
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of Janus nanoparticles from block copolymer thin films using triazolinedione chemistry†
Elio Poggia,
Wolfgang Ouvrya,
Bruno Ernoulda,
Jean-Pierre Bourgeoisa,
Subrata Chattopadhyaybc,
Filip Du Prez b and
J.-F. Gohy*a
b and
J.-F. Gohy*a
aInstitute of Condensed Matter and Nanosciences, Division of Bio and Soft Matter, Université catholique de Louvain, Place L. Pasteur 1, bte L4.01.01, B-1348 Louvain-la-Neuve, Belgium. E-mail: jean-francois.gohy@uclouvain.be
bPolymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4bis, B-9000 Ghent, Belgium
cDepartment of Chemistry, Indian Institute of Technology Patna, Patna 801103, Bihar, India
First published on 26th July 2017
Abstract
A nanostructured thin film obtained from a polystyrene-block-poly(4-vinylpyridine) diblock copolymer was used as a template to produce Janus-type nanoparticles. This was achieved by functionalizing the poly(4-vinylpyridine) microdomains with a diene molecule and by cross-linking them with diiodo compounds. A poly(methacrylate) chain end-functionalized by a triazolinedione group was selectively clicked onto the top of the poly(4-vinylpyridine) microdomains. The Janus nanoparticles were then released in solution by the dissolution of the thin film in a good solvent.
Introduction
Janus particles, named after the doubled faced Roman god “Janus”, are compartmentalized particles composed of two or more surface regions with different chemistries or polarities.1 This broken symmetry gives rise to unique properties such as the ability to display directional interactions, which makes this class of particles potentially useful in a wide range of applications, including as phase transfer agents,2 switchable devices,3 self-propelled carriers,4,5 theranostic agents,6,7 surfactants8–11 or catalysts.12,13 In this context, the ability of block copolymers to self-assemble into complex nanostructures makes them very interesting building blocks for the synthesis of such nano-sized particles.14–16 However, since direct self-assembly of block copolymers into anisotropic particles is challenging, dedicated strategies or peculiar systems are required to break the symmetry and generate anisotropic particles. Most of those strategies take advantage of triblock terpolymers forming particular morphologies or nanostructures, either in bulk17,18 or in selective solvents.19,20In a previous work, we reported on the formation of polymeric Janus nanoparticles by grafting of a homopolymer through copper(I)-catalysed alkyne–azide cycloaddition (CuAAC) on the top surface of previously cross-linked microdomains of a nanostructured block copolymer thin film.21 Although efficient, the CuAAC chemistry presents some drawbacks, the most obvious being the necessity to use a copper-based catalyst. This results in additional purification steps to remove the catalyst and often requires inert conditions during the process to avoid copper(I) oxidation. The use of click-like reactions that do not require the addition of a catalyst is therefore desirable in most applications.22 In this context, 1,2,4-triazoline-3,5-dione based chemistry represents a very interesting alternative to the classic CuAAC click reaction. Triazolinediones are indeed generally considered as the most reactive dienophile/enophile and can therefore react very quickly in mild conditions and without catalyst with electron-rich π-systems such as dienes as shown in Fig. 1.23
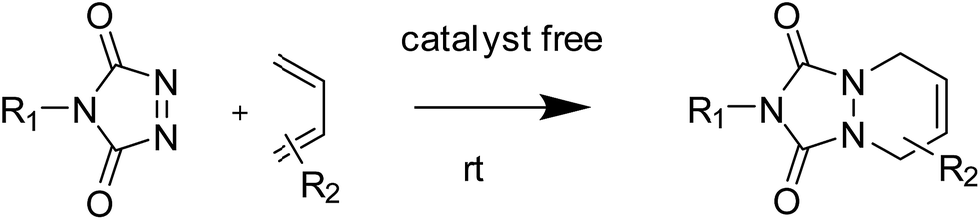 | ||
| Fig. 1 Reaction scheme between 1,2,4-triazoline-3,5-dione and dienes. | ||
TAD-ene chemistry has been successfully employed in organic synthesis since 1960's and has found many applications in polymer science, mainly for the chemical modification or cross-linking of polydienes, such as polyisoprene or polybutadiene.24 However, the difficulties to synthesise specific and functional TAD compounds has limited its widespread use. The spectacular development of click-chemistry during the last decade resulted in a strong revival of TAD chemistry. In recent years TAD-(di)ene click reactions were found very useful in the preparation of nanoparticles,25 polymer bioconjugates synthesis of block co-polymers,26 functionalization of polymers and materials etc.27–32 In an earlier report, while demonstrating the first example for the application of ‘triazolinedione-ene’ reactions to prepare nanoparticles, we have shown that this chemistry can be a useful platform to prepare spherical nanoparticles from multi-ene systems, such as plant oils, via a nanoprecipitation technique.25 However, such methods cannot be applied for the preparation of asymmetric structures such as Janus particles.
In the present report, the grafting of a TAD terminated polymer onto a diene functionalized block copolymers thin films has been used to break the symmetry and induce the formation of polymeric Janus nanoparticles upon dissolution of the film. The proposed strategy is highlighted in Fig. 2. In the first step, a nanostructured block copolymer PS-b-P4VP thin film with regularly spaced and nearly monodisperse P4VP microdomains is formed by spin-coating and solvent annealing. Afterwards, the P4VP microdomains are functionalized with diene functions and selectively cross-linked through quaternisation of the pyridine functions of the P4VP. The very high affinity of TAD toward diene is then used to efficiently graft, without addition of any catalyst and at ambient conditions, a TAD terminated homopolymer onto those functionalised microdomains. Finally, well-defined and asymmetric Janus nanoparticles are recovered by dissolution of the thin film since the microdomains are cross-linked and the grafting only occurs on top of them.
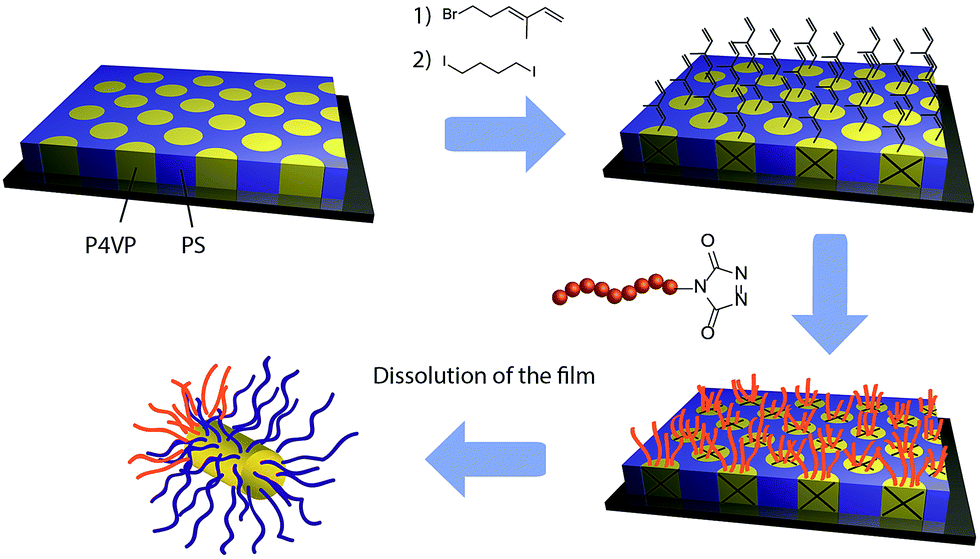 | ||
| Fig. 2 Schematic representation of the strategy to produce polymeric Janus nanoparticles via the grafting of a homopolymer onto the top of previously cross-linked microdomains of a nanostructured block copolymer thin film using TAD chemistry. | ||
Materials and methods
Materials and instrumentation
Poly(styrene)-block-poly(4-vinylpyridine) (PS-b-P4VP, number average molecular weight MPSn = 57.5 kg mol−1, MP4VPn = 18.5 kg mol−1, Đ = 1.15) and a poly(4-vinylpyridine) homopolymer (P4VP, Mn = 18.1 kg mol−1, Đ = 1.11) were purchased from Polymer Source. 6-Bromo-3-methylhexa-1,3-diene was synthesized following a two-step procedure adapted from the literature (see ESI for details†).33 All other chemicals were purchased from Acros or Aldrich and were of highest purity grade. All chemicals were used as received unless otherwise specified. Thick films of PS-b-P4VP, made by pouring 1 mL of a 20 g L−1 solution in CHCl3 into a small Petri dish and allowing the solvent to evaporate at room temperature, were used to estimate by FTIR the degree of cross-linking and functionalisation of P4VP in thin PS-b-P4VP films. FTIR spectra were recorded with a Shimadzu FTIR-8400S spectrometer from films deposited onto a ZnS crystal plate. Atomic force microscopy (AFM) was performed with a Digital Instruments Nanoscope V scanning force microscope in tapping mode using NCL cantilevers (Si, 48 N m−1, 330 kHz, Nanosensors). To determine the overall thickness of the thin films, the film was scratched with a razor blade and imaged with AFM. Water contact angle measurements were performed with an OCA-20 apparatus (Data-physics Instruments GmbH) in the sessile drop configuration. The results are the average of three values, each obtained with a droplet volume of 6 μL. The XPS analyses were performed on a SSX 100/206 photoelectron spectrometer from Surface Science Instruments (USA) equipped with a monochromatised micro focused Al X-ray source (powered at 20 mA and 10 kV). Samples were stuck onto sample holders with a double-face adhesive tape and then placed on an insulating homemade ceramic carousel (Macor, Switzerland). The spot size for irradiation was set at 800 μm × 1200 μm. The pressure in the analysis chamber was around 10−6 Pa. The angle between the surface normal and the axis of the analyser lens was 55°. The analysed area was approximately 1.4 mm2 and the pass energy was set at 150 eV. A flood gun set at 8 eV and a Ni grid placed 1 mm above the sample surface were used for charge stabilization. The C–(C,H) component of the C 1s peak of carbon has been fixed to 284.8 eV to set the binding energy scale. Data treatment was performed with the CasaXPS program (Casa Software Ltd, UK). Transmission electron microscopy (TEM) was performed on a LEO 922 microscope, operating at 120 kV accelerating voltage in bright field mode. Samples for TEM experiments were prepared by drop-casting a solution containing the particles onto TEM grid (carbon–copper 200 mesh). The grids were then dried in vacuum for 15 h. Dynamic light scattering (DLS) experiments were performed on a Malvern CGS-3 apparatus equipped with a He–Ne laser with a wavelength of 632.8 nm. The measurements were performed at a 90° angle and each sample was measured at least 5 times in order to check the reproducibility. The results were analysed using the CONTIN algorithm, a method based on a constraint inverse Laplace transformation of the data and which gives access to a size distribution histogram for the objects present in solution. The Stokes–Einstein approximation was used to convert diffusion coefficients into apparent hydrodynamic radii. The polydispersity index (PDI) of the micelles was estimated from the Γ2/Γ1 ratio in which Γ1 and Γ2 represent the first and second order moments calculated from the Cumulants method. Before measurements, the solutions were filtered (0.45 μm PTFE syringe filter) to remove dust.PS-b-P4VP thin film preparation
Silicon substrates were cleaned by a piranha solution (H2SO4 98%/H2O2 30% 3/1 v/v) for 30 min and thereafter rinsed several times using ultra-pure water. The substrates were dried with the spin-coater at a velocity of 4000 rpm for 20 s. Filtered solutions (0.45 μm) of polymer in chloroform were then spin-coated onto these substrates at 2000 rpm during 40 s. The thickness of the films was controlled by the solution concentration. The films were annealed in 1,4-dioxane vapours for 12 h at room temperature in a sealed desiccator containing 5 mL of 1,4-dioxane.Diene functionalisation of the PS-b-P4VP films
The functionalization of the P4VP domains with the diene was carried out at room temperature in a desiccator evacuated at 50 mbar containing the PS-b-P4VP films and 100 μL of 6-bromo-3-methylhexa-1,3-diene. After the desired reaction time, films were removed and dried under vacuum.Selective cross-linking of the PS-b-P4VP films
The chemical cross-linking of the P4VP domains was carried out at room temperature in a desiccator evacuated at 50 mbar containing the alkyne functionalized PS-b-P4VP films and 100 μL of 1,4-diiodobutane. After 24 h the films were removed and dried under vacuum.Synthesis of PMA–TAD
The synthesis of the ATRP-(1,2,4-triazoline-3,5-dione) initiator was carried out in four steps following a procedure reported by Du Prez et al.23 3 mL of methyl acrylate (MA, 36.68 mmol, 222 eq.) and 56.4 mg (1 eq.) of the initiator were added into a 5 mL flask and degassed with argon for one hour. In a separate flask, 5.2 mg of Cu(0) (0.5 eq.), 19.1 mg of tris[2-(dimethylamino)ethyl]amine (Me6TREN) (0.5 eq.) and 1.5 mL of methanol were degassed separately for 1 hour. The second flask was added to the first flask and the mixed flask was placed in an oil bath at 25 °C during 2.5 hours. The reaction was stopped by cooling with liquid nitrogen and the copper catalyst was removed by passing the reaction mixture over an Al2O3 column. The accordingly obtained end-functionalized poly(methacrylate) (PMA) was purified by precipitation in cold methanol and dried in a vacuum oven overnight. Oxidation of the TAD groups was carried out by reacting 100 mg of PMA with 3.4 mg of 1,4-diazabicyclo[2.2.2]octane bromine complex (DABCO-Br) in 1 mL of dichloromethane. After 3 hours, the DABCO-Br was filtered and the solvent evaporated under reduced pressure to recover the oxidized PMA–TAD.Grafting of PMA–TAD onto P4VP in solution
100 mg of PMA–TAD was dissolved in 0.5 mL of dimethylsulfoxide (DMSO). Diene-functionalized P4VP was dissolved in 0.5 mL of DMSO and the two solutions were mixed together.Grafting of PMA–TAD onto PS-b-P4VP thin films
The functionalized and cross-linked PS-b-P4VP thin films were placed in a solution of PMA–TAD in DMSO (100 mg mL−1). After the desired reaction time, the films were removed from the solution and carefully washed with neat DMSO and methanol.Dissolution of the PMA-grafted PS-b-P4VP thin films
The PMA-grafted PS-b-P4VP films were immersed into 1 mL of a good solvent of the PS matrix (chloroform, toluene or dimethylformamide) for 12 h in order to dissolve them completely and retrieve the nanoparticles. The solution containing the nanoparticles was filtered with a 1 μm poly(tetrafluoroethylene) syringe filter before characterization.Results and discussion
A relatively high molecular mass (76 kg mol−1) PS-b-P4VP block copolymer with a volume fraction of P4VP of 0.24 was used to produce the initial nanostructured thin film. A PS-b-P4VP copolymer was selected because the self-assembly of those type of copolymers is well known and well-controlled and because the presence of aromatic amines in the P4VP block allows for easy functionalization with a wide variety of molecules.34,35 Fig. 3 shows the atomic force microscopy (AFM) image of the film after spin-coating on a silicon wafer and annealing in vapours of 1,4-dioxane for 12 h, revealing the presence of relatively well ordered dot-like microdomains. As the thickness of the film (30 nm) lies in the same range as the size of the microdomains, the morphology of the film can be seen as P4VP dots that are regularly distributed in a PS matrix, as previously observed in the literature (Fig. 4).34,35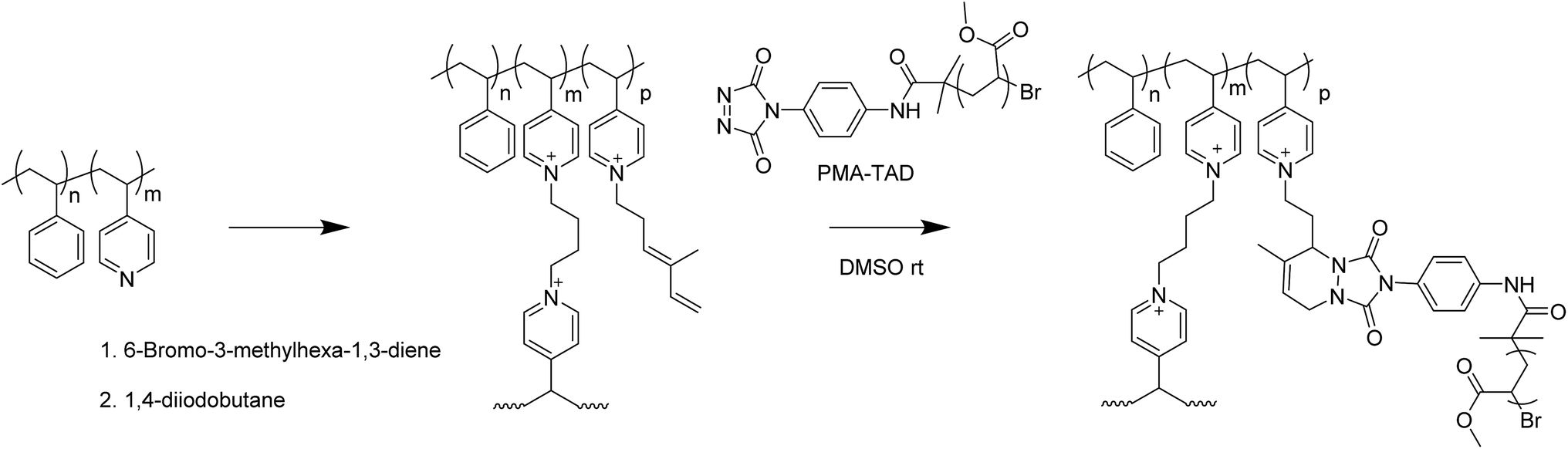 | ||
| Fig. 3 PS-b-P4VP functionalization by diene groups, cross-linking with a diiodo derivative and grafting of a TAD-terminated. | ||
 | ||
| Fig. 4 AFM images height and phase of PS-b-P4VP spin coated on a silicon wafer after solvent vapour annealing with 1,4-dioxane vapours for 12 h. | ||
As shown in Fig. 3, functionalization of the P4VP microdomains with a diene group, which further enables to react with a TAD-polymer is achieved by quaternisation of the pyridine moieties in the presence of 6-bromo-3-methylhexa-1,3-diene vapours. The functionalization of P4VP is monitored by FTIR spectroscopy of a PS-b-P4VP film, having a thickness of several micrometers and functionalized in the same conditions as for the spin-coated films. As shown in Fig. 5, the FTIR spectrum of PS-b-P4VP after exposition to vapours of 6-bromo-3-methylhexa-1,3-diene presents a peak at 1640 cm−1 that is characteristic for the vibration of quaternised pyridine rings.15–18
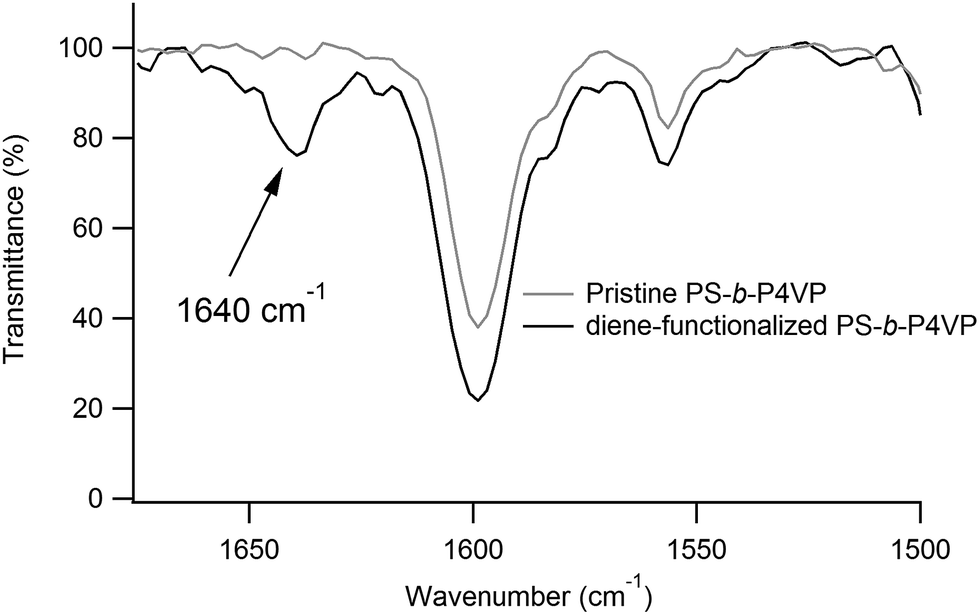 | ||
| Fig. 5 FTIR spectrum of pristine PS-b-P4VP and diene-functionalized PS-b-P4VP films. | ||
The degree of functionalization can be easily tuned by adjusting the reaction time as shown in Fig. 6. Since unreacted pyridine groups are required for the cross-linking step, the exposure time was fixed to 18 h in order to obtain a degree of quaternisation of around 35%.
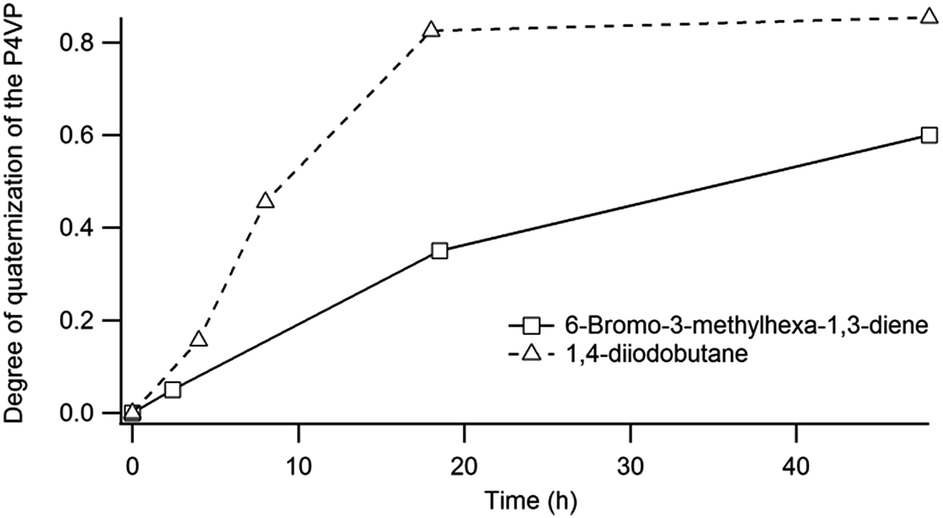 | ||
| Fig. 6 Evolution of the degree of quaternisation of P4VP by 6-bromo-3-methylhexa-1,3-diene and 1,4-diiodobutane as determined by FTIR. | ||
The unreacted pyridine units are then reacted with 1,4-diiodobutane in order to selectively chemically cross-link the P4VP microdomains (Fig. 3). As previously mentioned for the functionalization, the reaction is followed by FTIR spectroscopy and the extent of cross-linking can be adjusted by varying the time of exposure of the PS-b-P4VP films to the 1,4-diiodobutane vapours.
For the synthesis of PMA–TAD to be further grafted on the P4VP microdomains, PMA-Urazole (Ur) is first prepared via ATRP polymerization of MA using an initiator bearing an urazole group. After 2.5 h of polymerization, the Cu catalyst is removed and pure PMA-Ur is obtained after precipitation in cold methanol. The molar mass (Mn) of the recovered polymer is 17.6 kg mol−1, as determined by 1H NMR (Fig. S2b†) and Đ = 1.4 (Fig. S2a†). The Ur moieties are oxidized in the presence of DABCO-Br to obtain the reactive TAD functionalities, just before the grafting step, to ensure their stability. To understand the efficiency of the reaction between diene functionalized P4VP and PMA–TAD, the grafting is initially performed in solution. To achieve this, a homopolymer of P4VP, functionalized with 6-bromo-3-methylhexa-1,3-diene is dissolved in a DMSO solution containing the PMA–TAD. As expected, the reaction occurs within a few minutes as indicated by the change of colour from red (corresponding to the colour of an oxidised PMA–TAD solution) to slightly yellow. The efficiency of the coupling is further confirmed by SEC analysis that shows a clear shift of the P4VP-signal towards higher molar mass and the decrease of the PMA–TAD peak (Fig. 7). However, significant traces of PMA polymers are still noticed in the coupled polymer, which revealed a non-complete grafting under those reaction conditions. The steric hindrance caused by the already grafted PMA chains could explain why it is impossible to functionalize all the 4VP groups. Nevertheless, one should point out that this model compound is actually a graft P4VP-g-PMA copolymer. Such types of copolymers have been scarcely reported in the scientific literature, essentially using the macromonomer approach.36
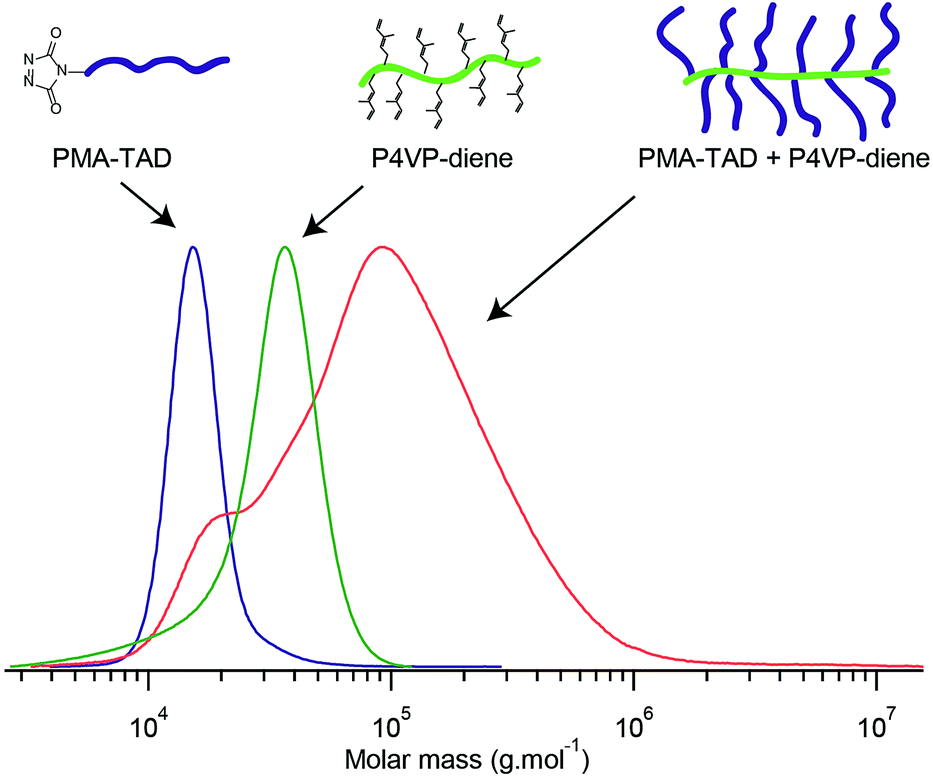 | ||
| Fig. 7 SEC curves of PMA–TAD, diene-functionalized P4VP and PMA-g-P4VP after 1 h of reaction. | ||
The grafting of PMA–TAD onto diene-functionalized PS-b-P4VP thin films has been carried out by immersing the films into a DMSO solution containing the PMA–TAD. DMSO was selected to avoid the dissolution of the film since it is a non-solvent of the PS matrix and the P4VP domains are chemically cross-linked and thus only swell in this solvent. After the desired elapsed time, the PS-b-P4VP films are removed from the solution and characterized by AFM, water contact angle measurements and XPS. AFM shows a distinct change in the film surface morphology (Fig. 8a and b).
 | ||
| Fig. 8 AFM height (A) and phase (B) pictures of a diene-functionalized PS-b-P4VP thin film after grafting of PMA–TAD. (C) Static water contact angles of diene-functionalized PS-b-P4VP and PMA-grafted PS-b-P4VP thin films. | ||
Such a change is not caused by the exposure of the film to the solvent as no changes in the morphology are observed when the films are immersed in pure DMSO or in the presence of non-functionalised polymer. The modification can thus be ascribed to the grafting of PMA chains onto the P4VP domains.
A change in the wetting properties of the films after grafting of PMA–TAD is also observed by static water contact angle measurements as illustrated in Fig. 8c. Indeed, the water contact angle shifts from 87° for the diene-functionalized PS-b-P4VP films to 64° after the grafting of PMA–TAD, which is in agreement with the values of pure PMA reported in the literature.37
The presence of PMA onto the film after the grafting step is further confirmed by XPS analysis that shows a significant increase (from 0.09 to 0.23) in the O/C ratio after grafting and the presence of oxidized carbon in the C 1s peak at 289 eV arising from the carbonyl functions of the PMA (Fig. 9).
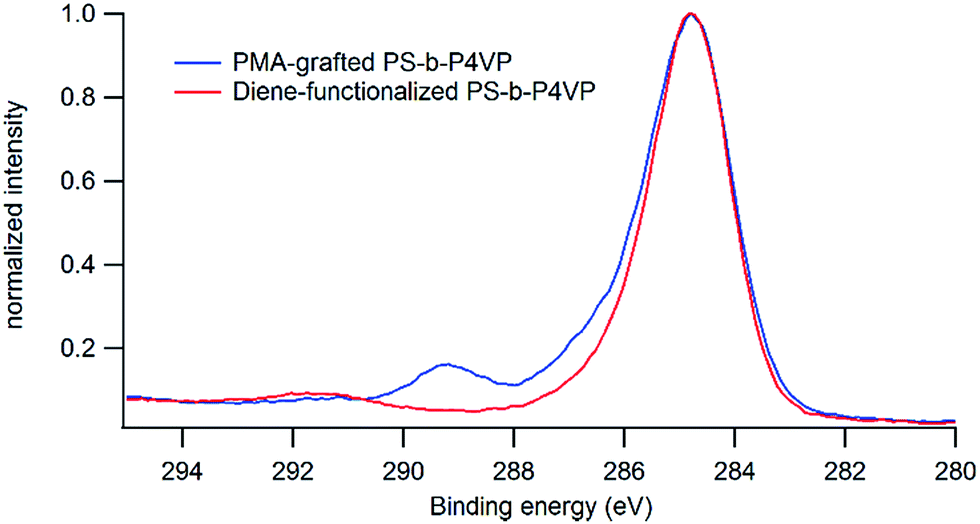 | ||
| Fig. 9 XPS spectra of the C 1s area of PS-b-P4VP thin films before and after grafting of PMA–TAD. | ||
In order to recover the Janus particles, the PMA-grafted PS-b-P4VP films are dissolved in DMF, a good solvent of PS and PMA, and the resulting solution is analysed by DLS and TEM. DLS analysis reveals the presence of well-defined (PDI = 0.1) objects in solution with an average apparent hydrodynamic radius of 90 nm (Fig. 10).
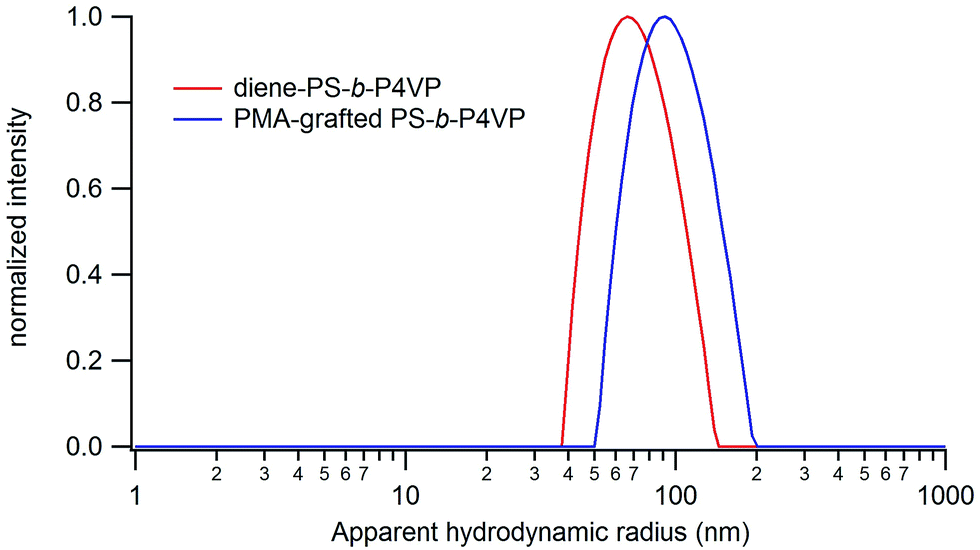 | ||
| Fig. 10 Size distributions of the particles obtained from PMA-grafted PS-b-P4VP and non-grafted PS-b-P4VP films in DMF solution. | ||
A first evidence of the Janus structure of the accordingly obtained nanoparticles is obtained by adding DMSO, a non-solvent of PS, to the particles in solution in DMF. The aggregation of the Janus nanoparticles into larger structures with an apparent hydrodynamic radius of 270 nm is then observed due to the insolubilisation of the PS coronal chains. The asymmetric architecture of the obtained nanoparticles is further demonstrated by TEM images. Indeed, a less contrasted area, compatible with PMA, is visibly attached on only one side of the cross-linked P4VP core, which appears dark as a result of the presence of iodine and bromine atoms in the structure (Fig. 11). In comparison, no asymmetric structure is observed in TEM pictures of nanoparticles obtained from non-grafted PS-b-P4VP films. TEM images are recorded on dried nanoparticles, which explains why the characteristic sizes of the objects imaged by TEM are significantly smaller than the hydrodynamic sizes measured by DLS for which nanoparticles with solvated PS and PMA chains are measured.
 | ||
| Fig. 11 TEM pictures of nanoparticles obtained after the dissolution of PMA-grafted PS-b-P4VP thin films in DMF. The nanoparticles were stained with RuO4 before imaging. | ||
Conclusions
In summary, we have demonstrated that TAD-based click chemistry can be used to efficiently graft a TAD terminated polymer onto diene functionalized and selectively cross-linked microdomains of a nanostructured block copolymer thin film. The dissolution of the whole assembly led to the formation of nanoparticles presenting a narrow size distribution and displaying an asymmetric structure with a cross-linked core of P4VP with an asymmetric shell of PS and PMA. Compared to other approaches based on the use of triblock copolymers, our approach allows an easy tuning of the chemical nature of the grafted third block and guarantees the formation of the asymmetric structure. As main drawback, our strategy produces a limited amount of Janus particles. The use of additive free TAD-based click chemistry allowed us to avoid catalyst addition to perform the grafting step. Moreover, the reaction between TAD and diene is known to proceed faster, which could be useful to reduce the preparation time of the Janus particles. Finally, this paper confirms the versatility of TAD chemistry, which can be used either in solution to prepare graft copolymers or at the surface of a nanostructured polymer film to produce well-defined Janus nanoparticles.Acknowledgements
E. P. is grateful to FRIA for financial support. F. D. P. and J. F. G. acknowledge the Belgian Program on Interuniversity Attraction Poles initiated by the Belgian State, the Prime Minister's office (P7/05).Notes and references
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- Y. Song and S. Chen, Langmuir, 2014, 30, 6389–6397 CrossRef CAS PubMed.
- S. Berger, A. Synytska, L. Ionov, K.-J. Eichhorn and M. Stamm, Macromolecules, 2008, 41, 9669–9676 CrossRef CAS.
- Y. Wu, X. Lin, Z. Wu, H. Möhwald and Q. He, ACS Appl. Mater. Interfaces, 2014, 6, 10476–10481 CAS.
- W. Gao, X. Feng, A. Pei, Y. Gu, J. Li and J. Wang, Nanoscale, 2013, 5, 4696–4700 RSC.
- L. Y. Wu, B. M. Ross, S. Hong and L. P. Lee, Small, 2010, 6, 503–507 CrossRef CAS PubMed.
- F. Wang, G. M. Pauletti, J. Wang, J. Zhang, R. C. Ewing, Y. Wang and D. Shi, Adv. Mater., 2013, 25, 3485–3489 CrossRef CAS PubMed.
- A. Walther, M. Hoffmann and A. H. E. Müller, Angew. Chem., Int. Ed., 2008, 47, 711–714 CrossRef CAS PubMed.
- R. Bahrami, T. I. Löbling, A. H. Gröschel, H. Schmalz, A. H. E. Müller and V. Altstädt, ACS Nano, 2014, 8, 10048–10056 CrossRef CAS PubMed.
- F. Tu and D. Lee, J. Am. Chem. Soc., 2014, 136, 9999–10006 CrossRef CAS PubMed.
- N. Glaser, D. J. Adams, A. Böker and G. Krausch, Langmuir, 2006, 22, 5227–5229 CrossRef CAS PubMed.
- J. Faria, M. P. Ruiz and D. E. Resasco, Adv. Synth. Catal., 2010, 352, 2359–2364 CrossRef CAS.
- A. Kirillova, C. Schliebe, G. Stoychev, A. Jakob, H. Lang and A. Synytska, ACS Appl. Mater. Interfaces, 2015, 7, 21218–21225 CAS.
- R. Deng, F. Liang, J. Zhu and Z. Yang, Mater. Chem. Front., 2017, 1, 431–443 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. H. Groschel and A. H. E. Muller, Nanoscale, 2015, 7, 11841–11876 RSC.
- A. Walther, X. André, M. Drechsler, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 6187–6198 CrossRef CAS PubMed.
- R. Erhardt, A. Böker and H. Zettl, Macromolecules, 2001, 1069–1075 CrossRef CAS.
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed.
- A. H. Gröschel, A. Walther, T. I. Löbling, J. Schmelz, A. Hanisch, H. Schmalz and A. H. E. Müller, J. Am. Chem. Soc., 2012, 134, 13850–13860 CrossRef PubMed.
- E. Poggi, J.-P. Bourgeois, B. Ernould and J.-F. Gohy, RSC Adv., 2015, 5, 44218–44221 RSC.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed.
- S. Chattopadhyay and F. Du Prez, Eur. Polym. J., 2016, 81, 77–85 CrossRef CAS.
- P. Wilke, T. Kunde, S. Chattopadhyay, N. ten Brummelhuis, F. E. Du Prez and H. G. Borner, Chem. Commun., 2017, 53, 593–596 RSC.
- B. Vonhören, O. Roling, K. De Bruycker, R. Calvo, F. E. Du Prez and B. J. Ravoo, ACS Macro Lett., 2015, 4, 331–334 CrossRef.
- S. van der Heijden, K. De Bruycker, R. Simal, F. Du Prez and K. De Clerck, Macromolecules, 2015, 48, 6474–6481 CrossRef CAS.
- Z. Wang, L. Yuan, N. M. Trenor, L. Vlaminck, S. Billiet, A. Sarkar, F. E. Du Prez, M. Stefik and C. Tang, Green Chem., 2015, 17, 3806–3818 RSC.
- S. van der Heijden, L. Daelemans, K. De Bruycker, R. Simal, I. De Baere, W. Van Paepegem, H. Rahier and K. De Clerck, Compos. Struct., 2017, 159, 12–20 CrossRef.
- S. Vandewalle, S. Billiet, F. Driessen and F. E. Du Prez, ACS Macro Lett., 2016, 5, 766–771 CrossRef CAS.
- L. Vlaminck, K. De Bruycker, O. Turunc and F. E. Du Prez, Polym. Chem., 2016, 7, 5655–5663 RSC.
- I. H. Wang, G. R. Dobson and P. R. Jones, Organometallics, 1990, 9, 2510–2513 CrossRef CAS.
- B. Bharatiya, J.-M. Schumers, E. Poggi and J.-F. Gohy, Polymers, 2013, 5, 679–695 CrossRef.
- E. Bhoje Gowd, B. Nandan, M. K. Vyas, N. C. Bigall, A. Eychmüller, H. Schlörb and M. Stamm, Nanotechnology, 2009, 20, 415302 CrossRef CAS PubMed.
- K. Ishizu and K. Mitsutani, J. Polym. Sci., Part C: Polym. Lett., 1988, 26, 511–516 CrossRef CAS.
- A. M. Granville, S. G. Boyes, B. Akgun, M. D. Foster and W. J. Brittain, Macromolecules, 2004, 37, 2790–2796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07924a |
| This journal is © The Royal Society of Chemistry 2017 |