
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous high specific surface area-activated carbon with co-doping N, S and P for high-performance supercapacitors†
Mengjiao Xu,
Danyang Li,
Yan Yan,
Teng Guo,
Huan Pang * and
Huaiguo Xue*
* and
Huaiguo Xue*
School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, Jiangsu, China. E-mail: huanpangchem@hotmail.com; panghuan@yzu.edu.cn; chhgxue@yzu.edu.cn
First published on 11th September 2017
Abstract
Starting from the renewable biomass material palm kernel shell (PKS), nitrogen, sulfur and phosphorus co-doped high specific surface area porous activated carbon materials were prepared through carbonization and KOH chemical activation and applied to the supercapacitor electrode materials. PKS is a low-cost carbon source byproduct of biomass, which is rich in cellulose, lignin, and moisture, and its other components also provide nitrogen, sulfur, phosphorus and other heteroatoms for the final product. After further activation with different ratios of alkali and carbon, porous activated carbon (PAC) with different pore sizes could be obtained. The presence of heteroatoms not only provides redox pseudocapacitance, but doping heteroatoms is also beneficial to suppress irreversible changes of the oxygen-containing functional groups on the carbon material surface during the charging and discharging process. In 3 mol L−1 KOH electrolyte solution, PAC showed an excellent electrochemical performance with good cycling stability, and its maximum specific capacity could reach 380 F g−l.
Introduction
Traditionally, activated carbon (AC) is a commercially functional material that is available from petroleum coke, bitumen and coal.1–3 At present, AC has attracted a lot of industrial applications, including electrochemical supercapacitors (SCs), lithium-ion batteries, hydrogen storage, catalysts, adsorbents, and medical components.4–10 Its production process is simple, and hence it is widely used in various fields. For electric double layer capacitor (EDLC) applications, AC exhibits a great demand for due to the advantages of high conductivity, chemical stability and low cost. The AC electrodes, through the accumulation of electrostatic charges at the electrode–electrolyte interface, store the charges and do not involve any chemical charge during the charging and discharging process, which is the basis of EDLC.11–16 Moreover, the specific capacitance of the AC-based SCs can be enhanced by the incorporation of heteroatoms (e.g., nitrogen, oxygen, sulfur, phosphorus, etc.) into the carbon framework, which is also directly proportional to the specific surface area of the PAC.17–21Because raw materials from coal, asphalt and coke that are used for the preparation of AC are not renewable and are expensive, research is now focused on finding low-cost, resource-rich and renewable natural biomass carbon sources (including hemp,22 fungus mushroom,23 pine cone,24 litchi shells,25 pumpkin,26 coconut shells27 and bamboo28) to produce AC, which is a very promising candidate as the SCs electrode. Palm Kernel Shell (PKS) as a dry solid waste, is produced after the extraction of palm oil.29–36 Because of the abundance of PKS, the recovery of value-added products is highly desirable. Therefore, the research and application of PKS' basic performance have been paid great attention to since it proves to be a promising material for the production of AC.33–36 The biggest challenge in PKS processing is the carbon activation process, which is a key factor that will have a direct impact on the performance of AC. For developing vast pore networks in carbon materials, various biomass-derived ACs are typically developed using physical activation37,38 (e.g. O2, CO2 or steam) or chemical activation39–41 techniques (e.g. KOH, H3PO4, ZnCl2, etc.) for EDLC applications. Among them, potassium hydroxide (KOH), because of its better activation effect, has become a widely used activator since it can promote the activated carbon to create a larger specific surface area and stronger adsorption performance.42–44
Recent studies have shown that the surface chemical properties of activated carbon materials are the primary factors that affect the specific capacitance of supercapacitors. A large number of biomass materials are being studied for supercapacitors, particularly after conducting carbonization and activation processes. The main advantages based on the PKS research method are that: (1) the nature of the discarded biomass material of PKS.29–32 PKS, as biomass precursors with a carbon content of more than 60% by weight, can be easily dispersed in the KOH aqueous solution and its sizes can reach nanoscale. Unique natural composites are generally complexes of organic chain macromolecules, including various ionic groups such as carboxylic acids, phenols and alcohols. If used in the preparation of porous carbon materials, cheap and environment friendly carbon precursors have a rich production effect, which can further optimize the pore structure and improve its capacitance performance. (2) A variety of functional groups and heteroatoms for doping (including N, S, O and P) could be formed.17–21 By a series of effective methods of carbonization and activation, PKS can not only form a carbon structure and provide more active sites to promote the formation of porous structures, but also improve the capacitance performance of porous carbon electrode material. Thus, PKS has a crucial effect on the preparation of porous activated carbon materials and their application in supercapacitors for improved capacitance.
However, the nature of carbon from the biomass will vary significantly, and hence it is a challenge to optimize the raw materials required for the process. Obviously, the pore microstructure and element composition of AC depends on the carbon source used or the activation parameters including the ratio of alkali to carbon and activation temperature. Here, by two steps of carbonization and subsequent KOH activation, it was mainly studied on the properties of activated carbon prepared by PKS and the performance effects applied to EDLC. The specific capacitance of 380 F g−1 could be achieved via the EDLC electrode based on PAC. Furthermore, electrochemical studies have shown that AC electrodes have satisfactory charge–discharge cycling stability to realize ultra-high power/energy density for EDLC, which is much higher than that achieved by various other biomass-derived AC electrodes. The specific capacitance (Cs) of the electrode materials prepared from similar activation methods of the various biomass precursors reported in the literature is summarized in Table 1 for comparison with our results. Based on the results obtained, it could be inferred that the excellent performance of biomass-derived porous activated carbon would make it the most attractive tool for high performance supercapacitors and low cost energy storage devices.
| Biomass precursor | Activation method | SBET (m2 g−1) | Cs (F g−1) | Current density | Electrolyte | Ref. |
|---|---|---|---|---|---|---|
| Lotus seedpods | KOH | 1813 | 402 | 0.5 | 6 M KOH | 11 |
| Amygdalus pcdunculata shell | H2SO4 | 2059 | 358.4 | 0.5 | 1 M Na2SO4 | 12 |
| Rotten potatoes | KOH | 960 | 269 | 1 | 6 M KOH | 13 |
| Prawn shells | KOH | 1917 | 357 | 0.5 | 6 M KOH | 17 |
| Tobacco rods | KOH | 2115 | 286.6 | 0.5 | 6 M KOH | 18 |
| Pine cone | KOH | 1515 | 137 | 0.5 | 1 M Na2SO4 | 24 |
| Pumpkin | KOH | 2968 | 419 | 1 | 6 M KOH | 26 |
| Coconut leaves | CO2 | 492 | 133.4 | 0.2 | 6 M KOH | 37 |
| Particleboards | KOH | 1758 | 263 | 0.05 | 7 M KOH | 44 |
| Cabbage leaves | KOH | 3102 | 336 | 1 | 6 M KOH | 45 |
| Shiitakes | KOH | 2335 | 315 | 0.5 | 6 M KOH | 47 |
| Oil palm kernel shells | KOH | 727.3 | 210 | 0.5 | 1 M KOH | 49 |
| Popcorn | KOH | 1489 | 245 | 0.5 | 6 M KOH | 50 |
| Palm kernel shells | KOH | 2760 | 380 | 1 | 3 M KOH | Present study |
Experimental
Synthesis of the PKS-derived activated carbon
The waste PKSs were purchased from an online marketplace. First, to remove soil, dirt and surface impurities, we washed the PKSs thoroughly with clean water and repeatedly with distilled water, and then crushed them into pieces, followed by drying at 100 °C in an oven. For pre-carbonization, the dried PKSs were pyrolyzed at 500 °C in a vacuum tube furnace for 2 h under nitrogen atmosphere to remove the small amount of organic molecules. Subsequently, for improving the specific surface area by chemical activation, the pre-carbonated PKSs were dipped in KOH aqueous solution as the chemical activating agent with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and then stirred thoroughly for 6 h to ensure complete infiltration. Thereafter, the mixture was annealed at 900 °C for 1 h under N2 atmosphere. Finally, hydrochloric acid (HCl, 1 M) was used to wash the annealed sample repeatedly, followed by rinsing with excess of distilled water to obtaining a neutral pH. The final sample was obtained by drying at 80 °C in air and labeled as porous activated carbon (PAC). Fig. 1 shows the transformation of PKSs into porous activated carbons.
:4 and then stirred thoroughly for 6 h to ensure complete infiltration. Thereafter, the mixture was annealed at 900 °C for 1 h under N2 atmosphere. Finally, hydrochloric acid (HCl, 1 M) was used to wash the annealed sample repeatedly, followed by rinsing with excess of distilled water to obtaining a neutral pH. The final sample was obtained by drying at 80 °C in air and labeled as porous activated carbon (PAC). Fig. 1 shows the transformation of PKSs into porous activated carbons.
 | ||
| Fig. 1 Schematic for the synthesis of porous activated carbons from PKSs. | ||
In contrast, the product prepared without the addition of KOH using the same procedure was labeled as nonporous activated carbon (NAC).
Material characterizations
The crystal structure was measured by a Bruker D8 Advanced X-ray diffractometer (Germany, CuKα radiation: λ = 1.5416 Å) to analyze X-ray diffraction (XRD) patterns. The functional groups investigation and the chemical analysis of the samples were carried out using a Spectrum One (Perkin Elmer, USA) FTIR spectrometer and an Omicron NanoTechnology (ESCAL.ab250) X-ray photoelectron spectrometer, respectively. The morphologies of the prepared porous structure were characterized via field emission scanning electron microscopy (FESEM, Zeiss-Supra 55) and high resolution transmission electron microscopy (HRTEM, Tecnai G2 F30 S-TWIN) with energy dispersive X-ray spectroscopy (EDS) mapping, while the N2 adsorption–desorption isotherm and Brunauer–Emmett–Teller (BET) specific surface area of the samples were obtained at 77 K using an ASAP 2020 (Micromeritics, USA) apparatus. Thermal properties were determined using the PerkinElmer Pyris 1 thermogravimetric analyzer (TGA, USA), where the sample was heated to 500 °C at a heating rate of 5 °C min−1 under N2 flow. The Raman analysis of the samples was recorded using a DXRxi Raman spectrometer (Thermofisher, USA) with the laser power set at 1.5 mW to minimize the heating effect.Fabrication of working electrodes
In the electrochemical performance characterization, the working electrodes were fabricated for the mixture with a mass ratio of 8:1:1 of the dried AC, conductive carbon (Super P) and polytetrafluoroethylene (PTFE) binder in N-methylpyrrolidinone (NMP) solvent dispersant, followed by thorough stirring for 0.5 h using the agate mortar till a paste was obtained. This paste was uniformly coated onto a pre-cleaned nickel foam substrate with a square size of 1 cm × 1 cm. Then, the electrode coated with abovementioned fabricated paste was annealed at 80 °C for 12 h in an oven. Until the annealed electrode was cooled to room temperature, we used a hydraulic equipment at 10 MPa to press the final electrode to allow sufficient contact of the foam nickel with the active materials. The mass of the active substance was obtained according to the ratio for calculating the electrochemical properties of the materials.
Electrochemical measurements
To examine the electrochemical properties of the prepared electrodes, we conducted cyclic voltammetry (CV), galvanostatic charge–discharge cycling (CDC) and electrochemical impedance spectroscopy (EIS) analysis, which were carried out using a conventional three-electrode configuration on a CHI 760D electrochemical workstation (Shanghai Chenhua, China) under the premises in a potential range of −1.0–0.0 V. Herein, the performance of the prepared electrodes was measured by placing them in 3 M KOH electrolyte. The electrochemical workstation was equipped with a counter electrode (platinum), a reference electrode (Hg/HgO) and a working electrode just above the prepared AC electrode. Cyclic voltammetry and galvanostatic CDC were obtained at different scan rates of 5–100 mV s−1 and various current densities in the range 1–20 A g−1, respectively. EIS plots reveal the response of the EDLC electrode observed in the frequency range from 0.1 Hz to 105 Hz.Results and discussion
Fig. 2 exhibits the representative XRD patterns of PAC and NAC samples. Two intense and broad diffraction peaks are evidently generated at approximately 2θ = 24° and 44° of both of the PAC and NAC samples, corresponding to the (002) and (100) diffraction patterns of amorphous carbon structure, respectively. The intensity of the graphitic carbon structure at 2θ = 24° is reduced and further broadened upon carbonation and activation. A wider peak indicates that the carbon structure is amorphous due to the increase in distance between activated carbon and carbon atoms and the destruction of the graphite structure during carbonation and activation. A wide diffraction peak at 2θ = 44° indicates that PAC sample has a certain degree of graphitization, which facilitates the conductivity of AC.45 The higher diffraction intensity of the activated PKS at a lower 2θ value clearly indicates that AC has a porous structure.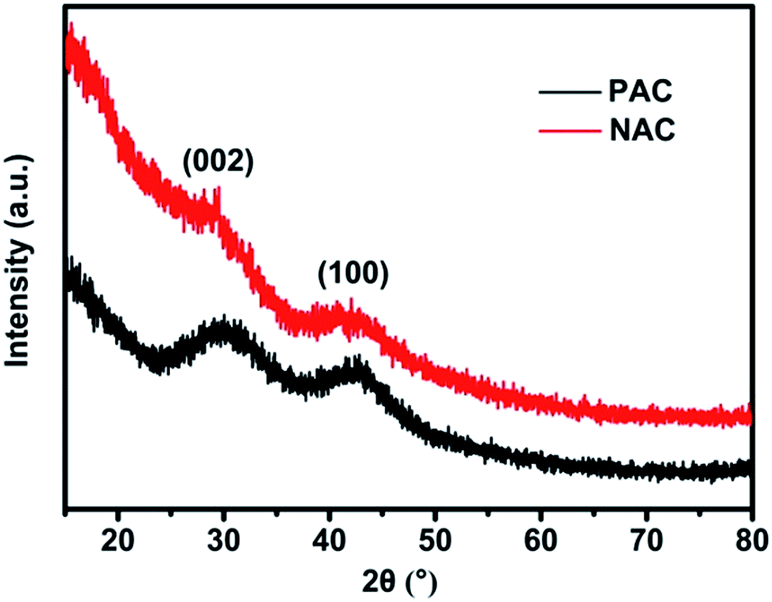 | ||
| Fig. 2 XRD patterns of PAC and NAC. | ||
Generally, we adopt the FTIR spectra to qualitatively analyze the surface functional groups of AC, and both infrared spectra patterns of AC samples before and after activation are observed to be nearly similar (Fig. S1†). The formation of the flexural vibration at 3415 cm−1 and 3700 cm−1 and bending vibration at 1560 cm−1 corresponding to –O–H stretching are attributed to physical adsorption of water molecules or hydroxyl groups domination. The two peaks that belong to asymmetric and symmetric C–H stretching are located at 2924 cm−1 and 2848 cm−1, respectively, while that of the C–H group with the deformation mode is located at 1382 cm−1. The carbonyl group on the AC creates two types of weak peaks, separately designated as the stretching mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1728 cm−1 and the stretching mode of CC at 1560 cm−1. Simultaneously, it is clearly observed that the C–O–C stretching mode is located at 1070 cm−1. Compared with NAC, the absorption peak of PAC shifted to a higher wavenumber due to KOH activation. From the FTIR analysis, it is observed that PAC and NAC exhibit an amorphous carbon structure with large amounts of oxygen-containing functional groups, the presence of which could provide additional pseudocapacitance and good conductivity. The numerous carboxyl and hydroxyl groups on AC surfaces also indicate the excellent capacitance properties of the material.
O at 1728 cm−1 and the stretching mode of CC at 1560 cm−1. Simultaneously, it is clearly observed that the C–O–C stretching mode is located at 1070 cm−1. Compared with NAC, the absorption peak of PAC shifted to a higher wavenumber due to KOH activation. From the FTIR analysis, it is observed that PAC and NAC exhibit an amorphous carbon structure with large amounts of oxygen-containing functional groups, the presence of which could provide additional pseudocapacitance and good conductivity. The numerous carboxyl and hydroxyl groups on AC surfaces also indicate the excellent capacitance properties of the material.
The PKS comprises organic macromolecules such as cellulose, hemicelluloses and lignin, which can absorb a large amount of elements from the soil during the growth process, including C, H, O, N, S, P, K, Na, and Mg. Most likely, these elements, in the form of inorganic matter, as the most important source of ash, still remain in the PKS carbonized material because of the pyrolysis in nitrogen environments. Presence of these elements is also observed during carbonization in nitrogen atmosphere. Therefore, it is highly probable that a large amount of N, S, P elements are introduced into the final carbon material during KOH activation.
The surface element composition of heteroatoms for PAC and NAC including carbon, nitrogen, oxygen, sulfur and phosphorus, which are clearly detected at 285 eV, 405 eV, 532 eV, 164 eV and 133 eV respectively, was analyzed using XPS (Fig. S2†). The amounts of different elements on the surface of the materials as calculated by XPS analysis are listed in Table 2, which show the content of N, S, P to be similar in NAC and PAC samples (the content of N, S, P for PAC only slightly higher than NAC). Furthermore, the central spectra of resolved peaks at N 1s, S 2p and P 2p for PAC are shown in Fig. 3. It is distinct that contents of nitrogen, sulfur and phosphorus elements of the prepared carbon material are low. In case of PAC, N doping accounts for 1.71% (atomic percentage), S doping accounts for 0.76% and P doping accounts for 0.57%. As presented in Fig. 3a, three peaks of the XPS spectrum for N 1s could be detected as pyridinic N1 (398.48 eV), pyrrolic N2 (400.48 eV) and pyridine-N-oxide N3 (403.08 eV). These N species produced may not only promote the formation of various nitrogen-containing functional groups on the porous carbon surface, but also can contribute to the efficient utilization of synergistic effect of heteroatoms and the rapid absorption for dissolved substances, thus enhancing cycling stability.46 Furthermore from Fig. 3b, the S 2p can be decomposed into two adjacent peaks between 168 eV and 170 eV, corresponding to some possible forms of the oxidation states arising from the strong interaction of sulfur and oxygen, which may be due to the sulfur element on the carbon surface adjacent to the oxygen molecules caused by oxidation.47 Finally, as observed in Fig. 3c, two peaks located at 134.48 eV and 133.48 eV are separated from P 2p. This occurs due to the possible phosphorus-oxygen functional groups, namely phosphates or C-PO3 phosphorus groups, which can moderately restrict irreversible reaction during discharge/charge process. Therefore, it can be inferred that the persistence of phosphorus in AC may contribute to the improvement of its electrochemical performances. Hence, a typical N, S, P co-doped activated carbon material is conducive to the formation of AC surface functionalities for EDLC effect in the charge storage mechanism.
| Surface elemental compositions of samples | |||||
|---|---|---|---|---|---|
| Samples | C (at%) | O (at%) | N (at%) | S (at%) | P (at%) |
| NAC | 87.12 | 10.01 | 1.69 | 0.65 | 0.53 |
| PAC | 89.04 | 7.92 | 1.71 | 0.76 | 0.57 |
 | ||
| Fig. 3 XPS spectra of (a) N 1s, (b) S 2p and (c) P 2p regions for PAC, respectively. | ||
In the case of pyrolysis, the decomposition of lignocellulose in PKS and the volatilization of some organic matter are carried out to obtain AC with a porous structure. Fig. S3c† is a SEM image of the PKS carbonized material obtained after carbonization at 500 °C. Fig. S3a and S3b† shows a SEM image of the NAC and PAC prepared from PKS carbonized material. A developed and irregular porous structure appeared in the PAC after KOH activation. After increasing the magnification, we could clearly see the surface of the AC inside the large holes with a large number of mesopores and micropores, as displayed in Fig. S3d;† hence in is confirmed that the material for the porous structure shows irregular block structures. It is taken for granted that the activation agent (KOH) scattered into the layered porous structure could sufficiently facilitate activation. NAC was found to exhibit only a few pores.
As for the KOH activation, the carbon framework containing various potassium compounds is partially etched by the carbon activation reaction between KOH and the feedstock. Consequently, the carbon lattice expands and the formed metal (K) can be effectively embedded in the carbon lattice of the carbon matrix, while the production of H2O and CO2 also contributes to the formation of a well-formed pore network in the resulting product during the activating process. Moreover, after washing to remove the resulting potassium salts and excess KOH, the pores that were present at the etched location could not be restored to their previous construction, thus resulting in a large number of PAC with high porosity.
As is depicted in Fig. 4, high-resolution TEM images of AC surface conditions exhibited almost similar morphologies, including the homologous five elemental mapping images of carbon, oxygen, nitrogen, sulfur and phosphorus for PAC. It is clear from the figure that there are numerous micropores and mesopores in AC, and the transparent and blank spots in the figure represent the pores of AC with pore size ranging from several nanometers to dozens of nanometers. As a result, the PAC material exhibits a larger specific surface area and a higher porosity. In addition, the sample is essentially made up of amorphous components with partially curved graphitized layers. In contrast, the NAC sample merely exerts the porous framework structure with only a few blank spots. Moreover, EDX spectra and the inset figures in Fig. S4† show detailed EDX elemental analysis of the PAC samples. It is clear from Fig. S4† that the contents of N, S, P measured by EDX mapping in the PAC after KOH activation are not significantly different from those for PAC summarized in Table 2 using XPS method, but slightly higher than the contents of N, S, P in the NAC observed using XPS method. Therefore, the higher N, S, P contents after the KOH treatment are conducive to the improvement of the supercapacitor performance.
 | ||
| Fig. 4 High resolution TEM images of (a) PAC and (b) NAC; STEM image (c) and the corresponding EDS mapping of C–K (d), N–K (e), O–K (f), S–K (g) and P–K (h) from PAC. | ||
The carbonized material without activation has a small amount of large pores and has a loose surface. This macroporous structure facilitates the rapid entry of the activator (KOH) into the activated pores, thus leading to the formation of ultra-high specific surface area. Moreover, large porous structure with large pore volume of the carbonized material enables the activator to etch with the carbon atoms faster for pore-formation and shorten the time of the activation reaction. This also indicates that the activation time has a minor effect on activated carbon. The formation of tunnels and pores during activation can facilitate ion adsorption and transportation in the charge storage mechanism, which makes the ion diffusion in the supercapacitor electrolyte much easier. Therefore, the porous surface (Fig. 4) indicates that PAC activated by KOH may achieve an ultra-high specific surface area and a large pore volume. This leads to a high specific capacitance, which is consistent with the nitrogen adsorption and desorption curve of the material.
Both curves of the nitrogen adsorption–desorption and pore size distributions for PAC and NAC measured at 77 K are shown in Fig. 5. It shows that the microporous structure of the N2 absorption increased greatly at P/P0 < 0.4. When P/P0 > 0.4, there is an evident hysteresis loop growing slowly for PAC. The emergence of this phenomenon indicates that PAC is composed of a large number of micropores and mesopores, in line with the combination of type I and IV isotherms.48 PAC consists of micropores (less than 2 nm), mesopores (2 nm to 50 nm) and macropores (greater than 50 nm), with the mesopores dominant in the sample. The specific surface area and pore volume of PAC are primarily as high as 2760 m2 g−1 and 1.6 cm3 g−1, respectively. At the same time, the pore size distribution is centered at 2.3 nm. On the contrary, NAC has a specific surface area of 1124 m2 g−1 and a pore volume 0.5 cm3 g−1; its pore size distribution is centered at 1.9 nm. The high specific surface area indicates that PAC has higher specific capacitance. Considering the comparative data above, it is easier to conclude that PKS precursors with KOH activation at high temperature could be obtained rapidly and produced ultra-high specific surface area and appropriate mesopore size, which is beneficial for efficient charge storage. The pore-rich surface and narrower pore size distribution of AC provide high ion accessibility through wider transport channels to micropores. Therefore, we expect that PAC would be a promising electroactive material for advanced SCs.
 | ||
| Fig. 5 (a) Nitrogen adsorption–desorption isotherms and (b) BJH pore size distributions of PAC and NAC. The inset in (a) shows the partial enlargement of nitrogen adsorption–desorption isotherms of NAC. | ||
To further investigate characteristics of PAC, Raman analysis and thermal properties, which were adopted to quantify the amount of AC that occurred during the heating of PKS, were performed, and the results obtained are shown in Fig. S5 and S6,† respectively. Clearly, as depicted in the Fig. S5,† the two prominent peaks shown in the Raman spectrum are located at 1342 cm−1 and 1587 cm−1, individually corresponding to D (defects and disorder)-band and G (graphitic)-band, which is attributed to the C–C graphitic lattice vibration mode. Because of the incorporation of heteroatom N, S and P in the PAC framework, some amount of disorder is caused that partially makes the G-band stronger. ID/IG, known as the ratio of intensities of “D-band” to “G-band”, is calculated to be about 0.88 and 0.95 for PAC and NAC, respectively, which could explain the higher graphitization degree with large amount of structurally ordered graphite crystallites for PAC. Furthermore, on observing the TG and DTG curves, three distinct decomposition periods of PAC, which occurred within the broad range of 200–900 °C, show different thermal degradation behaviors. It is essential for the degradation trend of PAC to compare with the one of lignocellulosic biomass; the weight loss primarily at 300–700 °C was probably due to the degradation of hemicelluloses (200–260 °C), cellulose (240–350 °C) and lignin (above 380 °C). In view of the TGA analysis data, 500 °C is optimized as the minimum pyrolysis temperature of PKS. Considering that the activation temperature plays an important role in regulating the electrochemical and textural properties of PKS derived activated carbons, a series of experiments were carried out to select the optimum activation temperature reaction conditions. In this study, the electrochemical and textural properties of synthetic porous activated carbons were quantitatively estimated by measuring their specific capacitance (Cs), BET SSA and pore volume. The figures of BET and Cs at different temperatures are listed in Table S1.† It could be clearly observed from the images that the excellent capacitive properties of PKS at 900 °C could be attributed to their relatively high BET SSA, optimum pore volume and the surface properties mentioned above.
To evaluate the electrochemical capacitance behavior of the PAC and NAC supercapacitor electrodes prepared above, the CV analysis was performed to probe in 3 M KOH solution, and the voltage window was −1.0 to 0.0 V. Both CV curves for PAC and NAC at a scan rate from 10 to 150 mV s−1 are depicted in Fig. 6a and b, respectively. Fig. 6c shows the comparison of CV curve for PAC and NAC at a scanning rate of 10 mV s−1, indicating that PAC activated with KOH has a good capacitance performance. As the activated carbon is composed of micropores, mesopores and macropores in the channel structure, the electrolyte ions penetrating into the pores of different sizes need to overcome different resistance. Due to the resistance, electrolyte ions have slower diffusion rate in the micropores compared to the macropores, so the response current may produce a certain relaxation time. Therefore, EDLC cyclic voltammetry curves show a quasi-rectangular shape, which indicates that the material with porous structure is stable. At the same time, it is further observed that the response current and the quasi-rectangular area of the CV curve for the PAC electrode, as compared with NAC, are much larger. In addition, in the range of the scan voltage, there is a slight polarization at the positive and negative potentials, which represent the complex reversible redox reactions occuring on the AC surface. The presence of polarization phenomenon is also attributed to the multiple functional groups on the AC surface.
 | ||
| Fig. 6 CV curves of (a) PAC and (b) NAC within the −0.1–0.0 V range at a scan rate from 10 to 150 mV s−1 in 3 M KOH; comparison of (c) CV curves for PAC and NAC at scan rate 10 mV s−1; CDC curves for (d) PAC and (e) NAC at various current densities of 1.0–10.0 A g−1 and (f) the area chart for CDC behavior of PAC and NAC electrodes in 3 M KOH electrolyte at 1.0 A g−1. | ||
Galvanostatic CDC cycling method is commonly used for studying the capacitance performance of electrode materials. Fig. 6d and e show galvanostatic CDC curves for PAC and NAC placed in 3 M KOH electrolyte at various current densities of 1.0–10.0 A g−1, respectively. Fig. 6f shows the comparison of the CDC behavior of PAC and NAC electrodes at 1 A g−1 current density, concluding that the charging and discharging time of PAC is significantly longer than that of NAC. The galvanostatic CDC curves of AC show a good linearity and a relatively symmetrical triangular shape, indicating that AC has a good EDLC behavior with excellent reversibility.
According to the following formula, we could calculate the specific capacitance of AC:

 | ||
| Fig. 7 (a) Specific capacitance of PAC and NAC electrode derived from the discharging curves at various current densities of 1.0–20.0 A g−1 and (b) the charge–discharge cycling stability test of PAC electrode at 1 A g−1. | ||
Studies have shown that PKS as a byproduct of biomass, not only is a low-cost carbon source, but also its rich cellulose, lignin, moisture and other major components provide nitrogen, sulfur, phosphorus and other heteroatoms for the final product. Due to the synergistic effect of the presence of various heteroatoms such as C, N, O, S, and P, the carbon material prepared exhibits larger pseudocapacitance. Moreover, heteroatom doping is beneficial to suppress an irreversible change of the oxygen-containing functional groups on AC surface during the charging and discharging process. It also shows that the reasonable porosity generated by KOH activation of AC is beneficial for the formation of dense double electric layer capacitance and rapid transmission in KOH electrolyte, which can significantly improve the capacitance behavior.49
Another important indicator for enhanced performance of supercapacitors is electrochemical stability. To check the actual expandability of these electrodes, a porous activated carbon was used, and a current density of 1 A g−1 was applied with a 3 M KOH electrolyte to perform 1000 cycles of the electrochemical stability; the result obtained is shown in Fig. 7b. The capacitance drops slowly in the first 500 cycles due to the reduction of the extra pseudocapacitance, which is caused by the gradual disappearance of the surface oxygen-containing functional groups of the porous activated carbon after the chemical activation. For 10000 cycles, the capacitance remained stable and the final specific capacitance retention rate was about 96%, which indicates that PAC has excellent cycle stability as an ideal EDLC electrode material.
The study of the ion transport mechanism and further demonstration of the performance for PAC and NAC electrodes were testified using electrochemical impedance spectroscopy. The EIS analysis carried out in the frequency range from 10 mHz to 105 Hz shows the Nyquist plot with a small semicircle and a nearly straight line at the high/low frequency region, respectively (Fig. 8), intuitively indicating porous carbon electrodes with a distinct capacitive property. From data analysis of the high frequency region, a small semicircle is generated relevant to charge transport phenomena (RCT) during the testing process at the electrode–electrolyte interface. Herein, the electrolyte ions could rapidly diffuse to the outer surface of the activated carbon electrode and the inner surface of the macropores or mesopores, thus concluding that there is an existence of Faraday impedance of charge transfer. Moreover, the ohmic resistance (RS) was estimated from which it could be deemed that the internal resistance of the carbon material is very small (less than 0.5 Ω), indicating that the carbon material is well conductive.50 It also manifested that the kinetics process of the electrochemical system is rapid and the ion transport plays a leading role. Viewing from the low frequency region where the inclined portion of the impedance rises sharply (slope close to 90°). The straight line shows that AC materials exhibit pure double electric layer capacitance behavior and the characteristics of rapid electrolyte ion transfer. The fundamental reason for this capacitive behavior is that the palm kernel shell-based porous activated carbon has a porous structure with low resistance, and thus is suitable for ion diffusion.
 | ||
| Fig. 8 Nyquist plots of PAC and NAC electrodes. | ||
Conclusions
In conclusion, N, S and P co-doped PAC was successfully prepared via pyrolysis and successive KOH activation using palm kernel shell as raw material. The specific surface area of activated PAC is about 2760 m2 g−1, which could be favorable to improve its EDLC applications. Along with the synergistic effect of N, P, S and other types of elements doping, the prepared carbon material exhibited excellent electrochemical capacitance characteristics. Furthermore, the presence of carbonyl and hydroxyl functional groups on PAC surface also provided the extra pseudocapacitance. In the 3 M KOH electrolyte, the specific capacitance is as high as 380 F g−1 at a current density of 1 A g−1. Subsequently, PAC shows excellent cycle performance. After 10000 charge and discharge cycles, the specific capacitance is still maintained at 96%. In this study, a variety of heteroatom-doped carbon materials was successfully prepared using biomass. The carbon material prepared showed a high specific surface area and exhibited excellent electrochemical properties. Therefore, the palm kernel shell is an excellent raw material for production of activated carbon.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The acknowledgements come at the end of an article after the conclusions and before the notes and references. This study was supported by the Program for New Century Excellent Talents of the University in China (NCET-13-0645) and the National Natural Science Foundation of China (NSFC-21201010, 21671170 and 21673203), Innovation Scientists and Technicians Troop Construction Projects of Henan Province (164200510018), Program for Innovative Research Team (in Science and Technology) in University of Henan Province (14IRTSTHN004), the Six Talent Plan (2015-XCL-030), and Qinglan Project. We also acknowledge the Priority Academic Program Development of Jiangsu Higher Education Institutions and the technical support we received at the Testing Center of Yangzhou University.Notes and references
- Y. Z. Ma, B. J. Yu, Y. Guo and C. Y. Wang, J. Solid State Electrochem., 2016, 20, 2231–2240 CrossRef CAS.
- Q. X. Xie, S. W. Zhou, A. R. Zheng, C. Xie, C. Y. Yin, S. H. Wu, Y. F. Zhang and P. Zhao, Electrochim. Acta, 2016, 189, 22–31 CrossRef CAS.
- X. J. Wei, X. Q. Jiang, J. S. Wei and S. Y. Gao, Chem. Mater., 2016, 28, 445–458 CrossRef CAS.
- Q. Jiang, Z. H. Zhang, S. Y. Yin, Z. P. Guo, S. Q. Wang and C. Q. Feng, Appl. Surf. Sci., 2016, 379, 73–82 CrossRef CAS.
- H. L. Wang, W. H. Yu, J. Shi, N. Mao, S. G. Chen and W. Liu, Electrochim. Acta, 2016, 188, 103–110 CrossRef CAS.
- A. Jain, R. Balasubramanian and M. P. Srinivasan, Chem. Eng. J., 2016, 283, 789–805 CrossRef CAS.
- S. Y. Zhu, Y. Q. Wang, J. C. Jiang, X. Yan, D. Sun, Y. C. Jin, C. W. Nan, H. Munakata and K. Kanamura, ACS Appl. Mater. Interfaces, 2016, 8, 17253–17259 CAS.
- D. L. Sivadas, S. Vijayan, R. Rajeev, K. N. Ninan and K. Prabhakaran, Carbon, 2016, 109, 7–18 CrossRef CAS.
- M. Zhou, F. Lu, X. S. Shen, W. W. Xia, H. He and X. H. Zeng, J. Mater. Chem. A, 2015, 3, 21201–21210 CAS.
- L. Fan, L. Tang, H. F. Gong, Z. H. Yao and R. Guo, J. Mater. Chem., 2012, 22, 16376–16381 RSC.
- B. Liu, X. H. Zhou, H. B. Chen, Y. J. Liu and H. M. Li, Electrochim. Acta, 2016, 208, 55–63 CrossRef CAS.
- W. C. Li, Y. Ding, W. Q. Zhang, Y. Shu, L. Zhang, F. C. Yang and Y. H. Shen, J. Taiwan Inst. Chem. Eng., 2016, 64, 166–172 CrossRef CAS.
- Y. X. Huang, L. L. Peng, Y. Liu, G. J. Zhao, J. Y. Chen and G. H. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 15205–15215 CAS.
- S. Q. Zhu, Q. L. Chen, Y. Y. Shi, Z. Y. Chen, R. Q. Bao, L. Zhou, L. R. Hou, K. N. Hui and C. Z. Yuan, J. Solid State Electrochem., 2016, 20, 713–723 CrossRef CAS.
- B. Li, M. B. Zheng, H. G. Xue and H. Pang, Inorg. Chem. Front., 2016, 47, 175–202 RSC.
- Y. Yan, H. Xu, W. Guo, Q. L. Huang, M. B. Zheng, H. Pang and H. G. Xue, Inorg. Chem. Front., 2016, 3, 791–797 RSC.
- F. Gao, J. Y. Qu, Z. B. Zhao, Z. Y. Wang and J. S. Qiu, Electrochim. Acta, 2016, 190, 1134–1141 CrossRef CAS.
- Y. Q. Zhao, M. Lu, P. Y. Tao, Y. J. Zhang, X. T. Gong, Z. Yang, G. Q. Zhang and H. L. Li, J. Power Sources, 2016, 307, 391–400 CrossRef CAS.
- W. Kiciński, M. Norek, A. Dziura and M. Polański, Microporous Mesoporous Mater., 2016, 225, 198–209 CrossRef.
- M. M. Wan, X. D. Sun, Y. Y. Li, J. Zhou, Y. Wang and J. H. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 1252–1263 CAS.
- L. L. Zhang, H. H. Li, Y. H. Shi, C. Y. Fan, X. L. Wu, H. F. Wang, H. Z. Sun and J. P. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 4233–4241 CAS.
- W. Sun, S. M. Lipka, C. Swartz, D. Williams and F. Q. Yang, Carbon, 2016, 103, 181–192 CrossRef CAS.
- H. L. Wu, J. R. Mou, L. Zhou, Q. J. Zheng, N. Jiang and D. M. Lin, Electrochim. Acta, 2016, 212, 1021–1030 CrossRef CAS.
- A. Bello, N. Manyala, F. Barzegar, A. A. Khaleed, D. Y. Momodu and J. K. Dangbegnon, RSC Adv., 2016, 6, 1800–1809 RSC.
- S. T. Zhang, M. B. Zheng, Z. X. Lin, N. W. Li, Y. J. Liu, B. Zhao, H. Pang, J. M. Cao, P. He and Y. Shi, J. Mater. Chem. A, 2014, 2, 15889–15896 CAS.
- S. Y. Bai, G. Q. Tan, X. Q. Li, Q. Zhao, Y. Meng, Y. J. Wang, Y. Z. Zhang and D. Xiao, Chem.–Asian J., 2016, 11, 1828–1836 CrossRef CAS PubMed.
- A. L. Cazetta, O. Pezoti, K. C. Bedin, T. L. Silva, A. P. Junior, T. Asefa and V. C. Almeida, ACS Sustainable Chem. Eng., 2016, 4, 1058–1068 CrossRef CAS.
- W. Chen, H. P. Yang, Y. Q. Chen, X. Chen, Y. Fang and H. Chen, J. Anal. Appl. Pyrolysis, 2016, 120, 186–193 CrossRef CAS.
- S. Nizamuddin, N. S. Jayakumar, J. N. Sahu, P. Ganesan, A. W. Bhutto and N. M. Mubarak, Korean J. Chem. Eng., 2015, 32, 1789–1797 CrossRef CAS.
- S. Nizamuddin, N. S. Jaya Kumar, J. N. Sahu, P. Ganesan, N. M. Mubarak and S. A. Mazari, Can. J. Chem. Eng., 2015, 93, 1916–1921 CrossRef CAS.
- S. Nizamuddin, N. M. Mubarak, M. Tiripathi, N. S. Jayakumar, J. N. Sahu and P. Ganesan, Fuel, 2016, 163, 88–97 CrossRef CAS.
- G. G. Choi, S. J. Oh, S. J. Lee and J. S. Kim, Bioresour. Technol., 2015, 178, 99–107 CrossRef CAS PubMed.
- A. Kundu, B. S. Gupta, M. A. Hashim and G. Redzwan, J. Cleaner Prod., 2015, 105, 420–427 CrossRef CAS.
- Z. Q. Ma, D. Y. Chen, J. Gu, B. F. Bao and Q. S. Zhang, Energy Convers. Manage., 2015, 89, 251–259 CrossRef CAS.
- J. R. García, U. Sedran, M. A. A. Zaini and Z. A. Zakaria, Environ. Sci. Pollut. Res., 2017, 1–10 Search PubMed.
- J. Y. Jeong, U. D. Lee, W. S. Chang and S. H. Jeong, Biosci. Rep., 2016, 219, 357–364 CAS.
- K. S. Sulaiman, A. Mat and A. K. Arof, Ionics, 2016, 22, 911–918 CrossRef CAS.
- M. Shoaib and H. M. Al-Swaidan, Hem. Ind., 2016, 70, 151–157 CrossRef.
- J. Yin, Y. Q. Zhu, X. Yue, L. Wang, H. Zhu and C. Y. Wang, Electrochim. Acta, 2016, 201, 96–105 CrossRef CAS.
- C. Laginhas, J. M. V. Nabais and M. M. Titirici, Microporous Mesoporous Mater., 2016, 226, 125–132 CrossRef CAS.
- Y. Q. Zhao, M. Lu, P. Y. Tao, Y. J. Zhang, X. T. Gong, Z. Yang, G. Q. Zhan and H. L. Li, J. Power Sources, 2016, 307, 391–400 CrossRef CAS.
- J. Li, K. Li, T. H. Zhang, S. Wang, Y. Jiang, Y. Bao and M. Y. Tie, Fibers Polym., 2016, 17, 880–887 CrossRef CAS.
- J. C. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- T. X. Shang and X. J. Jin, J. Solid State Electrochem., 2016, 20, 2029–2036 CrossRef CAS.
- P. Y. Wang, Q. L. Wang, G. H. Zhang, H. Y. Jiao, X. Y. Deng and L. W. Liu, J. Solid State Electrochem., 2016, 20, 319–325 CrossRef CAS.
- X. R. Li, S. Y. Ding, X. Xiao, J. Y. Shao, J. L. Wei, Y. Yu and H. Pang, J. Mater. Chem. A, 2017, 5, 12774–12781 CAS.
- H. Cao, Z. Y. Chen, Q. L. Chen, C. Yang, L. R. Hou, M. Rehan, L. N. Tong and C. Z. Yuan, RSC Adv., 2016, 6, 81527–81533 RSC.
- B. L. Xing, G. X. Huang, L. J. Chen, H. Guo, C. X. Zhang, W. Xie and Z. F. Chen, J. Porous Mater., 2016, 23, 67–73 CrossRef CAS.
- I. I. Misnon, N. K. M. Zain, R. A. Aziz, B. Vidyadharan and R. Jose, Electrochim. Acta, 2015, 174, 78–86 CrossRef CAS.
- T. Liang, C. L. Chen, X. Li and J. Zhang, Langmuir, 2016, 32, 8042–8049 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07945a |
| This journal is © The Royal Society of Chemistry 2017 |