
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAggregation of atomically precise graphene nanoribbons
Mikhail Shekhirev a,
Timothy H. Voa,
Donna A. Kunkelb,
Alexey Lipatova,
Axel Endersbc and
Alexander Sinitskii*ac
a,
Timothy H. Voa,
Donna A. Kunkelb,
Alexey Lipatova,
Axel Endersbc and
Alexander Sinitskii*ac
aDepartment of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588, USA. E-mail: sinitskii@unl.edu
bDepartment of Physics and Astronomy, University of Nebraska – Lincoln, Lincoln, NE 68588, USA
cNebraska Center for Materials and Nanoscience, University of Nebraska – Lincoln, Lincoln, NE 68588, USA
First published on 27th November 2017
Abstract
Solution bottom-up approaches can be used to prepare bulk quantities of narrow atomically precise graphene nanoribbons (GNRs) with various widths and geometries. These GNRs are often considered as promising materials for electronic and optoelectronic applications. However, the handling and processing of nanoribbons for practical applications can be difficult because of their entanglement and aggregation, and thus poor solubility in conventional solvents. In this work, we studied the aggregation-dependent properties of solution-synthesized chevron GNRs in a variety of solvents. We demonstrate that the spectroscopic features observed in the experimentally measured absorbance spectra of chevron GNRs are in a good agreement with the theoretically predicted excitionic transitions. We also show that the absorbance spectra of GNRs evolve with aggregation time, which is important to consider for the spectroscopic determination of optical bandgaps of nanoribbons. We discuss two types of GNR assemblies: bulk aggregates of π–π stacked nanoribbons that form in a solution and rather long one-dimensional (1D) structures that were observed on a variety of surfaces, such as Au(111), mica and Si/SiO2. We demonstrate that the few-μm-long 1D GNR structures can be conveniently visualized by conventional microscopy techniques and used for the fabrication of electronic devices.
Introduction
Narrow graphene nanoribbons (GNRs) attract considerable attention because of their promising semiconductor properties and potential applications in electronics and photovoltaics.1 According to theoretical studies, the properties of GNRs strongly depend on their width, edge structure and termination.1 Precise control of the structural parameters is very important for achieving GNRs with well-defined electronic and optical properties. Because of recent advances in bottom-up solution synthesis of GNRs, nanoribbons with narrow widths (w < 2 nm) and atomically precise edges can now be produced in large quantities.2–14 However, handling and processing of nanoribbons for the fabrication of macroscopic structures, such as fibers or films, could be challenging because of the GNRs' tendency for entanglement, aggregation, and thus poor solubility in conventional solvents. This problem appears to be general for one-dimensional (1D) nanomaterials, such as metallic and semiconductor nanowires and carbon nanotubes (CNTs), whose precise nanoscale assembly and alignment present a formidable challenge.15–23 For example, exceptional thermal, electrical and mechanical properties of single-walled CNTs are often discussed in regard to numerous applications, such as high-strength conductive fibers and transparent conducting films.24 However, translating the unique characteristics of CNTs from nanoscale to macroscale remains a great challenge due to the difficulties with scalable and effective assembly of nanotubes. As a result, macroscopic CNT materials typically exhibit only a small fraction of CNTs' nanoscale properties.25In contrast to CNTs that are relatively stiff and often considered as a model system for nanorods, GNRs are very flexible objects.26 Also, while CNTs and GNRs share the same honeycomb arrangement of carbon atoms, the flat structure of nanoribbons should result in stronger π–π interaction between individual GNRs compared to CNTs that have curved surfaces. Poor solubility and aggregation of CNTs remained serious problems to solve for nearly two decades.23,27–29 Due to their greater flexibility and stronger π–π interactions, GNRs are expected to aggregate and entangle even more than CNTs, which makes their alignment and assembly an even greater challenge. At the same time, since the research on solution-synthesized GNRs is still in its infancy, little is actually known about aggregation of GNRs.
A number of papers have reported investigations of aggregation-related phenomena in solutions of other graphene-like materials and different approaches on decreasing aggregation of thereof. These materials include graphene,30–32 carbon nanotubes23 and polyaromatic hydrocarbons (PAHs),33–37 which due to their small size and atomically precise structure dictated by the bottom-up synthesis resemble solution-synthesized GNRs. For example, it was reported that hexabenzocoronene (HBC), a large PAH molecule, produces different absorbance spectra when dispersed in different organic solvents, which was attributed to the HBC aggregation.36,38 Similarly, properties of bulk GNR materials could be quite different from those of individual GNRs due to aggregation effects. Therefore, studies of aggregation of nanoribbons are important not only for their processing for practical applications but also for the fundamental understanding of the physical properties of GNRs.
In this work, we have studied aggregation of chevron GNRs; their structure is schematically shown in Fig. 1a. These nanoribbons already have been a subject of several theoretical39–41 and experimental studies.2,5,42–48 We investigated chevron GNRs in solution and found that their optical properties depend on their aggregation state. We also found that in addition to forming bulk aggregates of the π–π stacked nanoribbons, on surfaces GNRs can assemble to form long one-dimensional (1D) structures. The 1D assemblies can be conveniently visualized by conventional microscopy techniques and used for the fabrication of electronic devices.
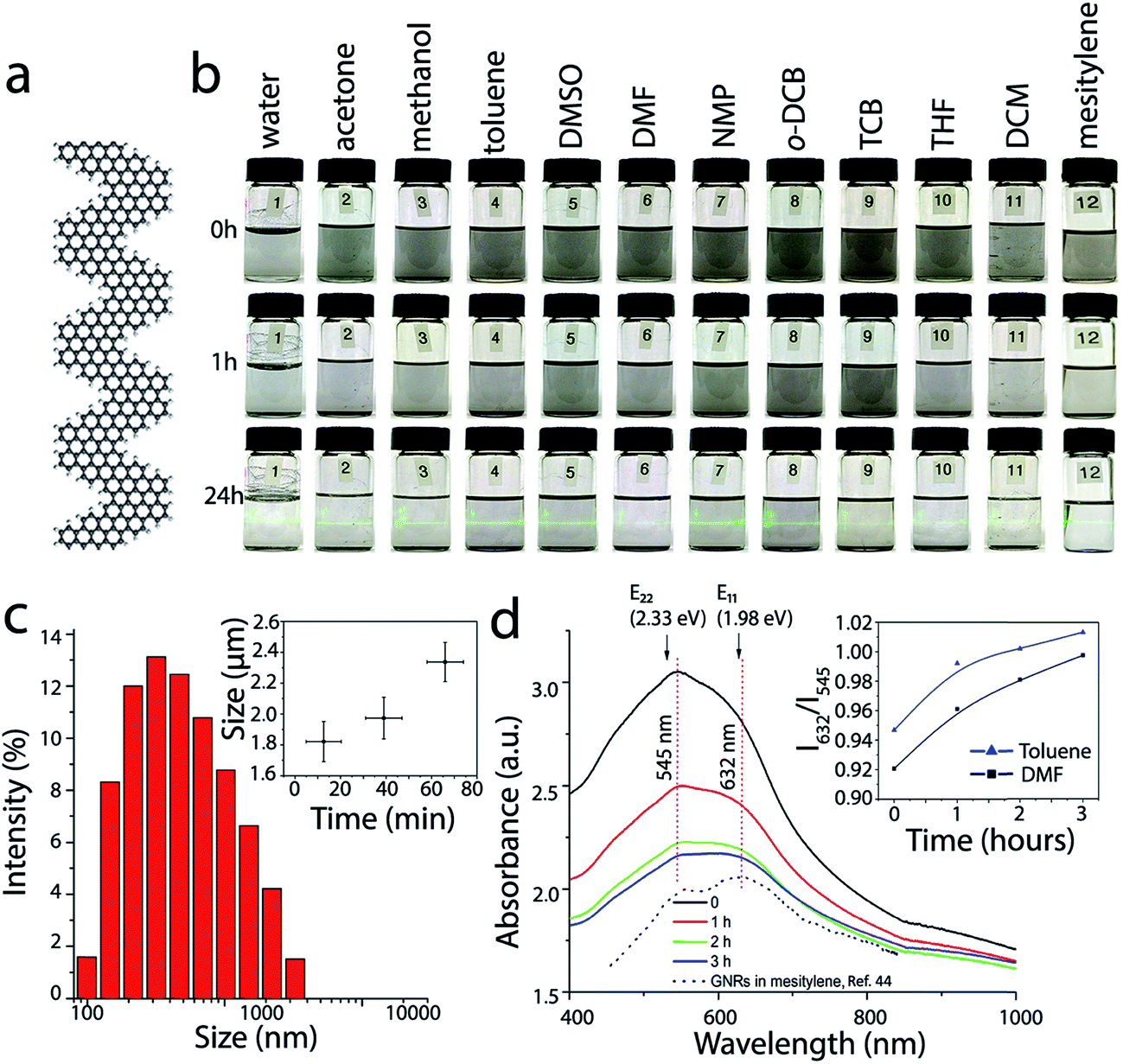 | ||
| Fig. 1 Dispersions of GNRs in conventional solvents. (a) Atomic structure of a fragment of chevron GNR. (b) Optical photographs of the vials with ∼0.1 mg mL−1 GNR suspensions in water (1), acetone (2), methanol (3), toluene (4), DMSO (5), DMF (6), NMP (7), o-DCB (8), TCB (9), THF (10), DCM (11) and mesitylene (12) taken immediately after sonication (top row), 1 h (middle row) and 24 h after sonication (bottom row). The bottom row of vials is illuminated with a green laser. (c) Typical size distribution of GNR aggregates in toluene measured by DLS. The inset represents time dependence of the average particle size. (d) UV-vis-NIR absorbance spectra of the dispersion of GNRs in DMF that were measured at different moments after sonication. The inset shows the time dependences of the ratios of the optical absorbance values at 632 and 545 nm for GNR suspensions in DMF (black) and toluene (blue). The black dotted spectrum shows the optical absorbance of chevron GNRs in mesitylene, which was reproduced from our previous work;44 the arbitrary absorbance units are different for GNR spectra in DMF and mesitlyene and should not be compared directly. E11 (1.98 eV) and E22 (2.33 eV) are the excitonic transitions calculated for the chevron GNR by first principles calculations using the GW approach in ref. 39. The E11 and E22 energies agree very well with the absorbance features in the experimental spectra of chevron GNRs in various solvents. | ||
Experimental
Materials
GNRs were synthesized according to the procedure that we reported in our previous work.2 Dimethyl sulfoxide (DMSO) and mesitylene were purchased from Sigma-Aldrich; o-dichlorobenzene (o-DCB) was purchased from TCI America. Toluene, methanol, 1,2,4-trichlorobenzene (TCB), N-methyl-2-pyrrolidone (NMP), and dimethylformamide (DMF) were purchased from Alfa Aesar. Acetone, tetrahydrofuran (THF) and dichloromethane (DCM) were purchased from Fisher Chemicals. All chemicals were used as received without any purification.TEM grids (Lacey carbon, 300 mesh) were purchased from Ted Pella Inc. Mica (V-4 grade) was purchased from SPI Supplies. Four-inch heavily doped silicon wafers with a 300 nm-thick layer of SiO2 were purchased from Silicon Quest International.
Methods
For UV-vis-NIR measurements GNR powder was sonicated in a solvent (DMF or toluene) for 3 h and the spectra were collected using a Jasco V-670 spectrophotometer. Raman spectroscopy of GNRs was performed using a Thermo Scientific DXR Raman microscope with a 532 nm laser.Atomic force microscopy (AFM) images were recorded using a Digital Instruments Nanoscope IIIa Dimension 3100 system and Bruker RTESPA AFM probes. An Omicron low-temperature scanning tunneling microscope (LT-STM) with an electrochemically etched W tip, kept at a base pressure of <10−10 Torr, was used for the scanning tunneling microscopy (STM). Scanning electron microscopy (SEM) was performed using a FEI Nova NanoSEM 450. For AFM, SEM and STM imaging GNR powder (approx. 0.5 mg) was sonicated for 1 min in a solvent (10 mL) and then heated to reflux. The hot suspension was sonicated for additional 30 s and heated back to reflux twice, then two drops of the suspension were deposited on a substrate (either mica, Au(111) or Si/SiO2) while still hot.
Solvents that were used to disperse GNRs for imaging included toluene, DMF, NMP, o-DCB, DMSO and mesitylene. Most of these solvents, especially DMF, NMP, DMSO, left residues after evaporation, which obstructed the imaging. Toluene was found to produce cleaner samples and therefore was primarily used for microscopy experiments, but 1D assemblies of GNRs were also observed via SEM using mesitylene or o-DCB as a solvent.
For TEM experiments GNR powder was sonicated in a solvent (DCM or toluene) for 30 s and deposited on a lacey carbon grid. In the case of toluene, the solution was heated to reflux before and after sonication and the grid was heated on a hot plate in order to accelerate the solvent evaporation. Both solvents showed similar results. TEM images of GNRs were obtained using a FEI Tecnai Osiris transmission electron microscope at the accelerating voltage of 200 kV.
Size distribution of GNR aggregates was measured using a Protein Solutions DynaPro DLS system. For the sample preparation GNR powder was sonicated in a solvent (toluene or DMF) for 1 min at a concentration of approx. 0.025 mg mL−1 and transferred to a cuvette for measurements.
XRD was recorded with Cu Kα radiation using a Rigaku Multiflex diffractometer. Gas adsorption isotherms were collected using a Micromeritics ASAP 2020-accelerated surface area and porosimetry analyzer.
Nanoribbon devices were fabricated by a standard electron beam lithography using a Zeiss Supra 40 field-emission scanning electron microscope and a Raith pattern generator to define electrodes on GNR nanostructures, and an AJA electron beam evaporation system to evaporate Cr and Au.
Results and discussion
Chevron GNRs were synthesized according to the previously reported procedure using Yamamoto coupling of molecular precursors followed by cyclodehydrogenation via Scholl reaction.2 Based on the STM analysis that we provide in our previous publications, these GNRs ranged from several to several tens of nm in length.2,45 Our prior studies on the solution-synthesized GNRs also include the results of their bulk characterization by spectroscopic techniques, such as XPS, EDX, NMR, UV-vis-NIR, FTIR and Raman spectroscopy.2,5,44 Unlike other solution-synthesized GNRs that have been recently reported,3,4,7,9–11,13,49 these chevron GNRs do not contain any alkyl groups to increase their solubility. In this paper we will not discuss in detail the advantages (mostly the increased solubility) and disadvantages (such as, for example, inferior electrical contacts between nanoribbons in bulk GNR materials and composites) of such alkyl-substituted GNRs. However, it is worth mentioning that the absence of solubilizing groups in chevron GNRs that were used in this study seems to be appropriate for better understanding of the intrinsic inter-ribbon interactions.Dispersions of GNRs and their optical properties
First, we compared dispersibility of the GNRs in several conventional solvents (Fig. 1b). Dry GNR powder was sonicated for 3 min in 12 different solvents, including water, acetone, methanol, toluene, DMSO, DMF, NMP, o-DCB, TCB, THF, DCM, and mesitylene, to yield GNR suspensions with nominal concentrations (concentration immediately after sonication) of ∼0.1 mg mL−1. Fig. 1b shows optical photographs of the vials with GNR suspensions in these solvents immediately (0 h), 1 h and 24 h after sonication. GNRs precipitate nearly completely from all of the suspensions and a thin layer of black precipitate appears at the bottom of each vial 1 h after sonication. However, even after 24 h after sonication a small amount of suspended GNRs is still observed in these vials, as evidenced by the Tyndall effect (see the bottom row of the vials in Fig. 1b). Based on the Tyndall effect and visual observations, GNR dispersions in toluene, DMSO, NMP and o-DCB appear to be most concentrated. These observations are in agreement with the results reported for other graphene-like materials; for example NMP and o-DCB were shown to be effective solvents for graphene,31,32,50 carbon nanotubes28,29,51 and HBC.38 These solvents, in which a certain amount of GNRs could be dispersed for hours, are promising for the solution processing of nanoribbons. Noteworthy, we recently demonstrated that another solvent that proved to be very effective for dispersing graphene and carbon nanotubes, chlorosulfonic acid,23,30 can be also be used to form stable solutions of GNRs14,46 at concentrations as high as 15 mg mL−1,46 but for some applications it is preferable to have GNRs dispersed in less corrosive solvents.In addition to the visual observations, the aggregation of GNRs can be monitored by dynamic light scattering (DLS) and optical spectroscopy; the latter was successfully used in studies of PAH molecules.38 Representative data illustrating the aggregation of GNRs in two solvents that were considered in this study, toluene and DMF, are shown in Fig. 1c and d. DLS data show a wide size distribution of particles in the suspension of GNRs in toluene immediately after sonication (Fig. 1c), confirming strong aggregation of nanoribbons. The inset in Fig. 1c shows that the aggregates grow with time (each data point in this graph represents averaged results for several DLS measurements). For UV-vis-NIR measurements, dry GNR powder was first dispersed in a solvent (DMF or toluene) by sonication to yield a GNR suspension with a concentration of ∼0.1 mg mL−1. A quartz cuvette with the suspension was placed in a UV-vis-IR spectrophotometer, and absorbance spectra were measured immediately and then 1, 2 and 3 h after the sonication (Fig. 1d), while the cuvette was undisturbed inside the instrument between the measurements. The spectra are shown in the same units, so the fact that the absorbance of the GNR suspension decreases with time indicates the precipitation of GNRs.
Optical properties of GNRs have been investigated in several theoretical studies.39,52,53 It was demonstrated that an accurate description of the optical properties of GNRs should include many-body effects.52,53 The optical spectra calculated for straight armchair GNRs52,53 as well as the chevron GNRs39 using the GW approximation were dominated by the absorption peaks corresponding to various excitonic transitions. In particular, for the chevron GNR the calculations predicted the first two lowest energy excitonic peaks at 1.98 and 2.34 eV, corresponding to the E11 and E22 transitions.39 Considering that the GW-calculated bandgap for the chevron GNR is 3.74 eV, the 1.98 eV exciton state has a remarkably large binding energy of 1.76 eV.39
Previously, we reported a UV-vis-NIR spectrum of the solution-synthesized chevron GNRs suspended in mesitylene, and pointed out a remarkable agreement between the observed absorbance peaks and the theoretically predicted energies of the E11 and E22 excitonic transitions.44 In Fig. 1d, we also include that spectrum from ref. 44 and show that the peak positions are indeed very close to the calculated E11 and E22 energies from ref. 39. Interestingly, studies of other nanoribbons, including N = 7 armchair GNRs,54 and cove-type GNRs,55 also showed an excellent agreement between the experimentally observed spectroscopic features and theoretically predicted excitonic transitions, further demonstrating the efficiency of the first-principle calculations for describing optical properties of atomically precise GNRs.
When GNRs are suspended in a different solvent, DMF, the excitonic peak structure looks slightly different and also evolves with time (Fig. 1d). As the GNRs aggregate after the initial sonication, the intensity of the broad shoulder at ∼632 nm clearly increases relative to the absorbance maximum at ∼545 nm. The inset in Fig. 1d shows that the ratio of the GNR absorbance values at 632 and 545 nm changes for both DMF and toluene in a very similar manner. Aggregation-dependent optical properties were previously reported for solutions of HBC and some conjugated polymers.38,56
Importantly, UV-vis-NIR absorption spectroscopy is often used to probe optoelectronic properties of synthetic GNRs and in particular, to measure their optical band gaps.2–5,7–9,49 While it is generally recognized that the optically measured band gaps of GNRs are smaller than their intrinsic band gaps because of the excitonic effects,54 understanding bulk properties of GNRs is important for various large-scale applications of nanoribbons within thin films, coatings and composites. The results presented in this study show that (1) the spectroscopic features observed in the experimentally measured absorbance spectra of chevron GNRs are in a good agreement with theoretically predicted excitionic transitions,39 and (2) the appearance of an absorbance spectrum could depend on the aggregation state of GNRs, which would affect the position of the absorbance edge and hence the measured optical band gap.
Aggregation of GNRs in bulk
We then studied the packing of nanoribbons in a powder form (Fig. 2a). Fig. 2b shows that the most pronounced feature in the X-ray diffraction (XRD) pattern of the GNR powder is the broad peak at 2Θ = 26°, which corresponds to d = 3.4 Å, the interlayer distance in graphite.57 This peak is very common for PAH molecules,58,59 graphene-based materials60,61 and GNRs synthesized by other methods,7,49,62 showing their tendency to form π–π stacked structures. Overall, the XRD results suggest that π–π stacking is the most favorable arrangement for GNRs in bulk. | ||
| Fig. 2 Aggregation of GNRs in bulk. (a) Optical photograph of a vial with a GNR powder. (b) XRD spectrum of a GNR powder. The dashed line indicates the position of the graphite [002] peak. (c) TEM image of a GNR aggregate. (d) Higher-resolution TEM image of a GNR aggregate showing π–π stacked nanoribbons. | ||
The high degree of GNR packing in the bulk form is consistent with the results of the Brunauer–Emmett–Teller (BET) surface area measurements. The theoretical surface area of a single sheet of graphene is 2670 m2 g−1,63 and the theoretical surface area of isolated nanoribbons should be even higher because of the large contribution of the GNR edges. The experimentally measured surface area of the as-synthesized chevron GNR powder is about 370 m2 g−1. This is comparable to the value reported for GNRs synthesized by the chemical unzipping of carbon nanotubes,62 although some other nanostructured graphene-based materials have been shown to have surface areas >700 m2 g−1.64–66 The relatively low surface area of the chevron GNR powder indicates dense packing of nanoribbons in bulk GNR aggregates.
We performed further investigation of GNR aggregates using transmission electron microscopy (TEM); representative TEM images are shown in Fig. 2c and d. The only periodic features that we consistently observed in these images were interpreted as π–π stacked GNRs. For example, the distance between the parallel lines in Fig. 2d is ∼4 Å, which is again close to the interlayer distance in graphite (3.4 Å).57 Thus, both XRD and TEM of GNR aggregates provide evidence for π–π stacking of nanoribbons in a powder form.
Aggregation of GNRs on surfaces
In order to gain further insights into the structure of GNR aggregates we performed microscopy studies of nanoribbons deposited from suspensions on various substrates, such as Si/SiO2, mica and Au(111). Scanning electron microscopy (SEM) images of GNRs deposited on Si/SiO2 substrate from a toluene dispersion are shown in Fig. 3a. In addition to bulky nanoparticles and aggregates, which have a substantial number of π–π stacked nanoribbons, we observed numerous elongated structures, which were several nm wide and a few μm long (Fig. 3a). Similar elongated structures were also observed by atomic force microscopy (AFM) on freshly cleaved mica substrates (Fig. 3b). Fig. 3c shows a height profile across this 1D structure indicating an apparent height of 3 to 4 Å, which is in a good agreement with the thickness of a monolayer graphene sheet (3.4 Å). For other AFM images of such 1D structures the apparent thickness values varied considerably depending on the substrate (mica or Si/SiO2) and the exact location where the measurement was taken. However, the heights of 3 to 15 Å observed from multiple scans are comparable with the range of values measured by AFM for monolayer graphene in other studies,67 which points at the uncertainty of such measurements due to the substrate's roughness, adsorbates, etc. The values at the lower end of this range, especially when obtained on a flat mica surface, suggest that in these elongated structures the GNRs could lay flat and thus arrange in a side-by-side fashion rather than stand perpendicularly to the substrate. | ||
| Fig. 3 Aggregation of GNRs on surfaces. (a) SEM images of GNRs deposited on Si/SiO2 substrate. (b) AFM image of GNRs deposited on mica. (c) Height profile measured along the blue line in panel (b). (d) STM image of GNRs on Au(111). Top panel shows the original image, and in the bottom panel one GNR is highlighted by green for the sake of clarity. The inset in the bottom panel shows the scheme of the arrangement of GNRs in this STM image. (e) Scheme of a possible alignment of GNRs on a surface. | ||
In general, the assembly of GNRs on a substrate should be determined by the interplay between the intermolecular interactions (in case of GNRs, primarily the π–π interactions) and the GNR-substrate interactions. Numerous studies have been performed to understand and control the packing of π-conjugated molecules on a variety of substrates.68 For example, pentacene molecules are known to lay flat on coinage metal surfaces, such as Au(111)69 and Cu(111),70 because of the interactions between the π-orbitals of pentacene and d orbitals of the metals, but stand on their edges on a SiO2 substrate due to weaker molecule–substrate interactions.71,72
While it is difficult to visualize the packing of GNRs on Si/SiO2 or mica, this could be done on a conductive substrate, such as Au(111), using scanning tunneling microscopy (STM), a technique that is capable of imaging GNRs with nearly atomic resolution.2,42,45,73,74 Fig. 3d shows STM image of GNRs on Au(111) substrate. The GNRs were drop-casted from a toluene solution and dried in air before the imaging. As a result, while the STM was performed in the ultrahigh vacuum (UHV) conditions, some residual toluene molecules and other surface adsorbates could still be present on the surface. The GNRs were found to arrange in a side-by-side fashion, which we also observed in our previous work.2 Top panel in Fig. 3d shows the original STM image, while in the bottom panel one nanoribbon is highlighted in green to better illustrate the GNR arrangement, in which GNRs are parallel to each other and the apices of one ribbon fit into the grooves of another one; the arrangement of these three GNRs is schematically shown in the inset. The side-by-side arrangement of GNRs observed in the STM images is consistent with the ∼3 Å thickness of larger elongated structures (Fig. 3c). It should also be noted, that the arrangement of GNRs in Fig. 3d is different from the side-by-side assembly of nitrogen-doped chevron GNRs (N-GNRs),44,48 where the apices of the adjacent N-GNRs face each other and the entire structure is likely stabilized by the hydrogen bonding.
Overall, the fact that the individual chevron GNRs lay flat on Au(111) is consistent with similar observations made by STM for many other PAH molecules on coinage metal surfaces. The assembly of GNRs in such structures (Fig. 3d), could be explained by the action of capillary forces upon drying a droplet of a GNR suspension on a surface (Fig. 3e). There are numerous precedents of self-assembly processes in evaporating solutions of colloidal nanoparticles,75 viruses76 and various one-dimensional nanocrystals.77 Fig. 3e shows that while the solvent evaporates, the GNRs could be drawn to the meniscus area by capillary flow and concentrate at the triple contact line. As the contact line moves along the substrate upon the droplet drying, structures of densely packed GNRs may form. While it is unclear whether the 1D structures of nanoribbons on Si/SiO2 and mica assume exactly the same GNR packing as on Au(111) because of the differences in physical properties of these substrates, overall these observations show the importance of the interplay between the GNR–GNR interactions and the GNR–substrate interactions in generating on-surface assemblies of nanoribbons that are different from their packing in bulk.
The 1D nanoribbon structures, which we will further refer to as GNR “nanobelts”, are interesting for several reasons. These results show that different arrangements of GNRs can be realized through the control of the assembly conditions. Interestingly, this idea was further extended in our recent work, in which in very different assembly conditions – on a surface of water – hydrophobic chevron GNRs exhibit a yet another arrangement, a monolayer film of π–π stacked nanoribbons in the “edge-on” geometry.46 Further work on assembly of nanoribbons in different conditions should result in new GNR-based structures with potentially interesting properties.
From the practical point of view, the formation of the GNR nanobelts could be useful for device fabrication and testing of the electronic properties of nanoribbons. Individual solution-synthesized chevron GNRs are too small for locating them on Si/SiO2 by microscopy techniques, such as SEM and AFM. However, once assembled into few-μm-long GNR nanobelts, they could be easily visualized (Fig. 3a and b). We located several GNR nanobelts that were >1 μm long on a Si/SiO2 substrate by SEM and used them to fabricate electronic devices by a standard electron beam lithography. Electrical contacts to GNRs were prepared by an electron beam evaporation of Cr (1 nm) and Au (15 nm). Fig. 4a shows SEM image of a typical device, where a GNR nanobelt, which is shown by the black arrow, bridges source (S) and drain (D) Cr/Au electrodes. The heavily doped p-type Si used as a gate (G) electrode was separated from the GNR by a 300 nm-thick layer of SiO2. The GNR-based electronic devices that were fabricated in this study had a separation between the S and D electrodes of 500 nm.
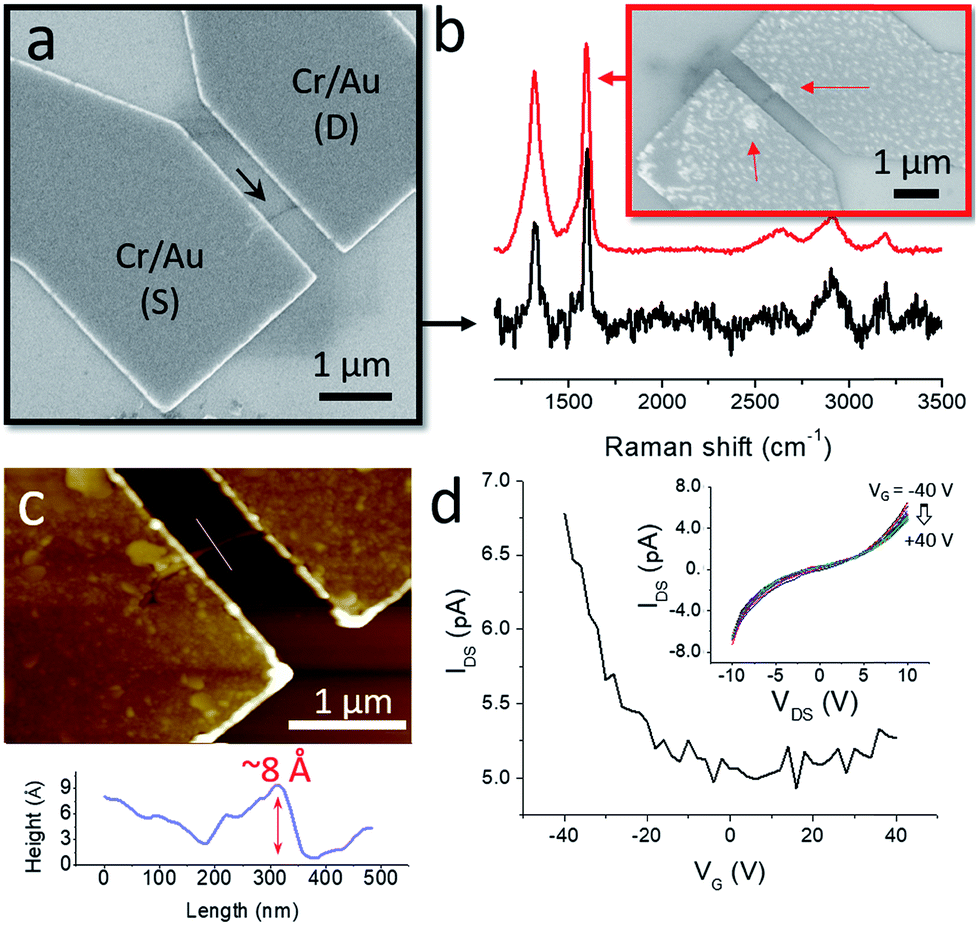 | ||
| Fig. 4 Electrical measurements of GNR nanobelts. (a) SEM image of a GNR device. The GNR nanobelt is shown by the black arrow. (b) Raman spectra of GNR nanobelts. Inset shows SEM image of another GNR device. The black spectrum represents the GNR nanobelt shown in panel (a), the red spectrum represents the GNR nanobelt shown in the inset. The arrows in the inset show the entangled ends of the GNR nanobelt. (c) AFM image of the device shown in the panel (a). The bottom panel shows the height profile across the GNR nanobelt measured along the white line in the AFM image. (d) IDS–VG dependence for a GNR device that was annealed at 300 °C in H2/Ar for 1 h. VDS = 10 V. The inset shows IDS–VDS dependencies for the same device that were measured at VG ranging from −40 to 40 V with the step of 20 V. | ||
We characterized nanobelts in the devices by Raman spectroscopy to confirm that they indeed consist of GNRs; representative Raman spectra are shown in Fig. 4b. Since the nanobelts contain small amounts of GNRs, the Raman spectra measured from them typically had low intensities and, as a result, low signal-to-noise ratios. But even in such spectra the characteristic D and G bands at about 1300 and 1600 nm−1, respectively, were clearly distinguishable, which is illustrated by the black Raman spectrum in Fig. 4b that was measured for the GNR nanobelt in the device shown in Fig. 4a. For some other GNR nanobelts we could measure spectra of higher quality, as shown by the red Raman spectrum in Fig. 4b (the corresponding device is shown in the inset), which is consistent with the Raman spectroscopy data that we reported for bulk chevron GNRs in our previous studies.2,5 In this particular case, the higher intensity of the spectrum and the improved signal-to-noise ratio can likely be explained by the fact that the ends of this nanobelt are entangled forming two GNR aggregates that can be recognized through the metal electrodes (see the red arrows in the inset in Fig. 4b). Such aggregates were often observed for other GNR nanobelts as well (Fig. 3a and b). Since the Raman measurements were performed from spots with a diameter of about 1 μm, the signals were collected from the entire nanobelts in Fig. 4a and b. In the second case, because of the discussed GNR aggregates, a larger amount of the GNR material was probed, which resulted in higher quality of the corresponding Raman spectrum. Spectra that were consistent with the previously published Raman spectra of chevron GNRs2,5 were collected for all fabricated devices.
AFM measurements of the device shown in Fig. 4a reveal that the GNR nanobelt has an apparent height of about 8 Å (Fig. 4c), which is within the range of thicknesses reported for graphene monolayers on Si/SiO2 in different studies.67 While this GNR nanobelt appears to be thicker than the one in Fig. 3b and c, the difference in apparent heights may also be attributed to different substrates, mica and Si/SiO2, on which these nanobelts were imaged,78 as well as the exact imaging parameters, such as the tip pressure.67
As-fabricated GNR devices showed very low electrical conductivities, which could be improved by annealing at 300 °C for 1 h in H2 (1 sccm)/Ar (75 sccm) atmosphere. Fig. 4d shows a representative drain–source current (IDS) – gate voltage (VG) dependence for an annealed GNR device, which was measured at the drain–source voltage (VDS) of 10 V. Similarly to the reported electrical measurements of thin films of chevron GNRs synthesized in solution46 and by chemical vapor deposition,79 these GNR nanobelts show a p-type behavior, low conductivities and on–off ratios.
It is important to point out that these electronic characteristics do not fully represent the intrinsic properties of chevron GNRs. Since the source–drain distance was significantly larger than individual GNRs, the electronic transport in these devices was affected by the inter-GNR junctions. Also, the nonlinear IDS–VDS dependencies shown in the inset in Fig. 4d indicate high contact resistances. Yet, these results demonstrate the possibility of fabricating electronic devices based on nanobelts of chevron GNRs, which is simplified by the convenience with which nanobelts can be located on surfaces by high-throughput microscopy techniques, such as SEM and AFM. With the device miniaturization, such that the source–drain distances would become comparable to the lengths of individual GNRs in nanobelts, and optimization of the contact resistances, these 1D assemblies may prove useful for the characterization of intrinsic electronic properties of nanoribbons.
Summary
In conclusion, using a combination of microscopic and spectroscopic techniques we investigated aggregation of solution-synthesized GNRs in different conditions. We studied the behavior of GNRs that were dispersed in variety of solvents and found that (1) the spectroscopic features observed in the experimentally measured absorbance spectra of chevron GNRs are in a good agreement with theoretically predicted excitonic transitions,39 and (2) the absorbance spectra evolve with aggregation time. We have shown two types of GNR assemblies: in addition to the π–π stacked structures that are typical for graphene-like materials and most favorable for nanoribbons in bulk, GNRs also form large 1D structures on surfaces. The 1D assemblies of GNRs can be observed on different substrates, such as Au(111), mica and Si/SiO2 wafers, by conventional microscopy techniques, which makes them promising objects for the future fabrication of electronic devices. Insights into the aggregation of GNRs are also important in the view of their potential bulk applications in conductive films, composite materials and as catalyst supports.80Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported by the Office of Naval Research (ONR, N00014-16-1-2899). Synthesis of graphene nanoribbons was supported by the National Science Foundation (NSF) through CHE-1455330. The materials characterization was performed in part the Nebraska Nanoscale Facility: National Nanotechnology Coordinated Infrastructure and the Nebraska Center for Materials and Nanoscience, which are supported by the NSF (ECCS-1542182) and the Nebraska Research Initiative.References
- O. V. Yazyev, Acc. Chem. Res., 2013, 46, 2319–2328 CrossRef CAS PubMed.
- T. H. Vo, M. Shekhirev, D. A. Kunkel, M. D. Morton, E. Berglund, L. Kong, P. M. Wilson, P. A. Dowben, A. Enders and A. Sinitskii, Nat. Commun., 2014, 5, 3189 Search PubMed.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS PubMed.
- M. G. Schwab, A. Narita, Y. Hernandez, T. Balandina, K. S. Mali, S. De Feyter, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 18169–18172 CrossRef CAS PubMed.
- T. H. Vo, M. Shekhirev, A. Lipatov, R. A. Korlacki and A. Sinitskii, Faraday Discuss., 2014, 173, 105–113 CAS.
- T. H. Vo, M. Shekhirev, D. A. Kunkel, F. Orange, M. J. F. Guinel, A. Enders and A. Sinitskii, Chem. Commun., 2014, 50, 4172–4174 RSC.
- K. T. Kim, J. W. Jung and W. H. Jo, Carbon, 2013, 63, 202–209 CrossRef CAS.
- A. Narita, I. A. Verzhbitskiy, W. Frederickx, K. S. Mali, S. A. Jensen, M. R. Hansen, M. Bonn, S. De Feyter, C. Casiraghi, X. Feng and K. Müllen, ACS Nano, 2014, 8, 11622–11630 CrossRef CAS PubMed.
- J. Gao, F. J. Uribe-Romo, J. D. Saathoff, H. Arslan, C. R. Crick, S. J. Hein, B. Itin, P. Clancy, W. R. Dichtel and Y.-L. Loo, ACS Nano, 2016, 10, 4847–4856 CrossRef CAS PubMed.
- G. Li, K.-Y. Yoon, X. Zhong, X. Zhu and G. Dong, Chem.–Eur. J., 2016, 22, 9116–9120 CrossRef CAS PubMed.
- R. S. Jordan, Y. Wang, R. D. McCurdy, M. T. Yeung, K. L. Marsh, S. I. Khan, R. B. Kaner and Y. Rubin, Chem, 2016, 1, 78–90 CAS.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef CAS PubMed.
- Y. Huang, Y. Mai, U. Beser, J. Teyssandier, G. Velpula, H. van Gorp, L. A. Straasø, M. R. Hansen, D. Rizzo, C. Casiraghi, R. Yang, G. Zhang, D. Wu, F. Zhang, D. Yan, S. De Feyter, K. Müllen and X. Feng, J. Am. Chem. Soc., 2016, 138, 10136–10139 CrossRef CAS PubMed.
- M. Mehdi Pour, A. Lashkov, A. Radocea, X. Liu, T. Sun, A. Lipatov, R. A. Korlacki, M. Shekhirev, N. R. Aluru, J. W. Lyding, V. Sysoev and A. Sinitskii, Nat. Commun., 2017, 8, 820 CrossRef PubMed.
- N. O. Weiss and X. Duan, Nat. Nanotechnol., 2013, 8, 312–313 CrossRef CAS PubMed.
- F. N. Ishikawa, H.-k. Chang, K. Ryu, P.-c. Chen, A. Badmaev, L. Gomez De Arco, G. Shen and C. Zhou, ACS Nano, 2009, 3, 73–79 CrossRef CAS PubMed.
- G. Yu, A. Cao and C. M. Lieber, Nat. Nanotechnol., 2007, 2, 372–377 CrossRef CAS PubMed.
- C. M. Hangarter and N. V. Myung, Chem. Mater., 2005, 17, 1320–1324 CrossRef CAS.
- Y. Huang, X. Duan, Q. Wei and C. M. Lieber, Science, 2001, 291, 630 CrossRef CAS PubMed.
- X. Duan, Y. Huang, Y. Cui, J. Wang and C. M. Lieber, Nature, 2001, 409, 66–69 CrossRef CAS PubMed.
- P. A. Smith, C. D. Nordquist, T. N. Jackson, T. S. Mayer, B. R. Martin, J. Mbindyo and T. E. Mallouk, Appl. Phys. Lett., 2000, 77, 1399–1401 CrossRef CAS.
- M. C. P. Wang and B. D. Gates, Mater. Today, 2009, 12, 34–43 CrossRef CAS.
- V. A. Davis, A. N. G. Parra-Vasquez, M. J. Green, P. K. Rai, N. Behabtu, V. Prieto, R. D. Booker, J. Schmidt, E. Kesselman, W. Zhou, H. Fan, W. W. Adams, R. H. Hauge, J. E. Fischer, Y. Cohen, Y. Talmon, R. E. Smalley and M. Pasquali, Nat. Nanotechnol., 2009, 4, 830–834 CrossRef CAS PubMed.
- R. H. Baughman, Science, 2000, 290, 1310 CrossRef CAS PubMed.
- N. Behabtu, C. C. Young, D. E. Tsentalovich, O. Kleinerman, X. Wang, A. W. K. Ma, E. A. Bengio, R. F. ter Waarbeek, J. J. de Jong, R. E. Hoogerwerf, S. B. Fairchild, J. B. Ferguson, B. Maruyama, J. Kono, Y. Talmon, Y. Cohen, M. J. Otto and M. Pasquali, Science, 2013, 339, 182 CrossRef CAS PubMed.
- K. V. Bets and B. I. Yakobson, Nano Res., 2009, 2, 161–166 CrossRef CAS.
- M. C. Hersam, Nat. Nanotechnol., 2008, 3, 387–394 CrossRef CAS PubMed.
- J. L. Bahr, E. T. Mickelson, M. J. Bronikowski, R. E. Smalley and J. M. Tour, Chem. Commun., 2001, 193–194 RSC.
- S. D. Bergin, V. Nicolosi, P. V. Streich, S. Giordani, Z. Sun, A. H. Windle, P. Ryan, N. P. P. Niraj, Z.-T. T. Wang, L. Carpenter, W. J. Blau, J. J. Boland, J. P. Hamilton and J. N. Coleman, Adv. Mater., 2008, 20, 1876–1881 CrossRef CAS.
- N. Behabtu, J. R. Lomeda, M. J. Green, A. L. Higginbotham, A. Sinitskii, D. V. Kosynkin, D. Tsentalovich, A. N. G. Parra-Vasquez, J. Schmidt, E. Kesselman, Y. Cohen, Y. Talmon, J. M. Tour and M. Pasquali, Nat. Nanotechnol., 2010, 5, 406–411 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- D. Wasserfallen, M. Kastler, W. Pisula, W. A. Hofer, Y. Fogel, Z. Wang and K. Müllen, J. Am. Chem. Soc., 2006, 128, 1334–1339 CrossRef CAS PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- P. Samorí, M. Keil, R. Friedlein, J. Birgerson, M. Watson, K. Müllen, W. R. Salaneck and J. P. Rabe, J. Phys. Chem. B, 2001, 105, 11114–11119 CrossRef.
- A. J. Fleming, J. N. Coleman, A. B. Dalton, A. Fechtenkötter, M. D. Watson, K. Müllen, H. J. Byrne and W. J. Blau, J. Phys. Chem. B, 2003, 107, 37–43 CrossRef CAS.
- M. Keil, P. Samorí, D. A. dos Santos, T. Kugler, S. Stafström, J. D. Brand, K. Müllen, J. L. Brédas, J. P. Rabe and W. R. Salaneck, J. Phys. Chem. B, 2000, 104, 3967–3975 CrossRef CAS.
- J. M. Hughes, Y. Hernandez, D. Aherne, L. Doessel, K. Müllen, B. Moreton, T. W. White, C. Partridge, G. Costantini, A. Shmeliov, M. Shannon, V. Nicolosi and J. N. Coleman, J. Am. Chem. Soc., 2012, 134, 12168–12179 CrossRef CAS PubMed.
- S. D. Wang and J. L. Wang, J. Phys. Chem. C, 2012, 116, 10193–10197 CAS.
- E. Costa Girão, L. Liang, E. Cruz-Silva, A. G. S. Filho and V. Meunier, Phys. Rev. Lett., 2011, 107, 135501 CrossRef PubMed.
- L. Liang and V. Meunier, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195404 CrossRef.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- S. Linden, D. Zhong, A. Timmer, N. Aghdassi, J. H. Franke, H. Zhang, X. Feng, K. Müllen, H. Fuchs, L. Chi and H. Zacharias, Phys. Rev. Lett., 2012, 108, 216801 CrossRef CAS PubMed.
- T. H. Vo, U. G. E. Perera, M. Shekhirev, M. Mehdi Pour, D. A. Kunkel, H. Lu, A. Gruverman, E. Sutter, M. Cotlet, D. Nykypanchuk, P. Zahl, A. Enders, A. Sinitskii and P. Sutter, Nano Lett., 2015, 15, 5770–5777 CrossRef CAS PubMed.
- A. Radocea, T. Sun, T. H. Vo, A. Sinitskii, N. R. Aluru and J. W. Lyding, Nano Lett., 2017, 17, 170–178 CrossRef CAS PubMed.
- M. Shekhirev, T. H. Vo, M. Mehdi Pour, A. Lipatov, S. Munukutla, J. W. Lyding and A. Sinitskii, ACS Appl. Mater. Interfaces, 2017, 9, 693–700 CAS.
- J. D. Teeter, P. S. Costa, M. Mehdi Pour, D. P. Miller, E. Zurek, A. Enders and A. Sinitskii, Chem. Commun., 2017, 53, 8463–8466 RSC.
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang, V. Meunier, R. Berger, R. Li, X. Feng, K. Müllen and R. Fasel, Nat. Nanotechnol., 2014, 9, 896–900 CrossRef CAS PubMed.
- K. T. Kim, J. W. Lee and W. H. Jo, Macromol. Chem. Phys., 2013, 214, 2768–2773 CrossRef CAS.
- C. E. Hamilton, J. R. Lomeda, Z. Sun, J. M. Tour and A. R. Barron, Nano Lett., 2009, 9, 3460–3462 CrossRef CAS PubMed.
- K. D. Ausman, R. Piner, O. Lourie, R. S. Ruoff and M. Korobov, J. Phys. Chem. B, 2000, 104, 8911–8915 CrossRef CAS.
- L. Yang, M. L. Cohen and S. G. Louie, Nano Lett., 2007, 7, 3112–3115 CrossRef CAS PubMed.
- D. Prezzi, D. Varsano, A. Ruini, A. Marini and E. Molinari, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 041404 CrossRef.
- R. Denk, M. Hohage, P. Zeppenfeld, J. Cai, C. A. Pignedoli, H. Söde, R. Fasel, X. Feng, K. Müllen, S. Wang, D. Prezzi, A. Ferretti, A. Ruini, E. Molinari and P. Ruffieux, Nat. Commun., 2014, 5, 4253 CAS.
- G. Soavi, S. Dal Conte, C. Manzoni, D. Viola, A. Narita, Y. Hu, X. Feng, U. Hohenester, E. Molinari, D. Prezzi, K. Müllen and G. Cerullo, Nat. Commun., 2016, 7, 11010 CrossRef CAS PubMed.
- M. Grell, D. D. C. Bradley, X. Long, T. Chamberlain, M. Inbasekaran, E. P. Woo and M. Soliman, Acta Polym., 1998, 49, 439–444 CrossRef CAS.
- R. Al-Jishi and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 4514–4522 CrossRef CAS.
- A. Fechtenkötter, K. Saalwächter, M. A. Harbison, K. Müllen and H. W. Spiess, Angew. Chem., Int. Ed., 1999, 38, 3039–3042 CrossRef.
- S. Ito, M. Wehmeier, J. D. Brand, C. Kübel, R. Epsch, J. P. Rabe and K. Müllen, Chemistry, 2000, 6, 4327–4342 CrossRef CAS PubMed.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 Search PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2013, 46, 1094–1103 CrossRef CAS PubMed.
- M. A. Worsley, S. O. Kucheyev, H. E. Mason, M. D. Merrill, B. P. Mayer, J. Lewicki, C. A. Valdez, M. E. Suss, M. Stadermann, P. J. Pauzauskie, J. H. Satcher, J. Biener and T. F. Baumann, Chem. Commun., 2012, 48, 8428–8430 RSC.
- V. H. Luan, H. N. Tien, L. T. Hoa, N. T. M. Hien, E.-S. Oh, J. Chung, E. J. Kim, W. M. Choi, B.-S. Kong and S. H. Hur, J. Mater. Chem. A, 2013, 1, 208–211 CAS.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 CrossRef PubMed.
- J. C. Shearer, D. A. Slattery, J. A. Stapleton, G. J. Shapter and T. C. Gibson, Nanotechnology, 2016, 27, 125704 CrossRef PubMed.
- S. R. Forrest, Chem. Rev., 1997, 97, 1793–1896 CrossRef CAS PubMed.
- C. B. France, P. G. Schroeder, J. C. Forsythe and B. A. Parkinson, Langmuir, 2003, 19, 1274–1281 CrossRef CAS.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- S. E. Fritz, S. M. Martin, C. D. Frisbie, M. D. Ward and M. F. Toney, J. Am. Chem. Soc., 2004, 126, 4084–4085 CrossRef CAS PubMed.
- R. Ruiz, B. Nickel, N. Koch, L. C. Feldman, R. F. Haglund, A. Kahn and G. Scoles, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 125406 CrossRef.
- Y.-C. Chen, D. G. de Oteyza, Z. Pedramrazi, C. Chen, F. R. Fischer and M. F. Crommie, ACS Nano, 2013, 7, 6123–6128 CrossRef CAS PubMed.
- L. Talirz, H. Söde, J. Cai, P. Ruffieux, S. Blankenburg, R. Jafaar, R. Berger, X. Feng, K. Müllen, D. Passerone, R. Fasel and C. A. Pignedoli, J. Am. Chem. Soc., 2013, 135, 2060–2063 CrossRef CAS PubMed.
- N. D. Denkov, O. D. Velev, P. A. Kralchevsky, I. B. Ivanov, H. Yoshimura and K. Nagayama, Nature, 1993, 361, 26 CrossRef.
- Z. Liu, J. Qiao, Z. Niu and Q. Wang, Chem. Soc. Rev., 2012, 41, 6178–6194 RSC.
- S.-Y. Zhang, M. D. Regulacio and M.-Y. Han, Chem. Soc. Rev., 2014, 43, 2301–2323 RSC.
- A. Sinitskii, D. V. Kosynkin, A. Dimiev and J. M. Tour, ACS Nano, 2010, 4, 3095–3102 CrossRef CAS PubMed.
- Z. Chen, W. Zhang, C.-A. Palma, A. Lodi Rizzini, B. Liu, A. Abbas, N. Richter, L. Martini, X.-Y. Wang, N. Cavani, H. Lu, N. Mishra, C. Coletti, R. Berger, F. Klappenberger, M. Kläui, A. Candini, M. Affronte, C. Zhou, V. De Renzi, U. del Pennino, J. V. Barth, H. J. Räder, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 15488–15496 CrossRef CAS PubMed.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |