
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of persulfate by MMIOC for highly efficient degradation of rhodamine B†
Huanling Xie
The School of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing, China 400054. E-mail: xhl2007@cqut.edu.cn; Tel: +86 13647638549
First published on 25th September 2017
Abstract
Mesoporous magnetic iron oxide composites (MMIOCs) were successfully prepared using one-step evaporation induced self-assembly using an organic ferrocene surface active agent as the iron source, and innovatively studied as a potential alternative to mesoporous Co3O4 for ammonium persulfate (PS) activation to generate active radicals. MMIOCs were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), N2 adsorption/desorption, transmission electron microscopy (TEM) and using a vibrating sample magnetometer (VSM). Among the different FexOy/PS systems tested, the MMIOC displayed the highest catalytic activity for PS activation, achieving almost complete degradation of 1 mM rhodamine B (RhB) within 120 minutes at 0.2 g L−1 catalyst and 40.0 mM PS. The superior catalytic performance of the MMIOC was ascribed to the combination of iron ions inlayed or embedded in the inner or outer surface, forming catalytically active sites and the magnetic mesoporous structure. Several factors played important roles in controlling the degradation of RhB. The equilibrium between ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe(II) and Fe(III) for PS activation in solution ensured continuous generation of the hydroxyl and sulfate radicals, and the latter made the predominant contribution for RhB degradation. Considering its outstanding catalytic activity, excellent reusability and long-term stability, MMIOC could be an ideal catalyst for the degradation of RhB. These results provide an applied solution for disposing of toxic organic wastewater in a relatively available environment, and a scientific foundation for solving refractory organic contaminant related to environmental management.
Fe(II) and Fe(III) for PS activation in solution ensured continuous generation of the hydroxyl and sulfate radicals, and the latter made the predominant contribution for RhB degradation. Considering its outstanding catalytic activity, excellent reusability and long-term stability, MMIOC could be an ideal catalyst for the degradation of RhB. These results provide an applied solution for disposing of toxic organic wastewater in a relatively available environment, and a scientific foundation for solving refractory organic contaminant related to environmental management.
1. Introduction
RhB, as a xanthene dye, is carcinogenic and may cause mutagenesis or teratogenesis.1 Hence, it is of great practical importance and socially beneficial to seek effective degradation techniques. However, certain challenges have been encountered in this research. Advanced oxidation process (AOPs) with O3,2,3 H2O2 (ref. 4) and PS,5 is used to produce free radicals for the degradation of these recalcitrant or hazardous organic pollutants to nontoxic small molecules, such as carbon dioxide, water and nitrogen. Further, a sulfate radical (SO4˙−) has become an alternative to traditional hydroxyl radicals (˙OH) and been widely studied due to their strong oxidizability, longer half-life and better selectivity.6–8At present, the activation methods of PS were diverse to generate sulfate radicals. Thermally or UV activated persulfate oxidation reactions generate sulfate radicals SO4˙−.9–12 Nevertheless, higher temperatures or UV lamps caused inconvenience to large-scale and high-chroma sewage systems.
Transition metal ions (Co2+, Cu2+, Ni2+, Fe2+, Ag+, Ru3+) are used for activation of PS to generate sulfate radicals. It has become a hotspot of research to synthesize transition metal oxides, for instance Co3O4 loaded on supports, LDH,13 graphitic carbon nitride14 and ordered mesoporous frameworks.5
Magnetic mesoporous composites (MMC) could be prepared using single-step or multi-step method. First, with inorganic ferric iron precursor, MMC is in situ synthesized by one-step hydrothermal method.15 Second, mesoporous materials must be prepared to produce mesoporous structure. Then, mesoporous materials are integrated with transition metal oxides via various pathways, such as sol–gel,16–18 solvothermal treatment19,20 and mixed modification.21 Third, Tao group have reported one-pot hydrothermal preparation for MMC with long chain containing hydrophobic ferrocene terminal group as both structure-directing agent and magnetic source.22,23 However, for example, Co contents generally leak from the catalysts to the solution, leading to heavy metal pollution and thus require additional recycling operation costs.1,24–26
To overcome the above mentioned disadvantages, herein, we selected iron ions, as active component. An improved strategy was reported for the one-step evaporation induced self-assembly synthesis for MMIOC. The key point of this strategy is that iron ions could be effectively scattered and inlaid or embedded in inner or outer surface of magnetic mesoporous materials, synthesized with ferrocene surfactant as structure-directing agents, which were applied as heterogeneous activator to degrade the RhB in the presence of PS. The objectives of the present study were to evaluate the degradation performance of MMIOC. Moreover, considerable efforts have been devoted to the synthesis of MMIOC to be free of active component leakage, and to be reused several times. Hence, herein, typical generated radicals in different MMIOC/PS systems are identified, and a reasonable RhB degradation mechanism is proposed.
2. Experimental
2.1 Reagents and materials
N,N-Dimethylaminomethylferrocene (C13H17FeN), triblock copolymer P123 (EO20PO70EO20) and F127 (EO106PO70EO106) were purchased from J&K Chemical Reagent Co. Ltd. 1-Bromohexadecane (C16H33Br), tetraethoxysilane (TEOS), ethanol, potassium chloride, ammonium ferrous sulfate, 1,3,5-trimethylbenzene (C9H12), hydrazine hydrate (N2H4 H2O), ferric nitrate (Fe(NO3)3·9H2O), ammonia solution (28%), ethanol, ammonium persulfate (PS) and RhB of analytical grades were purchased from Southwest Chemical Reagent Co. Ltd., China. All reagents were used without further purification.2.2 Preparation and characterization
Yield: 60%; bright yellow crystal.
1H NMR (MeOD): 4.518–4.441 (t, 5H), 4.299 (t, 2H), 4.299 (d, 2H), 3.35 (d, 2H), 3.215–3.181 (m, 2H), 2.955 (s, 6H), 1.825–1.81 (m, 2H), 1.397–1.321 (m, 26H), 0.934–0.907 (m, 2H).
Elemental analysis calcd (%) for C29H50FeBrN: C 63.52, H 9.13, N 2.56.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.13FcC16Br−N+:0.67KCl:33.3CH3CH2OH. Further, the reaction mixture was stirred at room temperature for 1 h, and then transferred into an autoclave and left undisturbed at 180 °C for 12 h. After hydrothermal treatment, the mixtures were evaporated and dried in a well ventilated space at room temperature. The samples were placed in a quartz boat and inserted into the quartz tube. Under a steady flow rate of O2, the temperature of the furnace was raised to 300 °C and maintained at this temperature for 6 h. Subsequently, the furnace was slowly cooled down to room temperature, and then MMIOC were obtained. This sample was named as MMS.:Si as 8/2 and 7/3. Hence, the samples were named as P8/2 and P7/3 with P123, while they were named as F8/2 and F7/3 with F127.000 Oe and at room temperature. X-ray photoelectron spectroscopy (XPS) measurements were performed on a ThermoFisher ESCALAB™ 250Xi System with Al Kα source. All binding energies were referenced to the C 1s peak at 284.6 eV of surface adventitious carbon. Electron paramagnetic resonance (EPR spectrometer, JES-FA200) was performed using 5,5-dimethyl-1-pyrrolidine-N-oxide (DMPO) as a spin-trapping agent. Metal content in the solution was determined by atomic absorption spectrometer (A8800) from American PE.
:0.13FcC16Br−N+:0.67KCl:33.3CH3CH2OH. Further, the reaction mixture was stirred at room temperature for 1 h, and then transferred into an autoclave and left undisturbed at 180 °C for 12 h. After hydrothermal treatment, the mixtures were evaporated and dried in a well ventilated space at room temperature. The samples were placed in a quartz boat and inserted into the quartz tube. Under a steady flow rate of O2, the temperature of the furnace was raised to 300 °C and maintained at this temperature for 6 h. Subsequently, the furnace was slowly cooled down to room temperature, and then MMIOC were obtained. This sample was named as MMS.:Si as 8/2 and 7/3. Hence, the samples were named as P8/2 and P7/3 with P123, while they were named as F8/2 and F7/3 with F127.000 Oe and at room temperature. X-ray photoelectron spectroscopy (XPS) measurements were performed on a ThermoFisher ESCALAB™ 250Xi System with Al Kα source. All binding energies were referenced to the C 1s peak at 284.6 eV of surface adventitious carbon. Electron paramagnetic resonance (EPR spectrometer, JES-FA200) was performed using 5,5-dimethyl-1-pyrrolidine-N-oxide (DMPO) as a spin-trapping agent. Metal content in the solution was determined by atomic absorption spectrometer (A8800) from American PE.2.3 Experimental procedures
Catalytic degradation experiments were performed in a series of 250 mL conical flasks on a controlled temperature shaking table. Typically, 2 g L−1 catalytic agent was added into 100 mL RhB (1 mM) stock solution and shaken to achieve uniform mixing. Through pre-adsorption experiment, catalytic experiment was initiated following the introduction of 0.9128 g PS. The initial pH of the solution was adjusted using 2 M HCl or NaOH. At a given time interval, a certain amount of sample was absorbed with methanol or TBA to quench primary radical species. The catalyst was separated from reaction fluid by centrifugation. The concentration of RhB was analyzed using TU-1901 automatic scanning UV-Vis spectrophotometer (Shanghai Analytical instruments Co. Ltd.) with a spectrometric quartz cell (1 cm). The value of COD was determined using spectrophotometric method. Degradation mechanism of RhB was analysed using an Agilent 7890A-5975 GC-MS analyzer. TOC data was acquired from a SHIMADZU TOC-V-CPH analyzer. All test performances were repeated at least three times to ensure smoothing error and correction data accuracy. A plot of C vs. t was obtained, where C0 was the initial concentration of RhB and C was the concentration at time t; decolorization efficiency was obtained based on (C0 − C)/C0.3. Results and discussion
3.1 Fabrication and characterization of MMIOC
The overall structure was designed as follows: given surfactant containing ferrocene as both template agent and iron precursors was used to devise desired MMIOC. During the formation of MMIOC, the hydrophobic ferrocene tails were closely packed in the center of the micelles, which were used as structure-directing agents for mesoporous channels. The iron resources were oxidized when ferrocene surfactant was removed by calcination in O2 atmosphere. Following this, highly dispersed iron ions were embedded in the channels. Clearly, this one-step self-assembly and oxidation process provided a simple and convenient strategy for preparing MMIOC.As shown in Fig. 1(a), the addition of ferrocene surfactant as template agent provokes one strong and two very weak diffraction peaks, which indicates that ferrocene terminal groups could penetrate into the hydrophobic core of surfactant micelle. Such a ferrocene terminal group leads to the micelle deformation into sphericity, which eventually assembles to form porous materials. The absence of diffraction peak would be ascribed to its non-porous structure.5 However, by low-angle XRD measurements, the diffractogram might be attributed to honeycomb mesoporous structure. Wide-angle X-ray diffraction (XRD) analyses were performed to corroborate the presence of metal oxides deposited on the support and its poor crystallinity. As shown in Fig. 1(b), diffraction signals at 2θ equal to 33.2, 35.7, 49.6, 54.2 and 62.6° could be assigned to magnetic α-Fe2O3 crystallites (JCPDS #84-0306).29
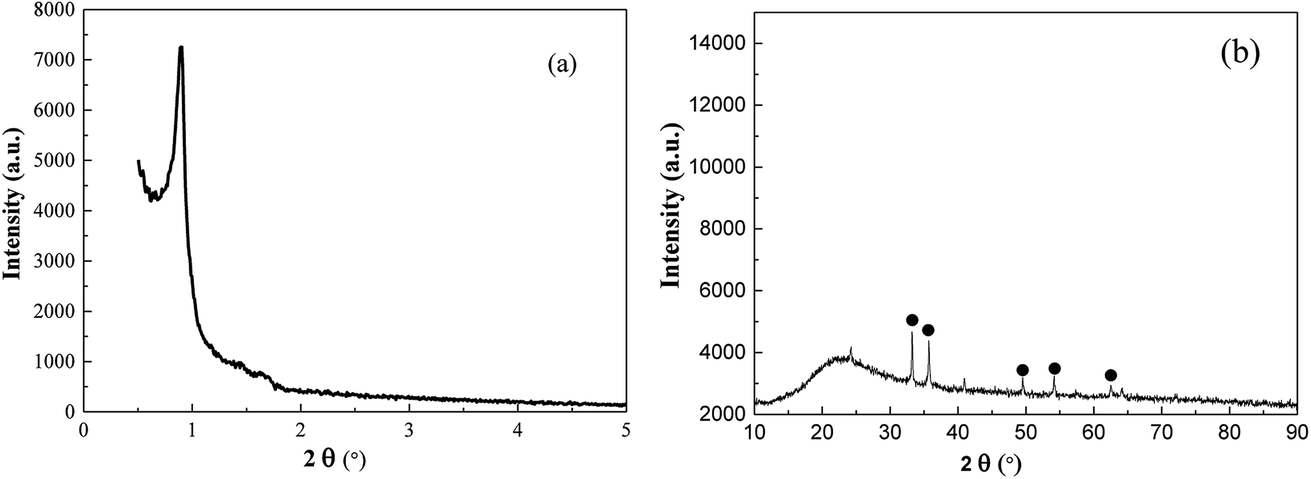 | ||
| Fig. 1 Small-angle (a) and wide-angle (b) XRD patterns for MMS. | ||
Transmission electron micrographs for MMS are shown in Fig. 2. Clearly, magnetic iron oxide was embedded in the mesoporous channel (Fig. 2(b)). Honeycomb mesoporous structure with pore diameter of about 2.0 nm displays local and less long-range order, in accordance with small-angle XRD patterns (Fig. 2(a)).30 Field dependence hysteresis loop for MMS at 293 K is shown in Fig. 3S.† The measured coercive force, Hc, was 316.23 Oe, the magnetization saturation value Ms was 0.8046 emu g−1 and the remanence was 0.2666 emu g−1. Hysteresis behavior is a consequence of surface spin disorder, showing soft ferromagnetic characteristics that are different from the reported antiferromagnetic characteristics.31
 | ||
| Fig. 2 TEM images of MMIOC for MMS. | ||
Further, the mesoporous structure was confirmed by nitrogen adsorption–desorption. It could be observed that MMS displayed Langmuir type IV isotherms (Fig. 3(a)), corresponding to typical mesoporous materials. The isotherms were clearly different from those of H1 hysteresis loop. Notably, there is still a large step observed at a relative pressure of 0.4–0.95, which is in agreement with characteristic results of TEM images, indicating the presence of honeycomb mesopores.32 However, type H1 hysteresis has been usually observed on solids with cylindrical mesopores.33 MMS have relatively high surface area (550.1 m2 g−1) in contrast to the composite reported in previous literature.5 Furthermore, the total pore volume was 0.5077 cm3 g−1. The mesoporous size distribution exhibits a sharp peak centered at about 2.4 nm (Fig. 3(b)).
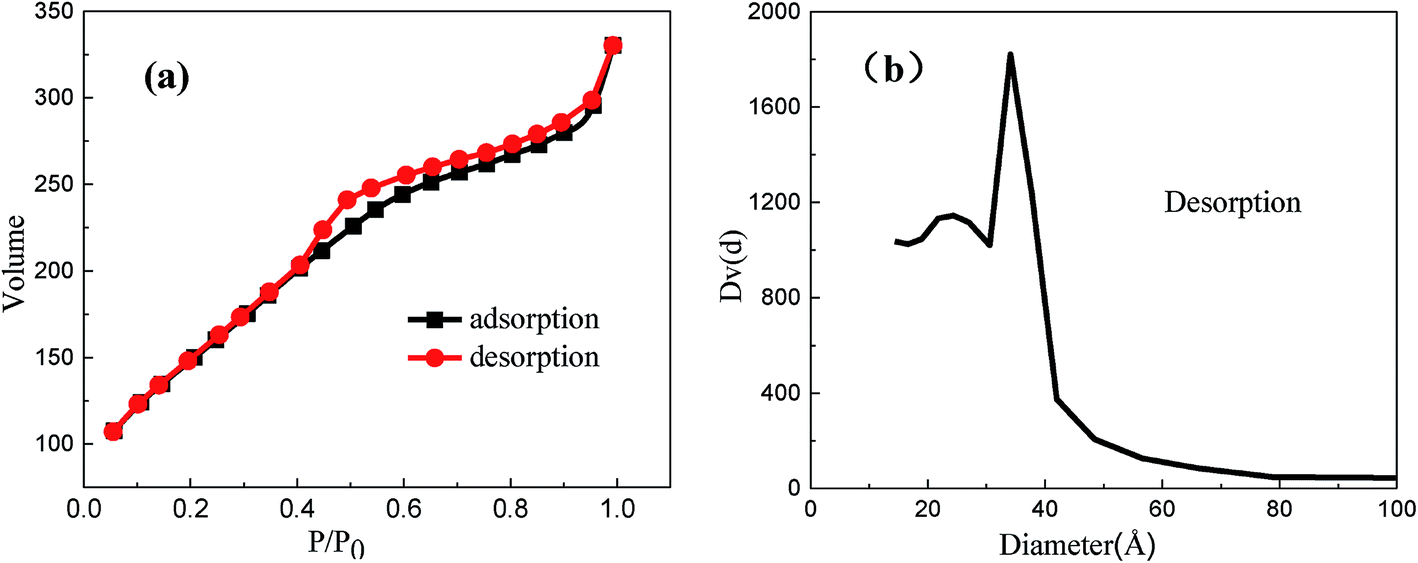 | ||
| Fig. 3 Nitrogen sorption isotherms (a) and pore size distribution (b) obtained from analysis of desorption branch of isotherms for the MMS. | ||
The MMS morphology showed no fixed shapes (Fig. 4(a)). Fig. 4(b–d) shows Fe, Si and O elements surface distribution, respectively. The EDS results shows that the atomic percentage of silicon, oxygen and iron are 16.9%, 79.9% and 3.2%, respectively (Fig. 5S†). The Fe elements map showed that iron oxides were highly dispersed in mesoporous framework in order to increase its specific surface area and improve its catalytic activity. Si and O elements were important components of the mesoporous framework.
 | ||
| Fig. 4 SEM photographs (a) element (b) Fe, (c) Si and (d) O mapping for MMS. | ||
X-ray photoelectron spectroscopy (XPS) is one of the most powerful tools to investigate surface element composition and their valence states; the XPS spectra of MMS and Fe3O4 samples are shown in Fig. 5. The full XPS spectrum clearly showed the presence of C, Si, O and Fe element on MMS sample surface, while only C, O and Fe elements were found on Fe3O4 sample surface (Fig. 5(A)). As shown in Fig. 5(B), XPS O (1s) of Fe3O4 sample envelope was deconvoluted into three parts, namely, lattice oxygen at 530.5 eV, hydroxylate oxygen at 531.8 eV and the oxygen adsorbed on H2O at 533.0 eV;34 for the spectrum O (1s) of the MMS, four single peaks with binding energies of 530.4, 531.4, 532.7 and 533.0 eV corresponded to lattice oxygen, hydroxylate oxygen, silica oxide and adsorbed water, respectively.35 Clearly, silica frameworks have been verified for MMS samples. Core-level Fe 2p spectra have evidently shown two kinds of binding energies of 711.36 and 724.68 eV ascribed to the Fe 2p3/2 and Fe 2p1/2, respectively, and the binding energy of 719.37 eV was ascribed to the satellite peak (denoted as Sat.) (Fig. 5(C)).36–38 The peak intensities of Fe 2p3/2 and Fe 2p1/2, after repeated use for 10 times, was weaker than that obtained before use, accompanied by broadening of their peak shapes. This suggested leakage of iron ions on the surface of the samples.36 However, peak intensity and broadening of Fe 2p3/2 and 2p1/2, and their satellite peaks have scarcely changed in case of Fe3O4 (Fig. 5(D)). This means that there was no change in surface element composition before and after use. Furthermore, the leakage of iron ions leads to a reduction of magnetic iron oxide blocks. In summary, the leakage of iron ions on the surface of the samples before and after use was confirmed.
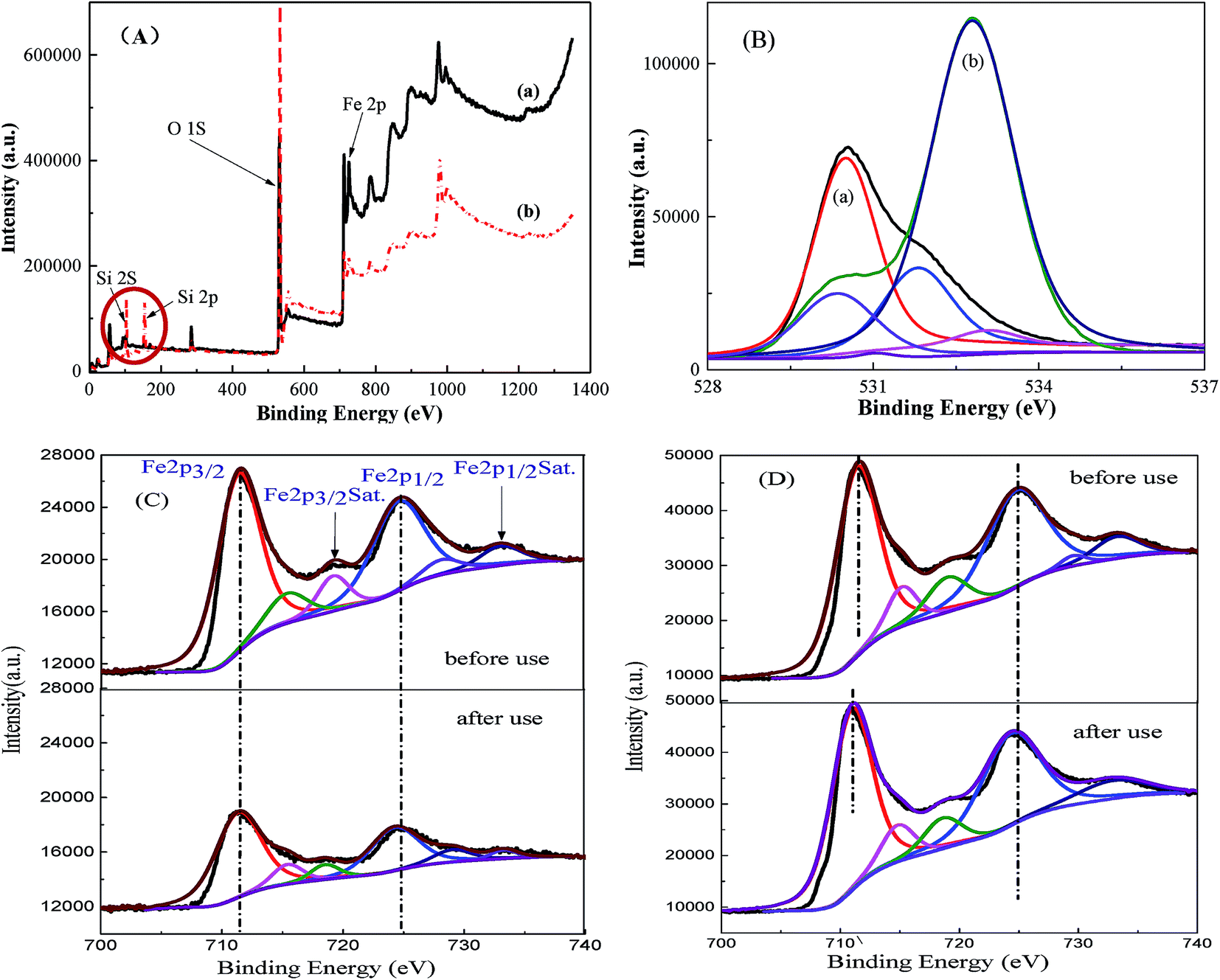 | ||
| Fig. 5 Full spectra (A) and O 1s XPS spectra (B) for (a) Fe3O4 and (b) MMS; Fe 2p XPS spectra for (C) MMS and (D) Fe3O4. | ||
Fe 2p3/2 spectra was divided into a strong peak (711.25 eV) and a small shoulder (710 eV) attributed to Fe(III) and Fe(II), respectively (Fig. 6S(A)†).39,40 The satellite signal (719.37 eV) suggested the presence of ferrous oxide. It was found that Fe(III):Fe(II) on the catalyst surface was 5.7 (Table 1S†). Owing to charge effect, the entire binding energy spectra shifted before and after use for the MMS and Fe3O4.41,42 This may be ascribed to iron ions partially dissolved during the degradation of RhB. However, these lattice positively-charged Fe groups could function as active sites for PS decomposition to generate the radicals and degrade the pollutants.
To elucidate the mechanism of PS activation on MMIOC systems of RhB degradation, in situ electron paramagnetic resonance (EPR) were performed, in which DMPO was used as spin trapping agent for SO4˙− and ˙OH radicals. Fig. 6 shows that the signals of both DMPO-SO4˙− and DMPO-˙OH have been easily detected in MMIOC/PS systems, meaning that the obtained MMIOC systems are effective for RhB degradation.
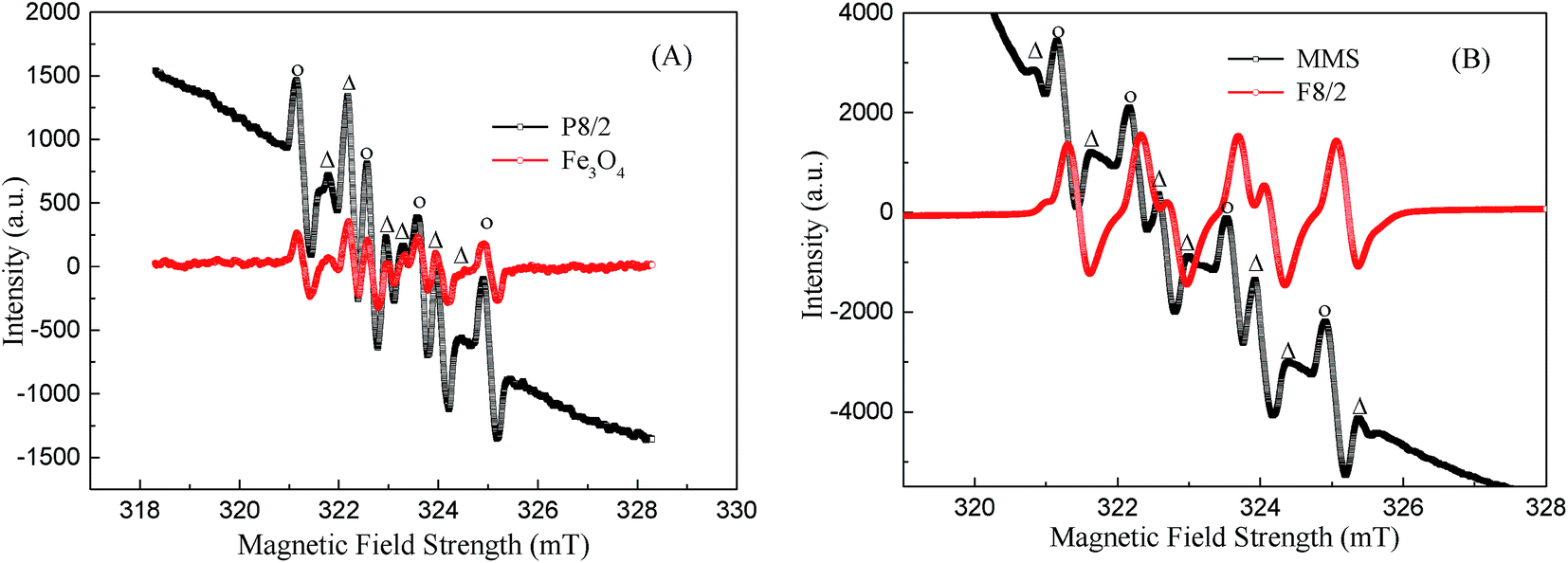 | ||
| Fig. 6 EPR spectra obtained through MMIOC systems: (A) P8/2 and Fe3O4; (B) MMS and F8/2; activated PS oxidative processes in the presence of DMPO. [PS] = 40 mM; [MMIOC or Fe3O4] = 5 g L−1; [DMPO] ≈ 0.1 M; pH = 7.0 ± 0.5; T = 25 °C. | ||
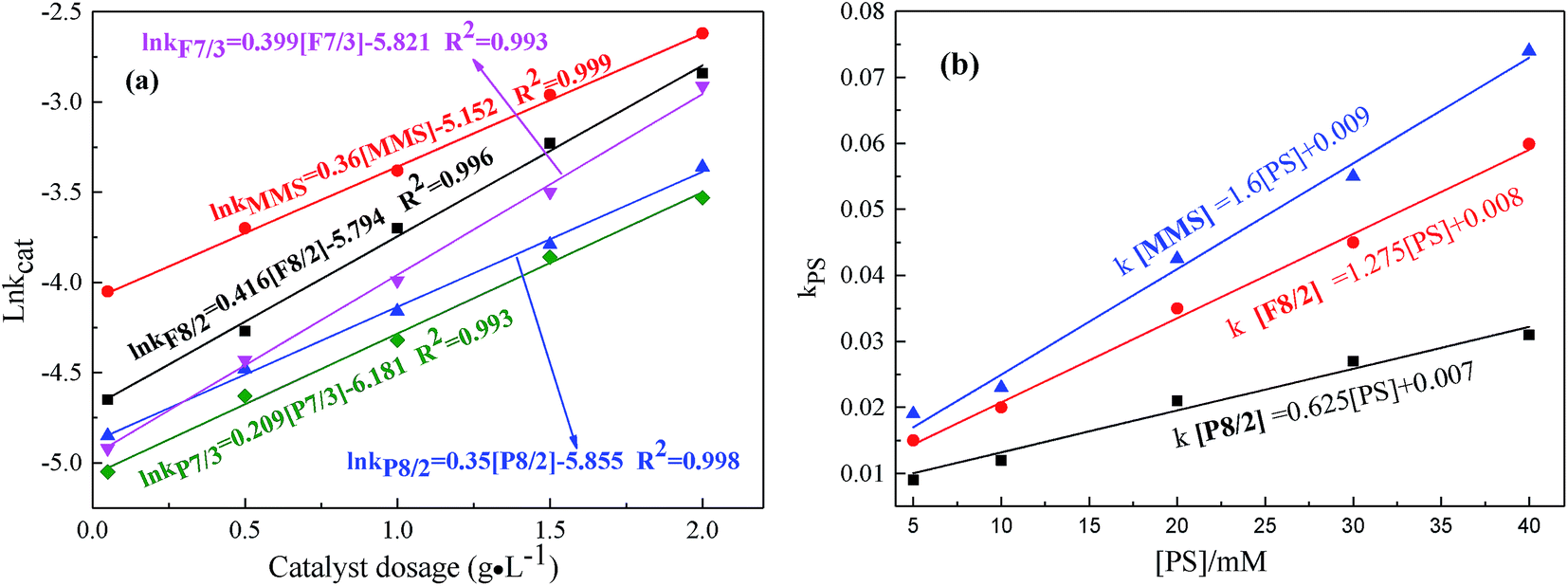 | ||
| Fig. 7 Effects of (a) catalyst dosage; (b) PS concentration on RhB degradation in different FexOy/PS systems. Expect investigated parameter, others fixed at [RhB] = 1 mM; initial pH = 7.0; T = 25 °C. | ||
The results obtained shows that MMS had excellent ability for catalytic activation of PS to generate active radicals. It has been demonstrated that PS could be activated by transition metal ions Fe2+ to generate sulfate radical (SO4˙−) (E0 = 2.6 eV). Furthermore, hydroxyl radical (˙OH−) (E0 = 2.8 eV). Fe3+ was reduced to Fe2+, to produce PS radical, as electron donor, on MMIOC surface (eqn (1)).43 However, the intensity of DMPO-OH signals was much stronger than that of DMPO-SO4 due to fast transformation from DMPO-SO4 to DMPO-OH via nucleophilic substitution (eqn (3) and (4)).44 These were the key steps in reaction system. This system could be applied in the degradation of RhB, as typical representative of dye wastewater (eqn (5)).
|
Fe(III) + S2O82− → Fe(II)+ S2O8˙−
| (1) |
|
Fe(II) + S2O82− → Fe(III)+ SO4˙− + SO42−
| (2) |
| SO4˙− + H2O → ˙OH + H+ + SO42− | (3) |
| SO4˙− + HO− → ˙OH + SO42− | (4) |
| RhB + SO4˙−/˙OH → CO2 + H2O + N2 | (5) |
As the iron component possesses a single electron in the 3d orbital, MMIOC could serve as a medium with high specific surface area and highly dispersed catalytic activity, to facilitate electron transfer from the iron to the PS to produce ˙OH and SO4˙− radicals. In contrast, bulk magnetic iron oxides presents relatively low catalytic effectiveness for PS activation,45 which could be due to partial electrons confined in crystalline structure. Thus, based on below-mentioned experimental results and detailed discussion, possible mechanism of PS activation on MMIOC systems of RhB degradation has been depicted in Scheme 1.
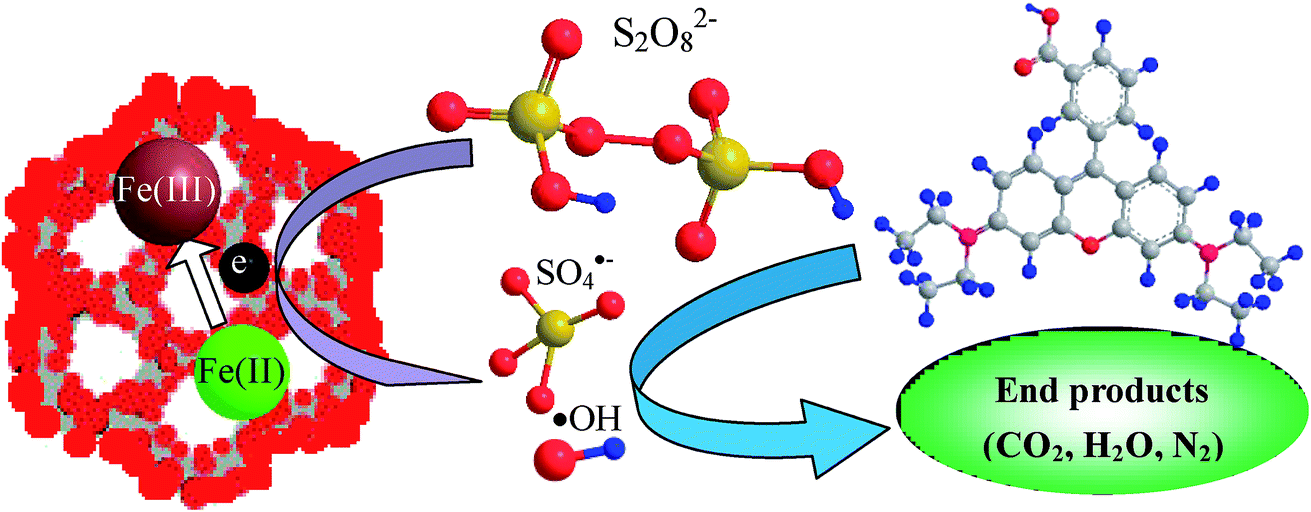 | ||
| Scheme 1 Activation mechanism of PS by MMS. | ||
3.2 Degradation performance of MMIOC
As shown in Fig. 7(a and b), the influences of catalyst dosage and PS concentration on RhB degradation were confirmed in different MMIOC/PS systems. As shown, the degradation rate constant is pseudo-first-order with respect to catalyst dosage concentration.34,46 Their correlation coefficients were higher than 99%. With the increase of the catalyst dosage from 0.05 to 2 g L−1, degradation effect was improved significantly. The apparent enhancement could be accounted for large number of surface active sites to activate PS and rapidly form active radicals on higher catalyst dosage.47 The ratio of natural logarithm of degradation rate constant and catalyst dosage was significantly different in different MMIOC/PS systems. This could be attributed to the difference between mesoporous structure, surface iron content and catalyst dosage. Further, it is concluded that the degradation rate constant of MMS catalyst dosage was highest. Superior catalytic performance could be ascribed to magnetic component embedded in inner or outer surface of magnetic mesoporous materials, forming catalytic active sites, thus providing a new strategy to modulate their electronic structure, resulting in generating more radicals for RhB degradation.48 Degradation rate constant of F type catalyst is higher than that of P type. Except iron content of samples, pore structure of F type is more favorable for molecular transport relative to P type. Moreover, mesoporous structure of MMS is most favorable to improve degradation efficiency. Pore structure of F type might be inferred to the formation of a cage-like mesopore,49–55 P type six square.56–59Similar to the behavior of catalyst dosage, degradation rate constants also linearly increase as PS concentration increases from 5 to 40 mM (Fig. 7(b)).34 In addition, degradation effect also improved significantly. With the increase of PS dosage, degradation ratio constant of MMS sample was the highest. At same PS dosage, the degradation ratio constant of F8/2 sample was higher than that of P8/2 sample. From a series of data it is inferred that, first, the pore structure corresponds to mass transportation, resulting in catalytic performance difference. Second, the specific surface area highly affects the degradation ratio constant.
The influence of initial pH from 3–11 on RhB degradation in different MMIOC/PS systems is displayed in Fig. 8(a). As observed, the best RhB degradation ratio occurs at pH 7–9, under or above which removal efficiency of RhB dramatically decreases. This revealed that PS could be effectively activated on as-prepared MMIOC surface and produce a large number of radicals to degrade RhB under neutral-alkaline conditions. Lower or higher pH value leads to lower degradation ratios. At higher pH, the lower degradation ratio is due to charge repulsion in active site of mesoporous surface. Nevertheless, the iron species deactivates PS at lower pH,60 resulting in the decrease of degradation ratio.34
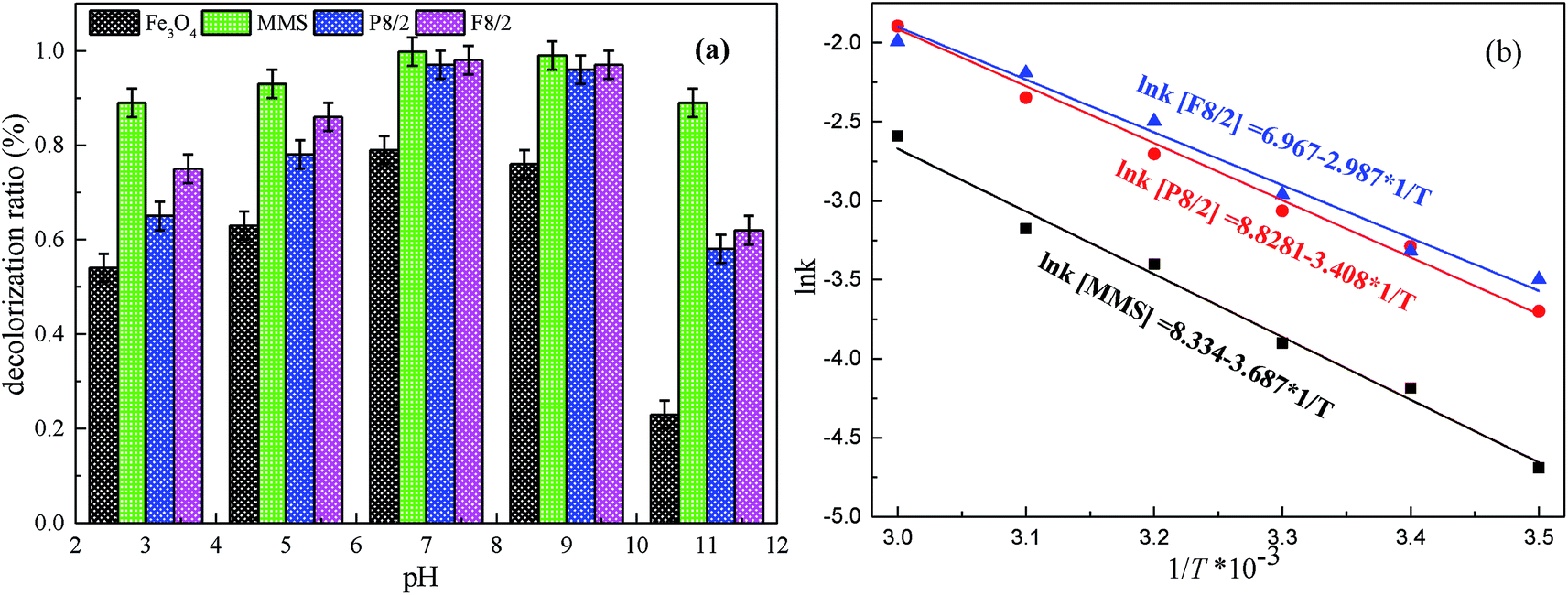 | ||
| Fig. 8 Effects of (a) pH, (b) temperature on RhB degradation in different FexOy/PS systems. Expect investigated parameter, others fixed at [RhB] = 1 mM; [catalyst] = 2.0 g L−1; [PS] = 40 mM. | ||
Fig. 8(b) presents the effect of reaction temperature on degradation ratios in various MMIOC/PS systems. As shown, the increase of reaction temperature accelerates the degradation ratio of the RhB, which could be caused by rapid breakdown of PS into ˙OH or SO4˙− under thermal activation.9–11,61 Moreover, the systems could cross reaction energy barrier easily at higher temperature.62 Furthermore, Arrhenius rate constants for various MMIOC/PS systems presented a good linear correlation with temperature. Calculated activation energy (Ea) values were 30.65, 28.33 and 24.84 kJ mol−1 for P8/2, F8/2 and MMS systems, respectively. Cai et al.63 reported a US/Fe–Co/SBA-15/PMS system for the degradation of orange II and measured the activation energy to be about 10.5 kJ mol−1. Activation energies of Co3O4 catalyst systems for phenol degradation are in the range of 47–70 kJ mol−1.64
The RhB degradation performance in aqueous solution was estimated by various FexOy/PS systems under optimum conditions. As shown in Fig. 7S and 8S,† complete degradation of 1 mM RhB was obtained within 120 minutes on various MMIOC/PS systems. Degradation performance could be basically fitted by first-order kinetic model.65,66 The RhB degradation ratio was below 80% on block Fe3O4/PS system in the absence of mesoporous structure, implying to eliminate the influence of diffusive mass transfer and adsorption.66
Recently, related papers have reported iron loading advanced oxidation process.63,67,68 The key problem was to solve iron leakage, catalyst deactivation, which caused additional contamination. Fig. 9 presents the stability and repetitive use rate of the catalyst by cyclic reuse on various FexOy/PS systems. In each test, the catalyst was separated by centrifugation; then, it was washed with an excess amount of deionized water and dried. The XPS spectra displayed chemical component change on catalyst surface before and after 10 times of repeated usage. The recycled catalyst maintained its original iron red color. Approximate 90% of degradation ratio was obtained for ten times of recycle on MMS/PS systems. MMS/PS systems showed a good regeneration and reusability, and so did the Fe/OMC system.69 Furthermore, MMS/PS system degraded organic pollutants by oxidation, while Fe/OMC system removed organic pollutants by adsorption. Therefore, Fe/OMC system required additional desorption steps to regenerate. For block Fe3O4/PS system, only 70% of the decolorization efficiency was obtained in the first five cycles. With the increase of cycles the degradation ratio decreased gradually, and for tenth cycle only 20% of degradation efficiency was observed. Small specific surface area of magnetic blocks and an iron leakage for the Fe3O4/PS system led to largely ineffective activation of PS. However, superior reusability of MMS could be ascribed to magnetic iron species inlayed or embedded in inner or outer surface of magnetic mesoporous materials, forming catalytic active site. The iron content of MMS catalysts was measured by EDS and Fe elements map. After removing the catalyst by centrifugation of MMS/PS system, iron content in centrifugal solution was determined by atomic adsorption. The catalyst was reused several times, and each time the iron content in centrifugal solution was less than 1 ppm.
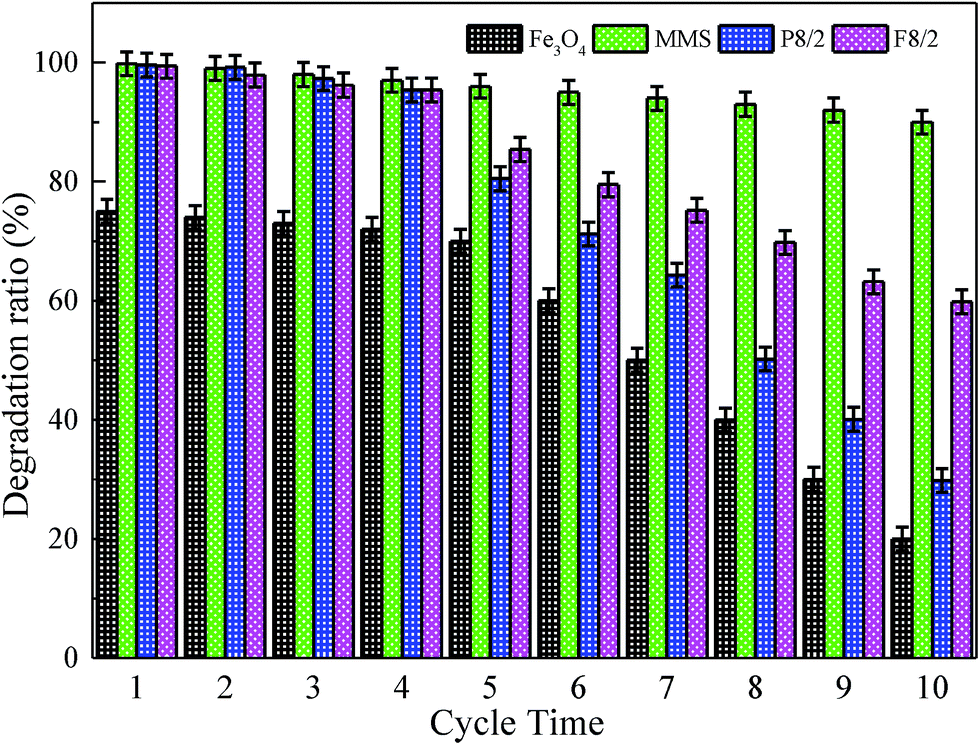 | ||
| Fig. 9 Effect of reusable times on RhB degradation in different FexOy/PS systems. Expect investigated parameter, others fixed at [RhB] = 1 mM; [catalyst] = 2.0 g L−1; [PS] = 40 mM; initial pH = 7.0; T = 25 °C. | ||
The variation of TOC and COD were determined to clarify the mineralization in MMS/PS systems. As shown in Fig. 10, the removal efficiencies of TOC and COD after 60 min reaction were 32% and 51%, respectively. This could be explained by the fact that the RhB was difficult to completely decolorize and the oxidation rate of intermediates could be much slower than RhB.63 Therefore, relatively more time was required to obtain a favorable mineralization, compared with RhB decolorization. When the reaction time was extended to 160 min, the removal efficiencies of TOC and COD increased to 42% and 65%, respectively. This showed the increase in the mineralization performance of the MMS/PS systems.
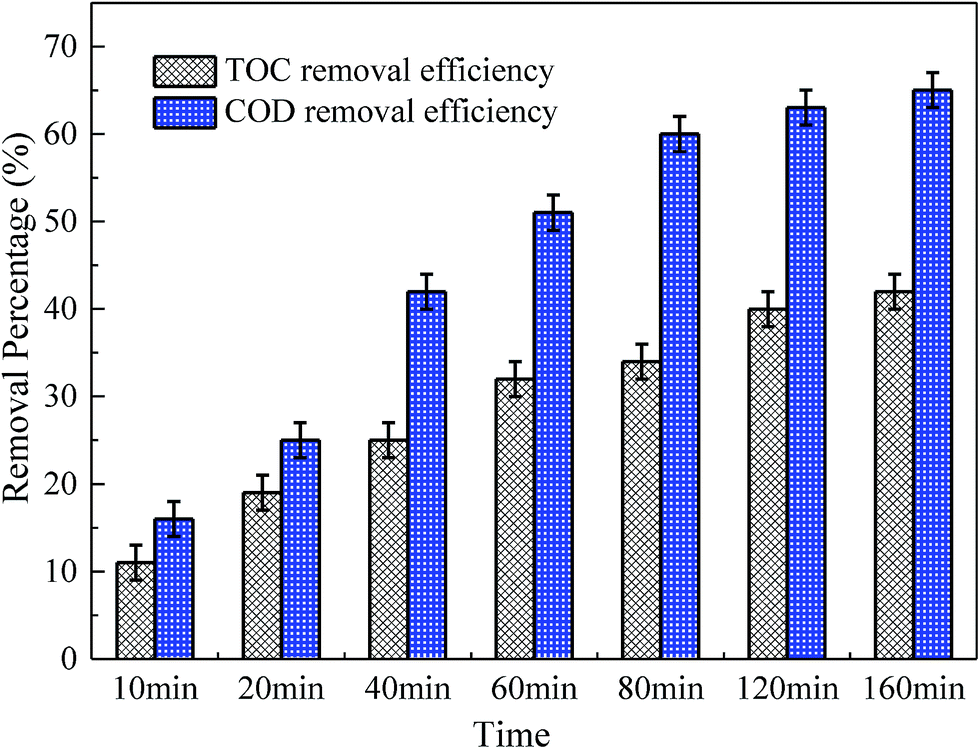 | ||
| Fig. 10 The variation of TOC and COD with reaction time on RhB degradation in MMS/PS systems. Expect investigated parameter, others fixed at [RhB] = 1 mM; [catalyst] = 2.0 g L−1; [PS] = 40 mM; initial pH = 7.0; T = 25 °C. | ||
On the MMS/PS systems, RhB degradation was divided into several stages. The molecular type at each stage of degradation was identified by an Agilent 7890A-5975 GC-MS analyzer. It has been reported previously that the introduction of ordered mesoporous graphitic carbon nitride could successfully and rapidly decolorize RhB.70 On the basis of free radicals formation, oxidative decarbonization and RhB degradation efficiency, this study generally analysed the mechanism of RhB degradation. Oxidation and rearrangement of the molecular structure on the MMS/PS systems are shown in Fig. 11. According to these organic intermediates, RhB degradation scheme containing three possible transformation pathways could be proposed. With respect to three degradation pathways initiated by ˙OH or SO4˙−, these unstable intermediates would open the rings and form low molecular organics, leading to mineralized inorganic compounds. The above result also evidenced that ˙OH and SO4˙−, which were generated by mesoporous embedded Fe3+ activated PS could chiefly participate in degradation of RhB in MMS/PS system.
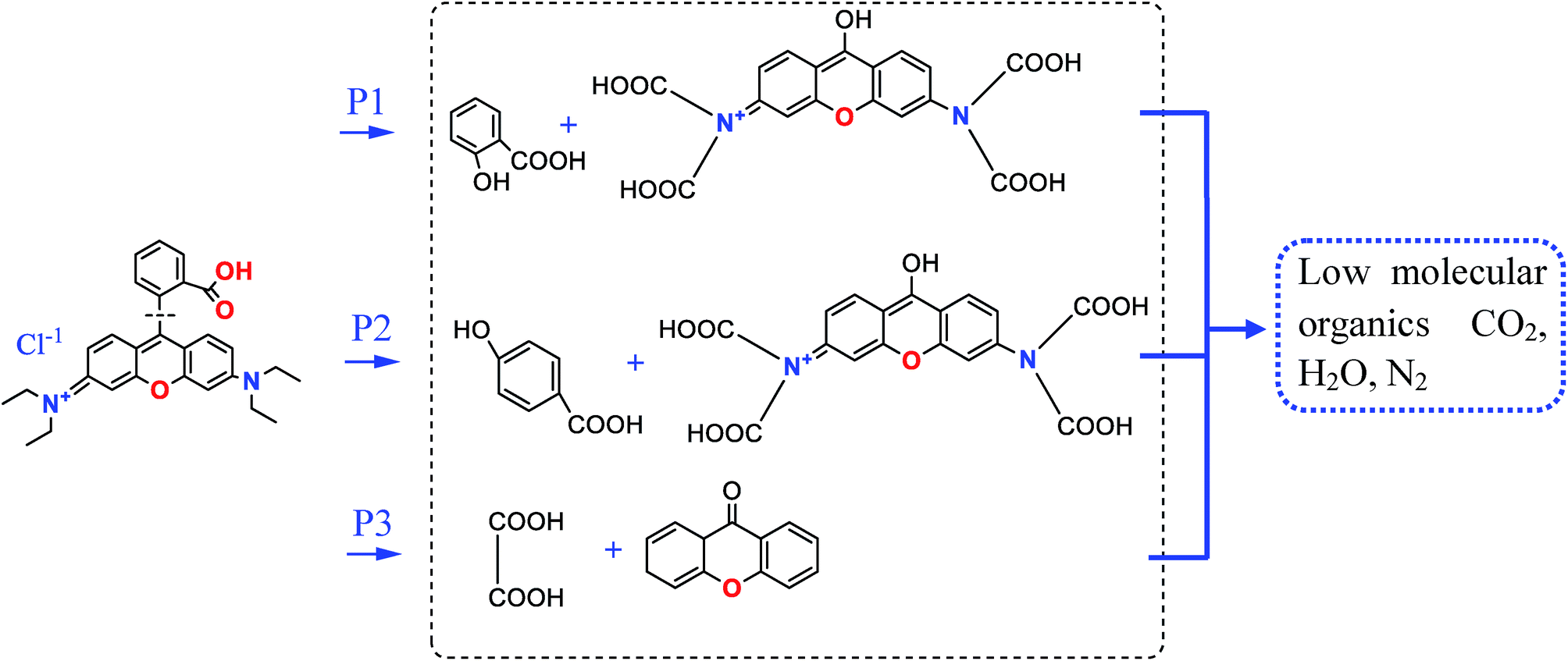 | ||
| Fig. 11 Degradation pathways of RhB in MMS/PS system. | ||
4. Conclusions
By employing hydrophobic ferrocene surface active agent as the micelles, MMIOC were successfully prepared via evaporation induced self-assembly and hydrothermal method. Iron ions were embedded in inner or outer surface, forming catalytic active sites that provide new strategy to modulate their electronic structure, resulting in generating radicals for efficient degradation of RhB. There is a linear relationship between various parameters, such as catalyst dose, PS dose, systematic temperature and degradation ratio constant. Under optimum conditions, degradation efficiency reached 99% after 60 min reaction, and increased to 99.8% when the reaction time was extended to 120 min. Degradation efficiency was compared between MMIOC/PS and Fe3O4/PS systems. Mesoporous structure and magnetism could synergistically improve degradation performance and increase the stability and reusability of the catalyst. Therefore, heterogeneous MMIOC/PS activation systems could be a potential technology for organic wastewater treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was financially supported by the National Young Natural Science Foundation of China (No. 21307169). The author wishes to thank the National Nature Science Foundation of China for their financial support.References
- A. Uygur and E. Kök, Color. Technol., 1999, 115, 350 CAS.
- S. Larouk, R. Ouargli, D. Shahidi, L. Olhund, T. C. Shiao, N. Chergui, T. Sehili, R. Roy and A. Azzouz, Chemosphere, 2017, 168, 1648 CrossRef CAS PubMed.
- R. F. Dantas, S. Contreras, C. Sans and S. Esplugas, J. Hazard. Mater., 2008, 150, 790 CrossRef CAS PubMed.
- A. G. Trovó, R. F. P. Nogueira, A. Agüera, C. Sirtori and A. R. Fernández-Alba, Chemosphere, 2009, 77, 1292 CrossRef PubMed.
- J. Deng, S. F. Feng, K. J. Zhang, J. Li, H. Y. Wang, T. Q. Zhang and X. Y. Ma, Chem. Eng. J., 2017, 308, 505 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705 CrossRef CAS PubMed.
- J. Deng, Y. S. Shao, N. Y. Gao, C. Q. Tan, S. Q. Zhou and X. H. Hu, J. Hazard. Mater., 2013, 262, 836 CrossRef CAS PubMed.
- C. Gong, F. Chen, Q. Yang, K. Luo, F. B. Yao, S. N. Wang, X. L. Wang, J. W. Wu, X. M. Li, D. B. Wang and G. M. Zeng, Chem. Eng. J., 2017, 321, 222 CrossRef CAS.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2012, 197, 483 CrossRef CAS.
- A. Ghauch and A. M. Tuqan, Chem. Eng. J., 2012, 183, 162 CrossRef CAS.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2015, 279, 861 CrossRef CAS.
- A. Ghauch, A. Baalbaki, M. Amasha, R. E. Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012 CrossRef CAS.
- Z. F. Ma, Y. Yang, Y. H. Jiang, B. D. Xi, T. X. Yang, X. Peng, X. Y. Lian, K. Yan and H. L. Liu, Chem. Eng. J., 2017, 311, 183 CrossRef CAS.
- Y. Li, Y. Li, S. Ma, P. Wang, Q. Hou, J. Han and S. Zhan, J. Hazard. Mater., 2017, 338, 33 CrossRef CAS PubMed.
- L. Tang, Y. Fang, Y. Pang, G. M. Zeng, J. J. Wang, Y. Y. Zhou, Y. C. Deng, G. D. Yang, Y. Cai and J. Chen, Chem. Eng. J., 2014, 254, 302 CrossRef CAS.
- W. Tanglumlert, T. Imae, T. J. White and S. Wongkasemjit, Mater. Lett., 2008, 62, 4545 CrossRef CAS.
- J. E. Chen, H.-Y. Lian, S. Duttacec, S. M. Alshehri, Y. Yamauchi, M. T. Nguyen, T. Yonezawa and K. C.-W. Wu, Phys. Chem. Chem. Phys., 2015, 17, 27653 RSC.
- J. C. Yan, W. G. Gao, M. G. Dong, L. Han, L. B. Qian, C. P. Nathanail and M. F. Chen, Chem. Eng. J., 2016, 295, 309 CrossRef CAS.
- B. Sahoo, S. K. Sahu, S. Nayak, D. Dhara and P. Pramanik, Catal. Sci. Technol., 2012, 2, 1367 CAS.
- N. Kato and N. Kato, Microporous Mesoporous Mater., 2016, 219, 230 CrossRef CAS.
- T. A. Arica, E. Ayas and M. Y. Arica, Microporous Mesoporous Mater., 2017, 243, 164 CrossRef CAS.
- C. Wang, S. Y. Tao and C. G. Meng, J. Mater. Chem., 2012, 22, 7179 RSC.
- S. Y. Tao, Z. Y. Zhu, C. G. Meng and C. Wang, Microporous Mesoporous Mater., 2013, 171, 94 CrossRef CAS.
- B. K. Vashisth, J. S. Bangruwa, A. Beniwal, S. P. Gairola, A. Kumar, N. Singh and V. Verma, J. Alloys Compd., 2017, 698, 699 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790 CrossRef CAS PubMed.
- J. Fernandez, P. Maruthamuthu, A. Renken and J. Kiwi, Appl. Catal., B, 2004, 49, 207 CrossRef CAS.
- H. L. Xie, W. He and G. Chen, J. Chin. Chem. Soc., 2015, 62, 1144 CrossRef.
- H. L. Xie and T. Y. Zhang, Int. J. Nanosci., 2015, 13, 1460010 CrossRef.
- S. Gomez, L. Lerici, C. Saux, A. L. Perez, C. D. Brondino, L. Pierella and L. Pizzio, Appl. Catal., B, 2017, 202, 580 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, D. Y. Zhao, P. D. Yang, B. F. Chemlka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254 CrossRef CAS.
- J. E. Chen, H.-Y. Lian, S. Duttacec, S. M. Alshehri, Y. Yamauchi, M. T. Nguyen, T. Yonezawa and K. C.-W. Wu, Phys. Chem. Chem. Phys., 2015, 17, 27653 RSC.
- S. Liu, H. Y. Zhang, X. J. Meng, Y. L. Zhang, L. M. Ren, F. Nawaz, J. Y. Liu, Z. Q. Li and F.-S. Xiao, Microporous Mesoporous Mater., 2010, 136, 126 CrossRef CAS.
- M. A. Betiha, A. M. Rabie, A. M. Elfadly and F. Z. Yehia, Microporous Mesoporous Mater., 2016, 222, 44 CrossRef CAS.
- Y. X. Wang, L. Zhou, X. G. Duan, H. Q. Sun, E. L. Tin, W. Q. Jin and S. B. Wang, Catal. Today, 2015, 258, 576 CrossRef CAS.
- M. Y. Wei, L. Gao, J. Li, J. Fang, W. X. Cai, X. X. Li and A. Xu, J. Hazard. Mater., 2016, 316, 60 CrossRef CAS PubMed.
- J. Conradie and E. Erasmus, Polyhedron, 2016, 119, 142 CrossRef CAS.
- C. Prasad, K. Sreenivasulu, S. Gangadhara and P. Venkateswarlu, J. Alloys Compd., 2017, 700, 252 CrossRef CAS.
- J. Liu, J. H. Zhou, Z. X. Ding, Z. W. Zhao, X. Xu and Z. D. Fang, Ultrason. Sonochem., 2017, 34, 953 CrossRef CAS PubMed.
- G. Nie, J. Huang, Y. Z. Hu, Y. B. Ding, X. Y. Han and H. Q. Tang, Chin. J. Catal., 2017, 38, 227 CrossRef CAS.
- Y. J. Yao, Y. M. Cai, G. D. Wu, F. Y. Wei, X. Y. Li, H. Chen and S. B. Wang, J. Hazard. Mater., 2015, 296, 128 CrossRef CAS PubMed.
- N. Y. He, C. Yang, P. F. Xiao, G. H. Wang, Y. J. Zhao, Z. H. Lu and C. W. Yuan, Stud. Surf. Sci. Catal., 2000, 129, 483 CrossRef CAS.
- J. Conradie and E. Erasmus, Polyhedron, 2016, 119, 142–150 CrossRef CAS.
- H. Z. Liu, T. A. Bruton, W. Li, J. V. Buren, C. Prasse, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2016, 50, 890–898 CrossRef CAS PubMed.
- C. Zhu, G. Fang, D. D. Dionysiou, C. Liu, J. Gao, W. Qin and D. Zhou, J. Hazard. Mater., 2016, 316, 232–241 CrossRef CAS PubMed.
- J. Deng, S. F. Feng, X. Ma, C. Tan, H. Wang, S. Zhou, T. Zhang and J. Li, Sep. Purif. Technol., 2016, 167, 181–189 CrossRef CAS.
- C.-H. Tsai, W.-C. Chang, D. Saikia, C.-E. Wu and H.-M. Kao, J. Hazard. Mater., 2016, 309, 236 CrossRef CAS PubMed.
- L. X. Hu, F. Yang, W. C. Lu, Y. Hao and H. Yuan, Appl. Catal., B, 2013, 134–135, 7 CrossRef CAS.
- Y. B. Wang, X. Zhao, D. Cao, Y. Wang and Y. F. Zhu, Appl. Catal., B, 2017, 211, 79 CrossRef CAS.
- C.-H. Tsai, W.-C. Chang, D. Saikia, C.-E. Wu and H.-M. Kao, J. Hazard. Mater., 2016, 309, 236 CrossRef CAS PubMed.
- S. Larouk, R. Ouargli, D. Shahidi, L. Olhund, T. C. Shiao, N. Chergui, T. Sehili, R. Roy and A. Azzouz, Chemosphere, 2017, 168, 1648 CrossRef CAS PubMed.
- Z. C. Bian, X. Meng, M. Tao, Y. H. Lv and Z. Xin, Fuel, 2016, 179, 193 CrossRef CAS.
- S. N. Azizi, S. Ghasemi, A. Samadi-Maybodi and M. Ranjbar-Azad, Sens. Actuators, B, 2015, 216, 271 CrossRef CAS.
- Y. L. Dong, X. L. Zhan, X. Y. Niu, J. Li, F. L. Yuan, Y. J. Zhu and H. G. Fu, Microporous Mesoporous Mater., 2014, 185, 97 CrossRef CAS.
- A. T. Shah, M. I. Din, F. N. Kanwal and M. L. Mirza, Arabian J. Chem., 2015, 8, 579 CrossRef CAS.
- S. N. Azizi, S. Ghasemi, A. Samadi-Maybodi and M. Ranjbar-Azad, Sens. Actuators, B, 2015, 216, 271 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS PubMed.
- W. R. Chen, X. K. Li, Z. Q. Pan, S. S. Ma and L. S. Li, Sep. Purif. Technol., 2017, 173, 99 CrossRef CAS.
- S. Karthikeyan, M. P. Pachamuthu, M. A. Isaacs, S. Kumar, A. F. Lee and G. Sekaran, Appl. Catal., B, 2016, 199, 323 CrossRef CAS.
- V. R. Elías, G. O. Ferrero, R. G. Oliveira and G. A. Eimer, Microporous Mesoporous Mater., 2016, 236, 218 CrossRef.
- C. Q. Tan, N. Y. Gao, Y. Deng, J. Deng, S. Q. Zhou, J. Li and X. Y. Xin, J. Hazard. Mater., 2014, 276, 452 CrossRef CAS PubMed.
- X. H. Li, W. L. Guo, Z. H. Liu, R. Q. Wang and H. Liu, Appl. Surf. Sci., 2016, 369, 130 CrossRef CAS.
- X. Y. Chen, X. L. Qiao, D. G. Wang, J. Lin and J. W. Chen, Chemosphere, 2007, 67, 802 CrossRef CAS PubMed.
- C. Cai, Z. Zhang and H. Zhang, J. Hazard. Mater., 2016, 313, 209 CrossRef CAS PubMed.
- E. Saputra, S. Muhammad, H. Q Sun, H.-M. Ang, M. O. Tadé and S. B. Wang, Appl. Catal., B, 2013, 142–143, 729 CrossRef CAS.
- J. K. Du, J. G. Bao, X. Y. Fu, C. H. Lu and S. H. Kim, Appl. Catal., B, 2016, 184, 132 CrossRef CAS.
- I. Mazilu, C. Ciotonea, A. Chirieac, B. Dragoi, C. Catrinescu, A. Ungureanu, S. Petit, S. Royer and E. Dumitriu, Microporous Mesoporous Mater., 2017, 241, 326 CrossRef CAS.
- M. J. Huang, T. Zhou, X. H. Wu and J. Mao, Water Res., 2017, 119, 47 CrossRef CAS PubMed.
- A. Ghauch, G. Ayoub and S. Naim, Chem. Eng. J., 2013, 228, 1168 CrossRef CAS.
- L. Tang, Z. H. Xie, G. M. Zeng, H. R. Dong, C. Z. Fan, Y. Y. Zhou, J. J. Wang, Y. C. Deng, J. J. Wang and X. Wei, RSC Adv., 2016, 6, 25724 RSC.
- L. Luo, A. Zhang, M. J. Janik, K. Li, C. Song and X. Guo, Appl. Surf. Sci., 2017, 396, 78 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07645b |
| This journal is © The Royal Society of Chemistry 2017 |