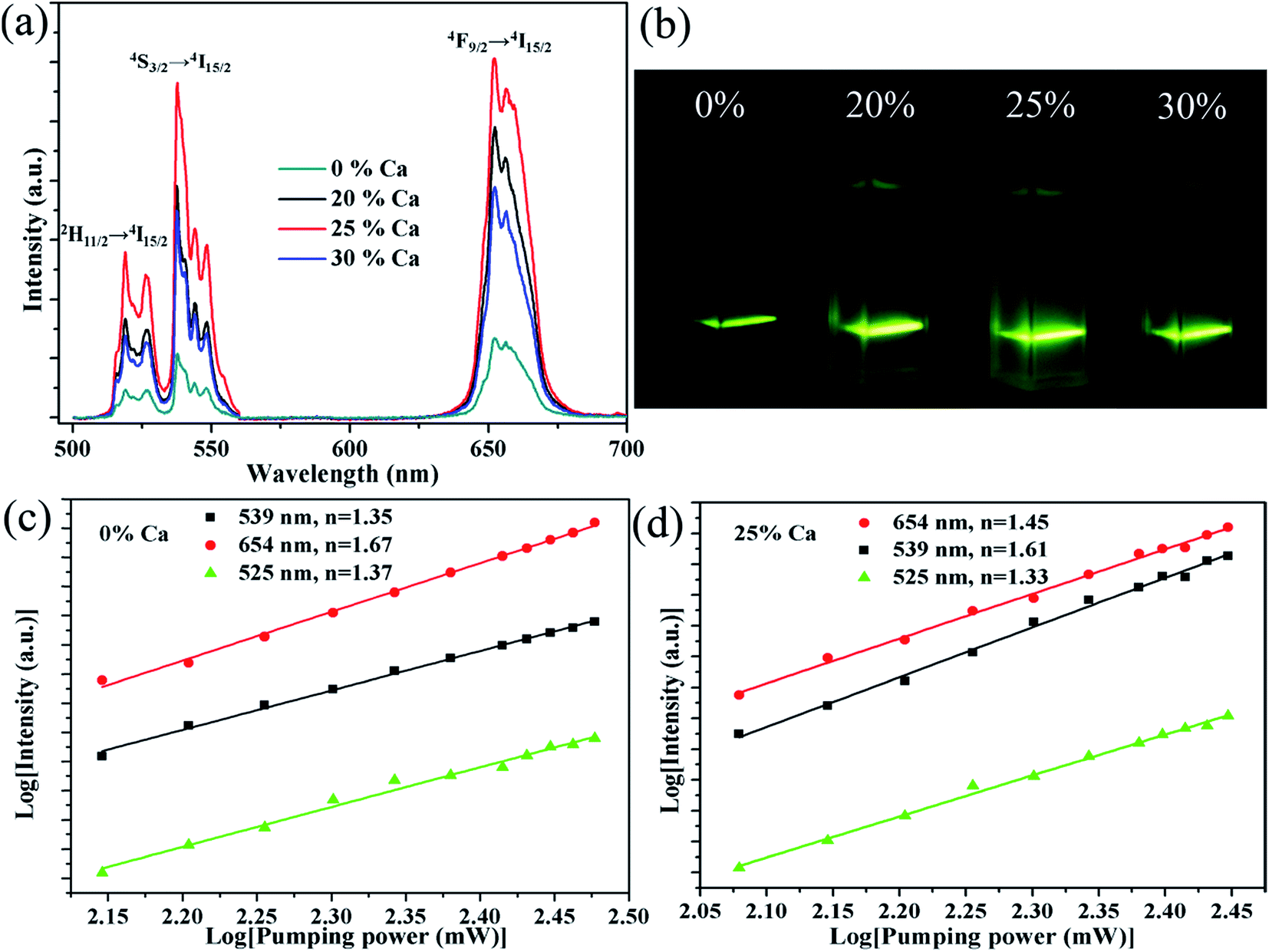 | ||
| Fig. 4 UC emission spectra of NaGdF4:20Yb/2Er/xCa (x = 0%, 20%, 25%, 30%) under 980 nm laser excitation. (b) The corresponding photographs of the submicrocrystals dispersed in water. log–log plots of the UC emission intensity against laser excitation power for NaGdF4:20Yb/2Er (c), and NaGdF4:25Ca/20Yb/2Er (d). | ||
Fig. 5 shows the NIR emission spectra of the submicrocrystals, which are recorded at the pump power of 270 mW. Obviously, the characteristic emission peak centered at 1535 nm is attributed to the 4I13/2 → 4I15/2 transitions of Er3+ ions. The NIR emission intensity increases with Ca2+ dopant concentrations from 0% to 25% before declining as Ca2+ dopant concentration is increased to 30% (Fig. 5a). The integrated NIR DC emission intensity shows an identical trend to that of the visible UC emission. Therefore, the optimum Ca2+ dopant concentration is determined to be 25%. Next, to further investigate the effect of Ce3+ concentration on the NIR emission properties, we systematically investigated the NIR emission spectra of NaGdF4:20Yb/2Er/xCe (x = 1%, 2%, 3%, 4%) crystals obtained upon excitation at 980 nm and the results are summarized in Fig. 5b. As the concentration of Ce3+ ions increases from 0 to 2%, the NIR emission significantly increases and reaches the most. The enhanced NIR emission arises from the efficient non-radiative phonon-assisted energy transfer between the 4I11/2 → 4I13/2 transition of Er3+ ions and the 2F5/2 → 2F7/2 transition of Ce3+ ions, which improves the branching ratio of NIR emission and suppress the visible emission by altered the UC pathways.26,27 All these transitions are illustrated in an energy levels diagram in Fig. 6.
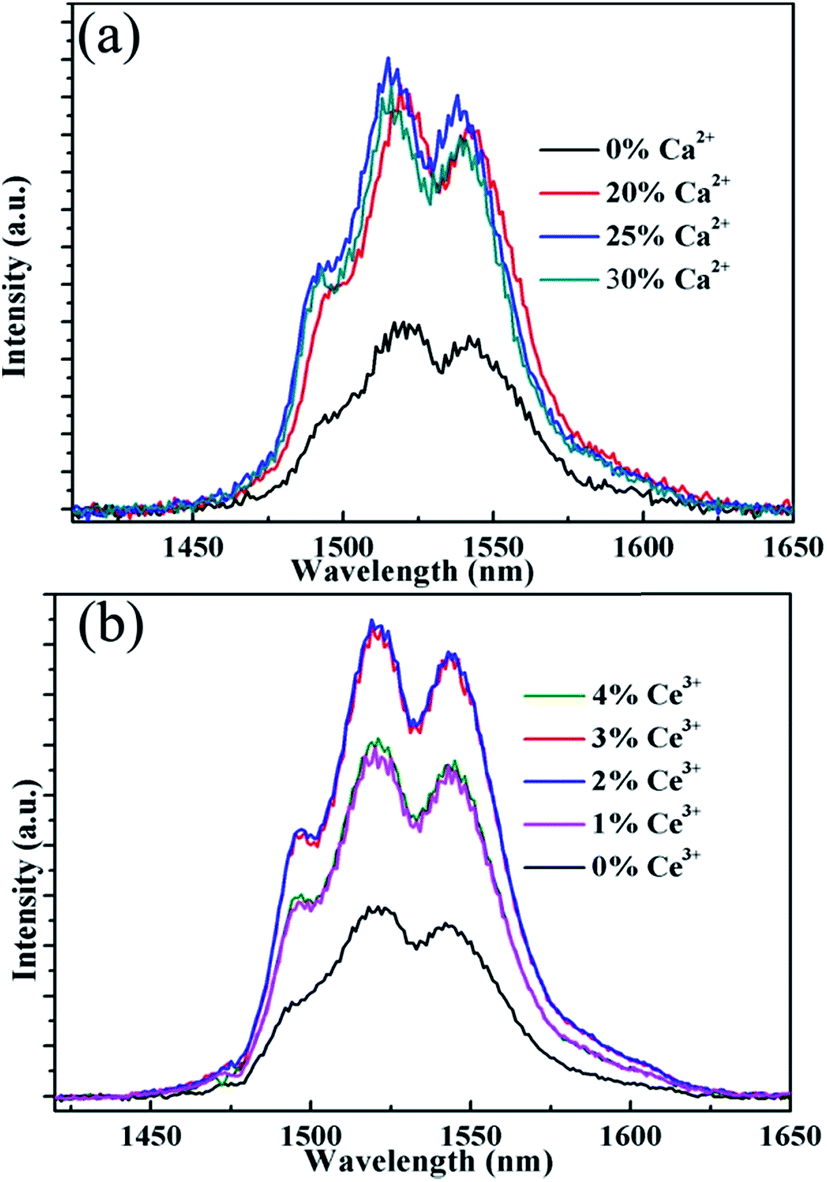 | ||
| Fig. 5 NIR emission spectra of the sample NaGdF4:xCa/yCe/20% Yb/2% Er, (a) x = 0%, 20%, 25%, 30%, y = 0; (b) x = 0, y = 1%, 2%, 3%, 4% under 980 nm laser excitation. | ||
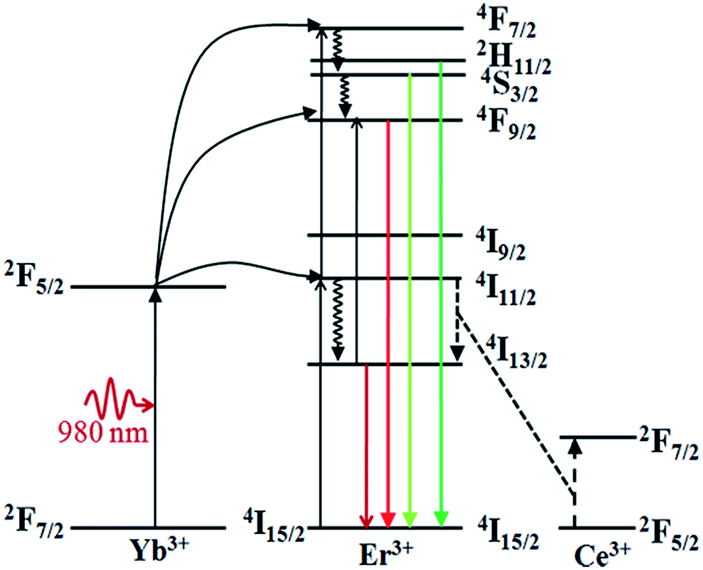 | ||
| Fig. 6 Energy level diagram of Yb3+ ion, Er3+ ion, Ce3+ ion, as well as the relevant transitions under the 980 nm laser excitation. | ||
It is essential to compare the NIR emission intensity of samples doped with the optimum Ca2+ and Ce3+ dopant concentration. Through measuring the integrated NIR emission intensity (Fig. 7a), the NIR emission of the 20Yb/2Er:NaGdF4 submicrorods doped with 25% Ca2+ and 2% Ce3+ is about 6.4 times higher than that of 20Yb/2Er:NaGdF4 submicrocrystals. Notably, the enhanced factor of the sample doped with 25% Ca2+ and 2% Ce3+ is only 2.3, 2.7, respectively. These results suggest that codoping Ca2+/Ce3+ can greatly enhance the NIR emission intensity. Fig. 7b shows the log–log plots of the NIR emission intensity against laser excitation power of the submicrocrystals doped with no extra ions, 25% Ca2+, 2% Ce3+, and coped with 25% Ca2+/2% Ce3+, the experimental data were fitted well with a linear relationship. The quasi-linear power dependence of 1530 nm emission in submicrocrystals doped without Ce3+ and quasi-quadratic power dependence in submicrocrystals doped with Ce3+ can be observed. The different character of the power-dependent luminescence for the 4I13/2 manifold is possibly due to the competition between UC and linear decay.40
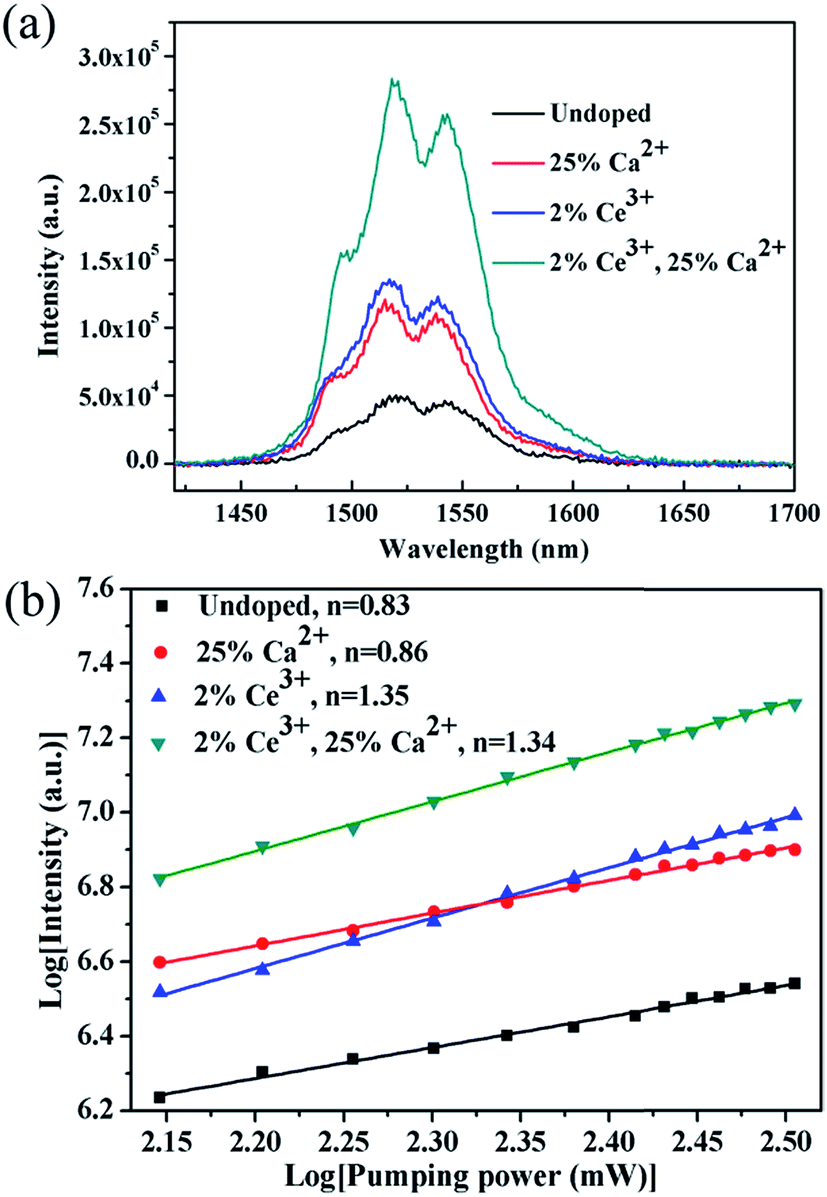 | ||
| Fig. 7 NIR emission spectra (a) and log–log plots of the NIR emission intensity against laser excitation power (b) of samples single doped with no extra ions, 25% Ca2+, 2% Ce3+, and coped with 25% Ca2+/2% Ce3+, the experimental data were fitted well with a linear relationship. | ||
To further characterize the NIR emitting properties of the obtained samples, the fluorescence decay curves of the 1530 nm emission for the crystals are shown in Fig. 8. To obtain the decay constants, the luminescence decay curves were fitted by a bi-exponential equation,
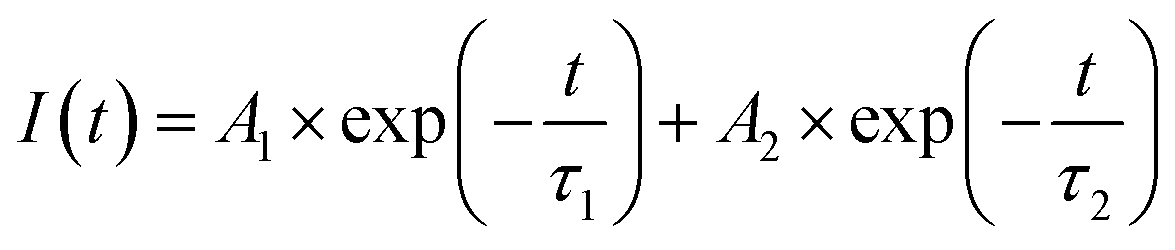 | (2) |
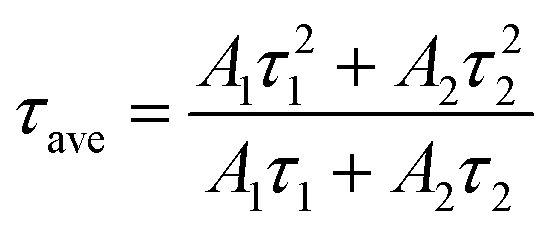 | (3) |
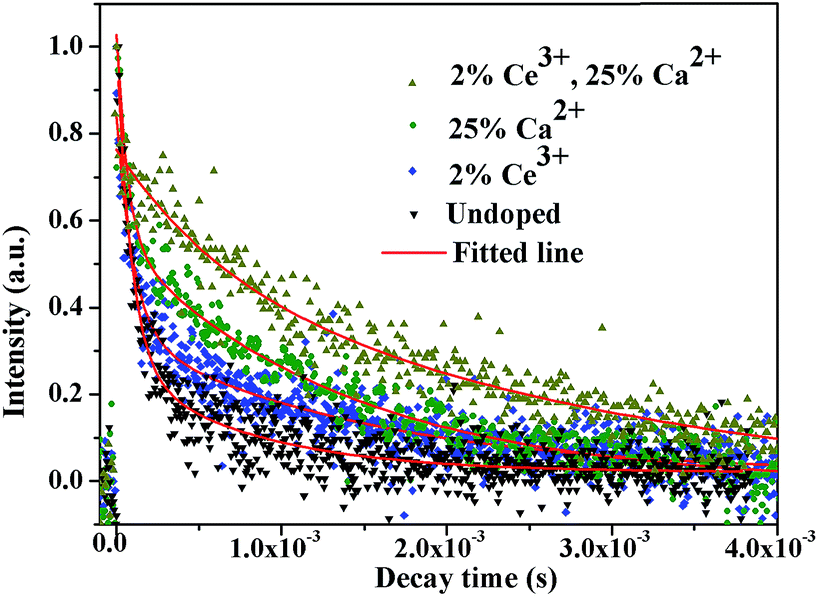 | ||
| Fig. 8 The decay curves of Er3+ 1530 nm emission wavelength upon 980 nm excitation. | ||
The estimated decay time increases from ∼609.2 μs for NaGdF4:20% Yb/2% Er to ∼1319.6 μs for crystals doped with 20% Ca2+, ∼1416.1 μs for crystals doped with 2% Ce3+, and ∼2942.5 μs for crystals doped with 20% Ca2+/2% Ce3+, respectively. These results show that Ca2+/Ce3+ codoping can effectively improve the NIR emission efficiency for NaGdF4:20% Yb/2% Er submicrorods.
4. Conclusions
In summary, we have disclosed morphology evolution mechanism and increased visible UC emission intensity of NaGdF4:20% Yb/2% Er submicrocrystals induced by Ca2+ doping. The influence of Ca2+ and Ce3+ doping concentration on the NIR emission intensity have been systematically investigated and the optimized Ca2+ and Ce3+ doping concentration was 25% and 2%, respectively. The NIR emission intensity was enhanced by ∼6.4 fold after introducing both of 25% Ca2+ and 2% Ce3+. The doping of Ca2+ mainly lowers the local crystal field symmetry around the rare earth and improves the morphology of NaGdF4:20% Yb/2% Er submicrocrystals. And the doping of Ce3+ facilitates the energy transfer between Ce3+ and Er3+. Hence, codoping Ca2+/Ce3+ synergistically enhance the NIR emission intensity. We believe that this novel strategy for simultaneous control of morphology and improving the NIR emission of rare earth doped nano/microcrystals may expand the application in IR-related miniaturization and integration of optoelectronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge funding support from the National Natural Science Foundation of China (NSFC) (11574221) and “the Fundamental Research Funds for the Central Universities” (2016SCU11003).Notes and references
- T. Soukka, K. Kuningas, T. Rantanen, V. Haaslahti and T. Lövgren, J. Fluoresc., 2005, 15, 513–528 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q.-H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS PubMed.
- X. Zhu, Q. Su, W. Feng and F. Li, Chem. Soc. Rev., 2017, 46, 1025–1039 RSC.
- T. Wang, H. Yu, C. K. Siu, J. Qiu, X. Xu and S. F. Yu, ACS Photonics, 2017, 4, 1539–1543 CrossRef CAS.
- R. Dey, A. Pandey and V. K. Rai, Spectrochim. Acta, Part A, 2014, 128, 508–513 CrossRef CAS PubMed.
- P. Du and J. S. Yu, Chem. Eng. J., 2017, 327, 109–119 CrossRef CAS.
- D. Chen, M. Xu and P. Huang, Sens. Actuators, B, 2016, 231, 576–583 CrossRef CAS.
- D. Chen, M. Xu, P. Huang, M. Ma, M. Ding and L. Lei, J. Mater. Chem. C, 2017, 5, 5434–5443 RSC.
- K. Korthout, P. F. Smet and D. Poelman, Appl. Phys. Lett., 2009, 94, 051104 CrossRef.
- J. D. Bradley and M. Pollnau, Laser Photonics Rev., 2011, 5, 368–403 CrossRef CAS.
- N. Mais, J. P. Reithmaier, A. Forchel, M. Kohls, L. Spanhel and G. Müller, Appl. Phys. Lett., 1999, 75, 2005–2007 CrossRef CAS.
- J. Wang, J. Hu, D. Tang, X. Liu and Z. Zhen, J. Mater. Chem., 2007, 17, 1597–1601 RSC.
- F. Rivera-López, P. Babu, L. Jyothi, U. R. Rodríguez-Mendoza, I. R. Martín, C. K. Jayasankar and V. Lavín, Opt. Mater., 2012, 34, 1235–1240 CrossRef.
- Y. P. Fang, A. W. Xu, L. P. You, R. Q. Song, J. C. Yu, H. X. Zhang, Q. Li and H. Q. Liu, Adv. Funct. Mater., 2003, 13, 955–960 CrossRef CAS.
- G. Gao, C. Zhang, Z. Zhou, X. Zhang, J. Ma, C. Li, W. Jin and D. Cui, Nanoscale, 2013, 5, 351–362 RSC.
- A. Shalav, B. S. Richards, T. Trupke, K. W. Krämer and H. U. Güdel, Appl. Phys. Lett., 2005, 86, 013505 CrossRef.
- D. Chen and P. Huang, Dalton Trans., 2014, 43, 11299–11304 RSC.
- M. Mondal, V. K. Rai and C. Srivastava, Chem. Eng. J., 2017, 327, 838–848 CrossRef CAS.
- D. Chen, L. Liu, P. Huang, M. Ding, J. Zhong and Z. Ji, J. Phys. Chem. Lett., 2015, 6, 2833–2840 CrossRef CAS PubMed.
- D. Chen and Y. Wang, Nanoscale, 2013, 5, 4621–4637 RSC.
- A. K. Singh, S. K. Singh and S. B. Rai, RSC Adv., 2014, 4, 27039–27061 RSC.
- X. Wang, Y. Bu, Y. Xiao, C. Kan, D. Lu and X. Yan, J. Mater. Chem. C, 2013, 1, 3158–3166 RSC.
- L. Lei, D. Chen, J. Xu, R. Zhang and Y. Wang, Chem.–Asian J., 2014, 9, 728–733 CrossRef CAS PubMed.
- L. Mukhopadhyay and V. K. Rai, New J. Chem., 2017, 41, 7650–7661 RSC.
- X. Gao, X. Liu, Q. Wen, X. Yang and S. Xiao, J. Appl. Phys., 2014, 116, 173105 CrossRef.
- X. Zhao and M. C. Tan, J. Mater. Chem. C, 2015, 3, 10207–10214 RSC.
- Y. Wang, R. Cai and Z. Liu, CrystEngComm, 2011, 13, 1772–1774 RSC.
- X. Liang, X. Wang, J. Zhuang, Q. Peng and Y. Li, Adv. Funct. Mater., 2007, 17, 2757–2765 CrossRef CAS.
- L. Tian, L. Tan, Q. Sun, S. Xiang, Q. Xiao, J. Tang and G. Zhu, J. Rare Earths, 2012, 30, 1260–1264 CrossRef CAS.
- T. Zhou, X. Jiang, C. Zhong, X. Tang, S. Ren, Y. Zhao, M. Liu, X. Lai, J. Bi and D. Gao, J. Lumin., 2016, 175, 1–8 CrossRef CAS.
- M. Karbowiak, A. Mech, A. Bednarkiewicz, W. Stręk and L. Kępiński, J. Phys. Chem. Solids, 2005, 66, 1008–1019 CrossRef CAS.
- F. Tao, F. Pan, Z. Wang, W. Cai and L. Yao, CrystEngComm, 2010, 12, 4263–4267 RSC.
- Z. Chen, Z. Liu, Y. Liu, K. Zheng and W. Qin, J. Fluorine Chem., 2012, 144, 157–164 CrossRef CAS.
- D. Chen, Y. Yu, F. Huang, P. Huang, A. Yang and Y. Wang, J. Am. Chem. Soc., 2010, 132, 9976–9978 CrossRef CAS PubMed.
- L. Lei, D. Chen, P. Huang, J. Xu, R. Zhang and Y. Wang, Nanoscale, 2013, 5, 11298–11305 RSC.
- X. Peng, J. Wickham and A. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS.
- M. Ding, D. Chen, S. Yin, Z. Ji, J. Zhong, Y. Ni, C. Lu and Z. Xu, Sci. Rep., 2015, 5, 12745 CrossRef CAS PubMed.
- Z. Xiong, Y. Yang and Y. Wang, RSC Adv., 2016, 6, 75664–75668 RSC.
- D. Xu, C. Liu, J. Yan, S. Yang and Y. Zhang, J. Phys. Chem. C, 2015, 119, 6852–6860 CAS.
- J. W. Stouwdam and F. C. J. M. van Veggel, Nano Lett., 2002, 2, 733–737 CrossRef CAS.
- J. Wu, S. Shi, X. Wang, J. Li, R. Zong and W. Chen, J. Mater. Chem. C, 2014, 2, 2786–2792 RSC.
- T. Fujii, K. Kodaira, O. Kawauchi, N. Tanaka, H. Yamashita and M. Anpo, J. Phys. Chem. B, 1997, 101, 10631–10637 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |