
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and catalytic activity of rare-earth metal amides incorporating cyclohexyl bridged bis(β-diketiminato) ligands†
Hui Miaoab,
Shaowu Wang *ac,
Xiancui Zhu*a,
Shuangliu Zhoua,
Yun Weia,
Qingbing Yuana and
Xiaolong Mua
*ac,
Xiancui Zhu*a,
Shuangliu Zhoua,
Yun Weia,
Qingbing Yuana and
Xiaolong Mua
aLaboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, Anhui 241000, P. R. China. E-mail: swwang@ahnu.edu.cn; zxc0805@ahnu.edu.cn; Fax: +86 553 3883517; Tel: +86 553 5910015
bSchool of Chemistry and Materials Engineering, Fuyang Normal College, Fuyang, Anhui 236037, P. R. China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 5th September 2017
Abstract
A series of organo rare-earth metal amides incorporating chiral cyclohexyl bridged bis(β-diketiminato) ligands with general formula LREN(SiMe3)2 (L1 = (1S,2S)-1,2-Cy[NC(Me)CHC(Me)NAr]2, Ar = 2, 6-Et2C6H3, RE = Nd (1a), Dy (1b), Yb (1c), Y (1d); L2 = (1R,2R)-1,2-Cy[NC(Me)CHC(Me)NAr]2, Ar = 2, 6-i-Pr2C6H3, RE = Nd (2a), Gd (2b), Dy (2c), Er (2d), Y (2e)) were synthesized in good yields via reactions of [(Me3Si)2N]3REIII(μ-Cl)Li(THF)3 with H2L1 and H2L2. All compounds were fully characterized by spectroscopic methods and elemental analyses. The complexes 1d and 2e were also characterized by 1H NMR and 13C NMR spectral analyses. The structures of complexes 1a–d were determined by single-crystal X-ray analyses. Investigation of the catalytic properties of the complexes indicated that all complexes exhibited a high catalytic activity towards the addition of diphenylphosphine oxide to β-nitroalkene and α,β-unsaturated carbonyl derivatives with an excellent regioselectivity.
Introduction
In the past decade, the β-diketiminato supported metal complexes, as some of the typical nonmetallocene complexes, have seen great development, because the monodeprotonated β-diketiminato ligand is isoelectronic with the cyclopentadienyl anion. Compared with the cyclopentadienyl ligand, the β-diketiminato ligand displayed different coordination modes,1 and tunable steric and electronic properties.1–3 The reactivities of rare-earth metal complexes incorporating different β-diketiminato ligands have also been widely investigated.4–6 Some of them displayed high catalytic activities in polymerization, such as olefins polymerization,1f,4a methyl methacrylate polymerization,1g,5b copolymerization of epoxide and CO2,1d,5d and ring-opening polymerization of ε-caprolactone or lactides.5f–5j Recently, we reported rare-earth metal complexes bearing cyclohexyl bridged β-diketiminato ligands.6 As a continuation of our own interest in cyclohexyl bridged β-diketiminato ligands, we further explored the chiral cyclohexyl bridged β-diketiminato supported rare-earth metal complexes to expand their catalytic applications. We report herein the synthesis and characterization of a series of the rare-earth metal amides incorporating chiral cyclohexyl bridged bis(β-diketiminato) ligands and their catalytic activity towards the hydrophosphination of β-nitroalkene and α,β-unsaturated carbonyl derivatives.Results and discussion
Synthesis and characterization of complexes
Treatment of H2L1 or H2L2 with 1 equiv. of the rare-earth metal amides [(Me3Si)2N]3REIII(μ-Cl)Li(THF)3 in toluene at 60–80 °C afforded the rare-earth metal complexes with general formula LREN(SiMe3)2 (L1 = (1S,2S)-1,2-Cy[NC(Me)CHC(Me)NAr]2, Ar = 2, 6-Et2C6H3, RE = Nd (1a), Dy (1b), Yb (1c), Y (1d); L2 = (1R,2R)-1,2-Cy[NC(Me)CHC(Me)NAr]2, Ar = 2, 6-i-Pr2C6H3, RE = Nd (2a), Gd (2b), Dy (2c), Er (2d), Y (2e)) (Scheme 1). The complexes are sensitive to air and moisture; and they have a good solubility in either polar solvents or nonpolar solvents. All complexes were fully characterized by IR and elemental analyses. The complexes 1d and 2e were also characterized by 1H NMR and 13C NMR spectra analyses. The structures of complexes 1a–d were determined by single-crystal X-ray analyses.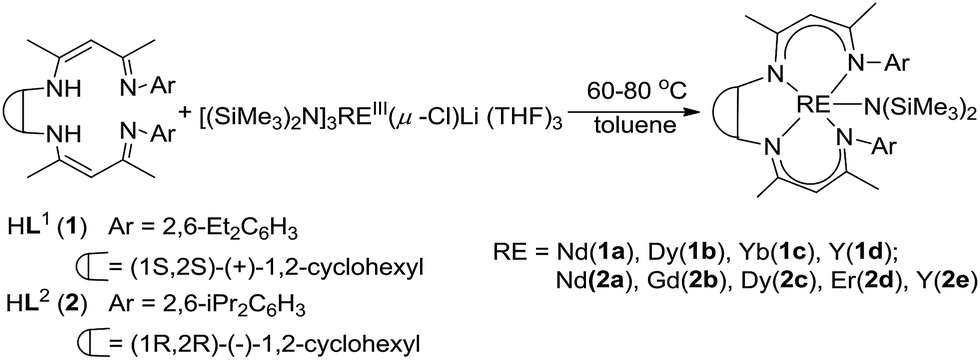 | ||
| Scheme 1 Preparation of the rare-earth metal complexes. | ||
Molecular structures of the complexes
X-ray structure analyses revealed that complexes 1a–d are mononuclear structures crystallized in the orthorhombic system with chiral space group P212121, they are isostructural and isomorphous. Representative structural diagram of complex 1d is shown in Fig. 1. The rare-earth metal adopts a distorted square pyramid, in which the N(SiMe3)2 ligand occupies the apical position and the four N atoms of the chiral bridged β-diketiminato moieties form the basis. The substituted aryl groups take the transoid fashion in all complexes with one aryl substituent pointing away from the amido group and the other aryl group pointing towards the amido group. The selected bond lengths and angles are listed in Table 1.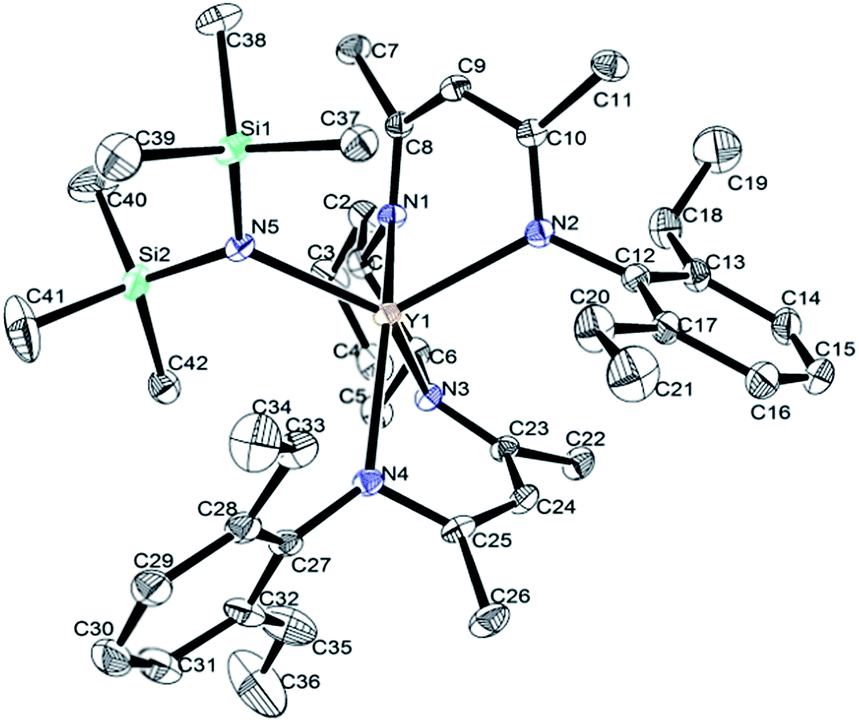 | ||
| Fig. 1 Representative structure of rare-earth metal amides incorporating chiral bridged bis(β-diketiminato) ligand. Hydrogen atoms were omitted for clarity. | ||
| 1a (RE = Nd) | 1b (RE = Dy) | 1c (RE = Yb) | 1d (RE = Y) | |
|---|---|---|---|---|
| RE(1)-N(1) | 2.421(3) | 2.332(3) | 2.294(6) | 2.324(3) |
| RE(1)-N(2) | 2.400(3) | 2.323(3) | 2.278(5) | 2.321(3) |
| RE(1)-N(3) | 2.385(3) | 2.323(2) | 2.227(5) | 2.320(3) |
| RE(1)-N(4) | 2.467(3) | 2.360(3) | 2.297(6) | 2.346(3) |
| RE(1)-N(5) | 2.364(2) | 2.284(3) | 2.240(5) | 2.273(3) |
| RE(1)-Nav. | 2.407(3) | 2.324(3) | 2.267(5) | 2.316(3) |
| RE(1)-C(8) | 2.950(3) | 2.997(3) | 3.015(5) | 2.998(3) |
| RE(1)-C(9) | 3.037(3) | 3.114(3) | 3.212(5) | 3.149(3) |
| RE(1)-C(10) | 3.054(3) | 3.042(3) | 3.049(5) | 3.059(3) |
| RE(1)-C(23) | 3.049(3) | 3.100(3) | 3.088(5) | 3.093(3) |
| RE(1)-C(24) | 3.169(3) | 3.259(3) | 3.254(5) | 3.245(3) |
| RE(1)-C(25) | 3.111(3) | 3.112(3) | 3.084(5) | 3.105(3) |
| RE(1)-Cav. | 3.062(3) | 3.104(3) | 3.117(5) | 3.108(3) |
| N(1)-RE(1)-N(2) | 73.38(11) | 76.15(10) | 78.0(2) | 77.14(11) |
| N(1)-RE(1)-N(3) | 65.96(12) | 68.20(10) | 69.4(2) | 68.28(11) |
| N(1)-RE(1)-N(4) | 138.60(12) | 144.13(10) | 146.9(2) | 144.33(12) |
| N(1)-RE(1)-N(5) | 95.50(11) | 93.70(10) | 93.8(2) | 94.03(11) |
| N(2)-RE(1)-N(3) | 96.34(9) | 95.43(10) | 94.9(2) | 95.71(12) |
| N(2)-RE(1)-N(4) | 125.32(10) | 121.41(10) | 117.2(2) | 120.36(12) |
| N(2)-RE(1)-N(5) | 126.89(8) | 122.73(10) | 121.4(2) | 122.31(12) |
| N(3)-RE(1)-N(4) | 75.61(10) | 78.54(10) | 79.8(2) | 78.63(11) |
| N(3)-RE(1)-N(5) | 130.04(10) | 133.15(10) | 136.4(2) | 133.98(12) |
| N(4)-RE(1)-N(5) | 96.74(11) | 99.64(10) | 100.8(2) | 99.76(12) |
From Table 1, we can see that the usual consequence of the ionic radius of the RE3+ ion when moving from Nd3+ to Yb3+ is clearly reflected by the average RE-N distances of 2.407(3) Å found in 1a, 2.324(2) Å found in 1b, 2.267(5) Å found in 1c, 2.316(3) Å found in 1d. The average RE-N distances of complexes 1a [2.407(3) Å], 1b [2.324(2) Å] and 1d [2.316(3) Å] were compared with the corresponding RE-N distances in the similar rare-earth complexes containing the cyclohexyl-linked bis(β-diketiminato) Cy[NC(Me)CHC(Me)NAr]2REN(SiMe3)2 (Ar = 2, 6-iPr2C6H3, RE = Nd [2.433(2) Å], Dy [2.350(2) Å], Y [2.340(2) Å]).6 The lanthanide contraction is contrary reflected by the average RE-C (C8–C10 and C23–C25) contacts of 3.062(3) Å in 1a, 3.104(3) Å in 1b, 3.117(5) Å in 1c, 3.108(3) Å in 1d, which is consistent with rare-earth complexes containing cyclohexyl-linked bis(β-diketiminato) Cy[NC(Me)CHC(Me)NAr]2REN(SiMe3)2 (Ar = 2, 6-iPr2C6H3), probably due to steric effects of ligands on the Ln–C interaction.6
Catalytic activities of the complexes
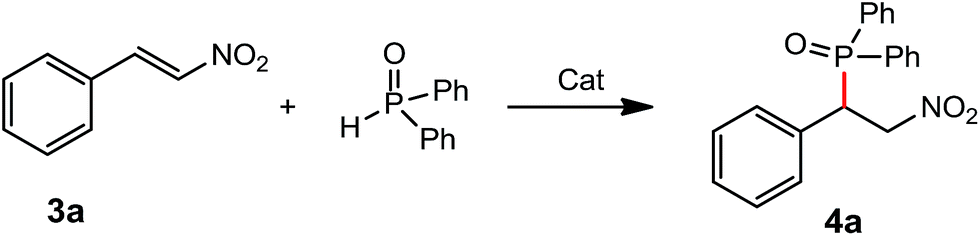
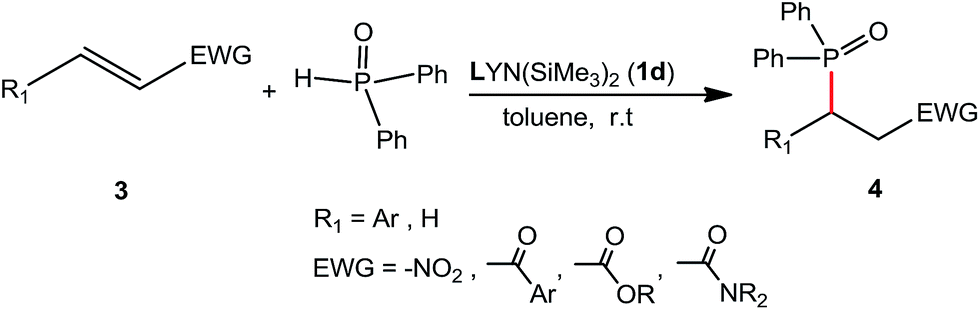
Development of new catalysts for hydrophosphination is of interest and importance due to 100% atom economy pathway through the additions of dialkyl phosphites, secondary phosphines and diarylphosphine oxides to the unsaturated substrates.7 In the addition of diarylphosphine oxide to the α,β-unsaturated derivatives, rare-earth metal complexes have advantages of lower catalysts loadings (3–5 mol%), available materials, solvents compatibility and easy preparation, compared with the reported chiral bicyclic guanidinate catalyst (10 mol% loading, Et2O as solvent) and chiral multifunctional amine-thiourea catalyst (10 mol% loading, CH2Cl2 as solvent).8 In our previous study, we have reported the rare-earth metal amido complexes as catalysts for various C–P bond formation.9a,b,f,g In order to further explore the chiral rare-earth metal amido complexes for the catalytic C–P bond formation, we examined the chiral rare-earth complexes as catalysts for the catalytic hydrophosphination of β-nitroalkene and α,β-unsaturated carbonyl derivatives.Addition of diphenyphosphine oxide to the β-nitrostyrene was tested as a template by employing complex 1d as a catalyst, and results were summarized in Table 2. Results showed that 3 mol% of the catalyst 1d could catalyze the hydrophosphination of β-nitrostyrene in solvents such as toluene, THF, diethyl ether and n-hexane at room temperature. The catalytic reaction was run in n-hexane to afford the product 4a in a 49% yield, probably due to solubility of catalyst. It is a pity that the addition products catalyzed by the above chiral rare-earth complexes was racemic under the conditions screened, probably due to the ligands' chiral backbone far from the metal centre which cannot control the stereochemistry of the product, but the reactions afforded the products with a high regioselectivity. The catalytic activities of the above different rare-earth metal amides on hydrophosphination of β-nitrostyrene were investigated with 3 mol% of catalyst loading using toluene as a solvent (Table 2, entries 4, 13–20). It is found that all complexes 1a–d, 2a–e exhibited high catalytic activities on the hydrophosphination of β-nitrostyrene, indicating that the ionic radii of the rare-earth metals and the substituent groups on phenyl ring have little influence on the catalytic activities of the catalysts.
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | Temp. (°C) | Time (h) | Yield (%) |
| a Reactions were performed with 0.5 mmol of diphenyphosphine oxide and 0.5 mmol of β-nitrostyrene in 5 mL of solvent. | |||||
| 1 | 1d (1%) | Toluene | r.t. | 24 | 58 |
| 2 | 1d (3%) | Toluene | r.t. | 24 | 96 |
| 3 | 1d (5%) | Toluene | r.t. | 24 | 96 |
| 4 | 1d (3%) | Toluene | r.t. | 6 | 95 |
| 5 | 1d (3%) | Toluene | r.t. | 1 | 57 |
| 6 | 1d (3%) | Toluene | r.t. | 3 | 78 |
| 7 | 1d (3%) | Toluene | 45 | 6 | 93 |
| 8 | 1d (3%) | Toluene | 0 | 6 | 67 |
| 9 | 1d (3%) | Toluene | −10 | 6 | 45 |
| 10 | 1d (3%) | THF | r.t. | 6 | 93 |
| 11 | 1d (3%) | Et2O | r.t. | 6 | 92 |
| 12 | 1d (3%) | n-Hexane | r.t. | 6 | 49 |
| 13 | 1a (3%) | Toluene | r.t. | 6 | 95 |
| 14 | 1b (3%) | Toluene | r.t. | 6 | 94 |
| 15 | 1c (3%) | Toluene | r.t. | 6 | 95 |
| 16 | 2a (3%) | Toluene | r.t. | 6 | 93 |
| 17 | 2b (3%) | Toluene | r.t. | 6 | 92 |
| 18 | 2c (3%) | Toluene | r.t. | 6 | 94 |
| 19 | 2d (3%) | Toluene | r.t. | 6 | 95 |
| 20 | 2e (3%) | Toluene | r.t. | 6 | 94 |
Under the optimized reaction conditions, we next examined the substrate scope of the catalytic addition of diphenyphosphine oxide to different β-nitroalkene employing 1d as a catalyst (Table 3). From Table 3, we can see that the complex 1d exhibited a high catalytic activity towards different β-nitroalkene despite of the electronic nature and the steric effect of the substituents on the aryl groups of the substrates (entries 1–8, Table 3). Furthermore, the additions of diphenyphosphine oxide to various α,β-unsaturated carbonyl derivatives have also examined. The catalytic system could suit well for the α,β-unsaturated amides, esters and chalcones producing the corresponding 1,4-regioselective addition products in high yields (entries 9–14, Table 3). In contrast to the imino-hydroxyquinolyl functionalized rare-earth metal catalysts,8c the catalytic loadings of new synthesized rare-earth metal complexes supported by chiral cyclohexyl bridged bis(β-diketiminato) ligand were reduced to 3 mol%, and the substrates could be different α,β-unsaturated amides, esters and chalcones, indicating ligands effects on the catalytic activity of the catalysts.
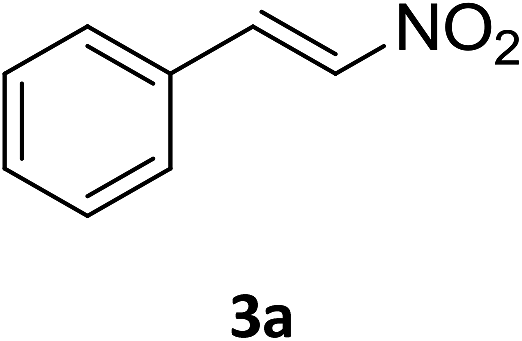
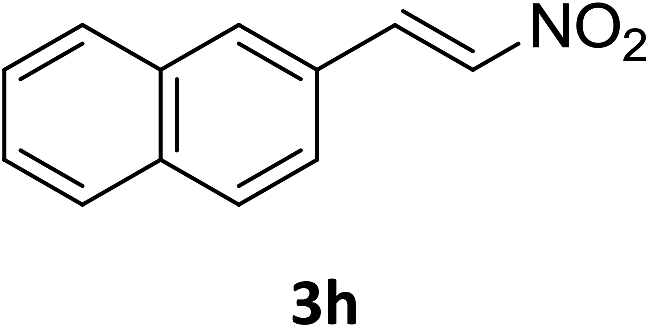
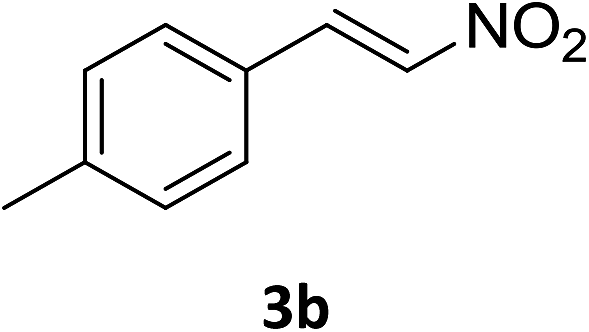
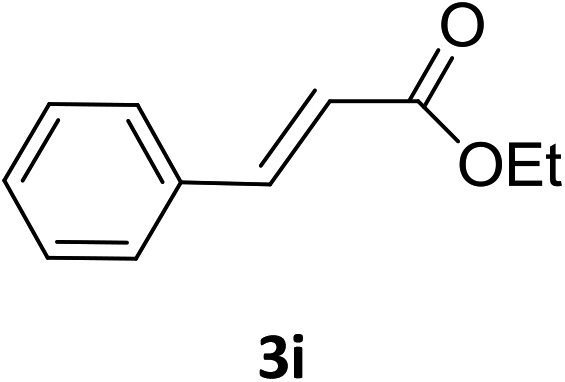
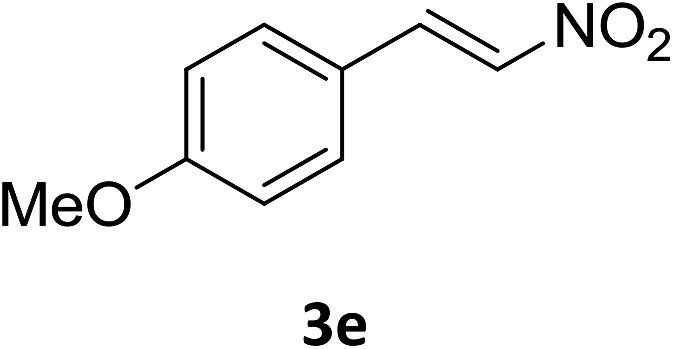
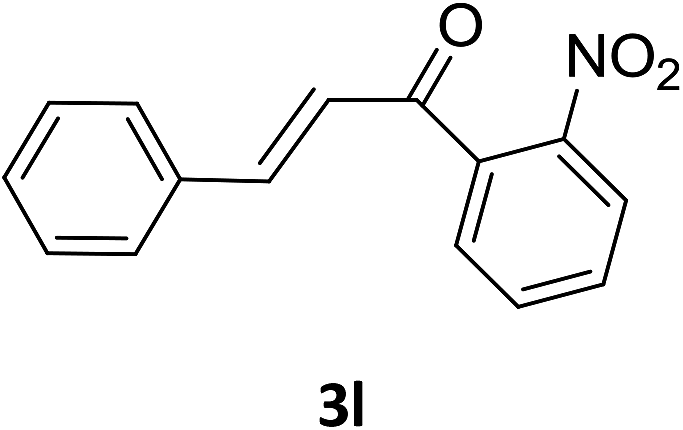
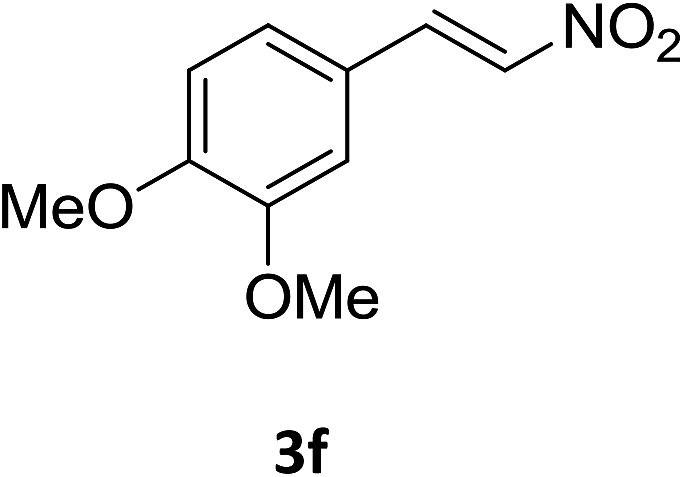
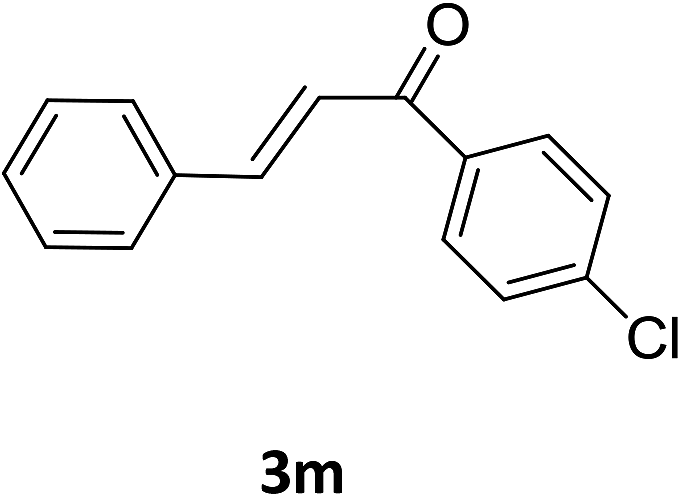
The catalytic addition mechanism of diphenylphosphine oxide to the β-nitrostyrene and α,β-unsaturated carbonyl derivatives is proposed (Scheme 2). Interaction of the catalyst with diphenyphosphine oxide produced the intermediate A via silylamine elimination, coordination of the substrate with A produced B, which subsequently underwent 1,4-conjugate addition to form C, C then interacted with diphenyphosphine oxide to regenerate intermediate A finishing a catalytic cycle. The stereocenters of the ligands are far from the reactive site in the intermediate B which controls the stereochemistry to produce racemic products. The metal center will have little influence on the catalytic activity.
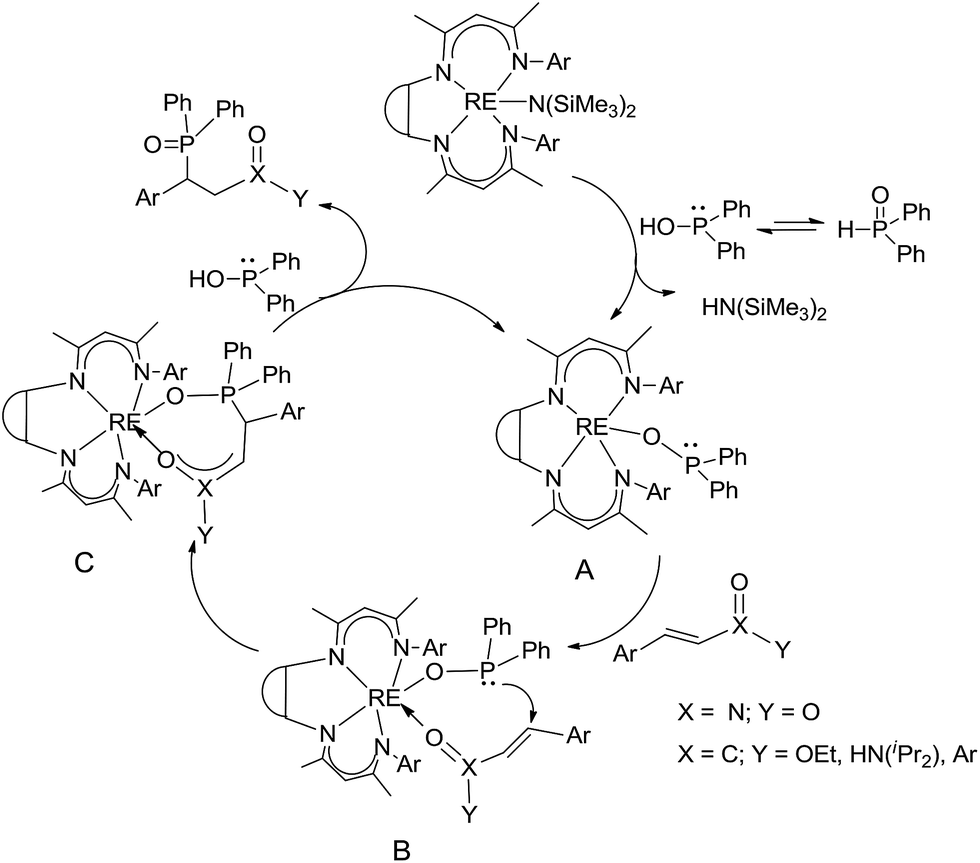 | ||
| Scheme 2 The proposed mechanism of the catalytic reaction. | ||
Experimental section
Materials and methods
All syntheses and manipulations of air- and moisture-sensitive materials were performed under dry argon and oxygen-free atmosphere using standard Schlenk techniques or in a glove box. All solvents were refluxed and distilled over sodium benzophenone ketyl under argon prior to use unless otherwise noted. [(Me3Si)2N]3REIII(μ-Cl)Li(THF)3 (RE = Nd, Gd, Dy, Er, Yb and Y) were prepared according to literature methods.9p (1S,2S)-(+)-1,2-diaminocyclohexane, 2,6-diethylaniline, (1R,2R)-(−)-1,2-diaminocyclohexane, 2,6-diisopropylaniline, acetylacetone were purchased and used without purification. 2-((2,6-Diisopropylphenyl)imido)-2-penten-4-one and 2-((2,6-diethylphenyl)imido)-2-penten-4-one were synthesized by following the literature procedures.10 Elemental analyses data were obtained on a Perkin-Elmer 2400 Series II elemental analyzer. 1H NMR and 13C NMR spectra for analyses of compounds were recorded on a Bruker AV-300 NMR spectrometer (300 MHz for 1H; 75.0 MHz for 13C) in C6D6 for lanthanide complexes and in CDCl3 for organic compounds. Chemical shifts (δ) were reported in ppm. J values are reported in Hz. IR spectra were recorded on a Perkin-Elmer 983(G) spectrometer (KBr pellet).General procedures for hydrophosphination of β-nitroalkene and α,β-unsaturated carbonyl derivatives (4a as an example)
A 30.0 mL Schlenk tube under dried argon was charged with complex 1d (11.8 mg, 0.015 mmol), diphenyphosphine oxide (0.101 g, 0.5 mmol), and 5.0 mL of toluene, and then β-nitrostyrene (0.075 g, 0.5 mmol) was added to the mixture. The mixture was stirred at room temperature for 6 hours. After the reaction was completed, the reaction mixture was hydrolyzed by water, extracted with ethyl ether, dried over anhydrous sodium sulfate, and then filtered. After the solvent was removed under reduced pressure, the final products were further purified by recrystallization from ethyl acetate or column chromatography. Compound 4a was isolated as a white solid (0.167 g, 95%).Conclusion
A series of novel rare-earth metal amides bearing chiral cyclohexyl bridged bis(β-diketiminato) ligands were synthesized via the reactions of [(Me3Si)2N]3REIII(μ-Cl)Li(THF)3 with the corresponding chiral cyclohexyl bridged bis(β-diketimines) in good yields. These complexes exhibited an excellent catalytic activity on the hydrophosphination of β-nitroalkene and α,β-unsaturated carbonyl derivatives with a high regioselectivity. The catalysts have the advantages of a high efficiency, a low catalysts loading, a wide of solvents and substrates compatibility, and mild conditions. The method provides a highly atomic efficient way for the preparation functionalized phosphine oxides, which can be easy transferred to useful phosphines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was co-supported by the National Natural Science Foundation of China (Grant No. 21432001, 21372010, 21402029, 21372009) and Foundation for Young Talents in College of Anhui Province (2013SQRL058ZD). We are also grateful for the assistance of Fuyang Teachers College.Notes and references
- (a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed; (b) V. C. Gibson and K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS PubMed; (c) P. M. Zeimentz, S. Arndt, B. R. Elvidge and J. Okuda, Chem. Rev., 2006, 106, 2404 CrossRef CAS PubMed; (d) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC; (e) M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194 CrossRef CAS PubMed; (f) M. Nishiura and Z. Hou, Nat. Chem., 2010, 2, 257 CrossRef CAS PubMed; (g) E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157 CrossRef CAS PubMed; (h) F. T. Edelmann, Coord. Chem. Rev., 2017, 338, 27 CrossRef CAS; (i) F. T. Edelmann, Coord. Chem. Rev., 2016, 318, 29 CrossRef CAS.
- (a) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239 CrossRef CAS PubMed; (b) F. Basuli, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2003, 42, 8003 CrossRef CAS PubMed; (c) X. Dai, P. Kapoor and T. H. Warren, J. Am. Chem. Soc., 2004, 126, 4798 CrossRef CAS PubMed; (d) E. Kogut, A. Zeller, T. H. Warren and T. Strassner, J. Am. Chem. Soc., 2004, 126, 11984 CrossRef CAS PubMed; (e) H. Y. Gao, W. J. Guo, F. Bao, G. Q. Gui, J. K. Zhang, F. M. Zhu and Q. Wu, Organometallics, 2004, 23, 6273 CrossRef CAS; (f) C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404 CrossRef CAS PubMed; (g) E. A. Gregory, R. J. Lachicotte and P. L. Holland, Organometallics, 2005, 24, 1803 CrossRef CAS; (h) A. M. Reynolds, E. A. Lewis, N. W. Aboelella and W. B. Tolman, Chem. Commun., 2005, 2014 RSC; (i) C. Shimokawa and S. Itoh, Inorg. Chem., 2005, 44, 3010 CrossRef CAS PubMed; (j) G. C. Bai, P. R. Wei, A. K. Das and D. W. Stephan, Dalton Trans., 2006, 1141 RSC; (k) Y. Yu, J. M. Smith, C. J. Flaschenriem and P. L. Holland, Inorg. Chem., 2006, 45, 5742 CrossRef CAS PubMed; (l) Y. M. Yao, Z. Q. Zhang, H. M. Peng, Y. Zhang, Q. Shen and J. Lin, Inorg. Chem., 2006, 45, 2175 CrossRef CAS PubMed; (m) E. C. Brown, B. N. Itsik, J. T. York, N. W. Aboelella and W. B. Tolman, Inorg. Chem., 2007, 46, 486 CrossRef CAS PubMed; (n) Y. C. Tsai, P. Y. Wang, K. M. Lin, S. A. Chen and J. M. Chen, Chem. Commun., 2008, 205 RSC; (o) Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624 CrossRef CAS PubMed; (p) S. Pfirrmann, C. Limberg and B. Ziemer, Dalton Trans., 2008, 6689 RSC; (q) M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Dalton Trans., 2008, 2854 RSC; (r) K. C. MacLeod, J. L. Conway, L. Tang, J. J. Smith, L. D. Corcoran, K. H. D. Ballem, B. O. Patrick and K. M. Smith, Organometallics, 2009, 28, 6798 CrossRef CAS; (s) K. Ding, T. R. Dugan, W. W. Brennessel, E. Bill and P. L. Holland, Organometallics, 2009, 28, 6650 CrossRef CAS; (t) R. E. Cowley, N. J. DeYonker, N. A. Eckert, T. R. Cundari, S. DeBeer, E. Bill, X. Ottenwaelder, C. Flaschenriem and P. L. Holland, Inorg. Chem., 2010, 49, 6172 CrossRef CAS PubMed; (u) K. M. Lin, P. Y. Wang, Y. J. Shieh, H. Z. Chen, T. S. Kuo and Y. C. Tsai, New J. Chem., 2010, 34, 1737 RSC; (v) Z. J. Tonzetich, F. Heroguel, L. H. Do and S. J. Lippard, Inorg. Chem., 2011, 50, 1570 CrossRef CAS PubMed; (w) S. Schulz, J. Spielmann, D. Bläser and C. Wölper, Chem. Commun., 2011, 47, 2676 RSC; (x) R. E. Cowley, N. A. Eckert, S. Vaddadi, T. M. Figg, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 9796 CrossRef CAS PubMed; (y) N. C. Tomson, J. Arnold and R. G. Bergman, Dalton Trans., 2011, 40, 7718 RSC; (z) K. C. Chang, C. F. Lu, P. Y. Wang, D. Y. Lu, H. Z. Chen, T. S. Kuo and Y. C. Tsai, Dalton Trans., 2011, 40, 2324 RSC.
- (a) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48 RSC; (b) H. M. El-Kaderi, M. J. Heeg and C. H. Winter, Organometallics, 2004, 23, 4995 CrossRef CAS; (c) M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042 CrossRef CAS PubMed; (d) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS PubMed; (e) Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed; (f) J. Chai, V. Jancik, S. Singh, H. Zhu, C. He, H. W. Roesky, H. G. Schmidt, M. Noltemeyer and N. S. Hosmane, J. Am. Chem. Soc., 2005, 127, 7521 CrossRef CAS PubMed; (g) Y. X. Cheng, P. B. Hitchcock, M. F. Lappert and M. Zhou, Chem. Commun., 2005, 752 RSC; (h) M. S. Hill, R. Pongtavornpinyo and P. B. Hitchcock, Chem. Commun., 2006, 3720 RSC; (i) A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 4076 CrossRef CAS; (j) A. Jana, H. W. Roesky and C. Schulzke, Inorg. Chem., 2009, 48, 9543 CrossRef CAS PubMed; (k) S. P. Sarish, H. W. Roesky, M. John, A. Ringe and J. Magull, Chem. Commun., 2009, 2390 RSC; (l) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Dalton Trans., 2009, 9715 RSC; (m) W. Y. Wang, S. Inoue, S. L. Yao and M. Driess, Chem. Commun., 2009, 2661 RSC; (n) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed; (o) M. R. Crimmin, M. S. Hill, P. B. Hitchcock and M. F. Mahon, New J. Chem., 2010, 34, 1572 RSC; (p) G. G. Sandra, J. Vojtech, A. D. R. Alma and M. C. Monica, Inorg. Chem., 2011, 50, 4226 CrossRef PubMed; (q) S. I. Diego, J. V. H. Miriam, H. L. Raul and J. Vojtech, Inorg. Chem., 2011, 50, 8907 CrossRef PubMed; (r) M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, K. K. Gabriele and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS; (s) F. Drouin, T. J. J. Whitehorne and F. Schaper, Dalton Trans., 2011, 40, 1396 RSC; (t) A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659 RSC.
- (a) P. G. Hayes, W. E. Piers and R. McDonald, J. Am. Chem. Soc., 2002, 124, 2132 CrossRef CAS PubMed; (b) P. G. Hayes, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2003, 125, 5622 CrossRef CAS PubMed; (c) O. Eisenstein, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert, L. Maron, L. Perrin and A. V. Protchenko, J. Am. Chem. Soc., 2003, 125, 10790 CrossRef CAS PubMed; (d) A. G. Avent, C. F. Caro, P. B. Hitchcock, M. F. Lappert, Z. Li and X. H. Wei, Dalton Trans., 2004, 1567 RSC; (e) L. K. Knight, W. E. Piers, P. Fleurat-Lessard, M. Parvez and R. McDonald, Organometallics, 2004, 23, 2087 CrossRef CAS; (f) P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2005, 951 RSC; (g) P. G. Hayes, W. E. Piers and M. Parvez, Organometallics, 2005, 24, 1173 CrossRef CAS; (h) S. Bambirra, H. Tsurugi, D. Leusen and B. Hessen, Dalton Trans., 2006, 1157 RSC; (i) S. K. Ibrahim, A. V. Khvostov, M. F. Lappert, L. Maron, L. Perrin, C. J. Pickett and A. V. Protchenko, Dalton Trans., 2006, 2591 RSC; (j) R. Jiao, X. D. Shen, M. Q. Xue, Y. Zhang, Y. M. Yao and Q. Shen, Chem. Commun., 2010, 46, 4118 RSC; (k) M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert and A. V. Protchenko, Dalton Trans., 2010, 39, 6426 RSC; (l) E. L. Lu, J. X. Chu, Y. F. Chen, M. V. Borzov and G. Y. Li, Chem. Commun., 2011, 47, 743 RSC; (m) W. Q. Mao, L. Xiang, C. A. Lamsfus, L. Maron, X. B. Leng and Y. F. Chen, J. Am. Chem. Soc., 2017, 139, 1081 CrossRef CAS PubMed; (n) M. Q. Xue, Y. Zheng, Y. B. Hong, Y. M. Yao, F. Xu, Y. Zhang and Q. Shen, Dalton Trans., 2015, 44, 20075 RSC.
- (a) Y. M. Yao, Y. Zhang, Q. Shen and K. B. Yu, Organometallics, 2002, 21, 819 CrossRef CAS; (b) Y. M. Yao, Y. Zhang, Z. Q. Zhang, Q. Shen and K. B. Yu, Organometallics, 2003, 22, 2876 CrossRef CAS; (c) F. Basuli, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Organometallics, 2003, 22, 4705 CrossRef CAS; (d) D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2005, 690, 5182 CrossRef CAS; (e) D. V. Vitanova, F. Hampel and K. C. Hultzsch, Dalton Trans., 2005, 1565 RSC; (f) M. Q. Xue, Y. M. Yao, Q. Shen and Y. Zhang, J. Organomet. Chem., 2005, 690, 4685 CrossRef CAS; (g) Y. L. Duan, J. X. He, W. Wang, J. J. Zhou, Y. Huang and Y. Yang, Dalton Trans., 2016, 26, 10807 RSC; (h) Y. M. Yao, Z. Q. Zhang, H. M. Peng, Y. Zhang, Q. Shen and J. Lin, Inorg. Chem., 2006, 45, 2175 CrossRef CAS PubMed; (i) X. Xu, X. Y. Xu, Y. F. Chen and J. Sun, Organometallics, 2008, 27, 758 CrossRef CAS; (j) M. Schmid, S. M. Guillaume and P. W. Roesky, Organometallics, 2014, 33, 5392 CrossRef CAS; (k) P. B. Hitchcock, A. V. Khvostov, M. F. Lappert and A. V. Protchenko, Dalton Trans., 2009, 2383 RSC; (l) E. L. Lu, Y. X. Li and Y. F. Chen, Chem. Commun., 2010, 46, 4469 RSC; (m) E. L. Lu, W. Gan and Y. F. Chen, Dalton Trans., 2011, 40, 2366 RSC; (n) C. Wang, J. L. Zhou, X. F. Zhao, L. Maron, X. B. Leng and Y. F. Chen, Chem.–Eur. J., 2016, 22, 1258 CrossRef CAS PubMed; (o) S. V. Klementyeva, M. Y. Afonin, A. S. Bogomyakov, P. W. Roesky and S. N. Konchenko, Eur. J. Inorg. Chem., 2016, 3666 CrossRef CAS.
- H. Miao, S. L. Zhou, S. W. Wang, L. J. Zhang, Y. Wei, S. Yang, F. H. Wang, Z. Chen, Y. Chen and Q. B. Yuan, Sci. China: Chem., 2013, 56, 329 CrossRef CAS.
- (a) D. Enders, A. Saint-Dizier, M. I. Lannou and A. Lenzen, Eur. J. Org. Chem., 2006, 29 CrossRef CAS; (b) A. D. Sadow and A. Togni, J. Am. Chem. Soc., 2005, 127, 17012 CrossRef CAS PubMed; (c) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS PubMed; (d) D. Zhao, Y. Yuan, A. S. C. Chan and R. Wang, Chem.–Eur. J., 2009, 15, 2738 CrossRef CAS PubMed.
- (a) X. Fu, Z. Jiang and C.-H. Tan, Chem. Commun., 2007, 47, 5058 RSC; (b) S. G. Wen, P. F. Li, H. B. Wu, F. Yu, X. M. Liang and J. X. Ye, Chem. Commun., 2010, 46, 4806 RSC; (c) Q. B. Yuan, S. L. Zhou, X. C. Zhu, Y. Wei, S. W. Wang, X. L. Mu, F. S. Yao, G. C. Zhang and Z. Chen, New J. Chem., 2015, 39, 7626 RSC; (d) K. Yamakoshi, S. J. Harwood, M. Kanai and M. Shibasaki, Tetrahedron Lett., 1999, 40, 2565 CrossRef CAS.
- (a) S. Yang, X. C. Zhu, S. L. Zhou, S. W. Wang, Z. J. Feng, Y. Wei, H. Miao, L. P. Guo, F. H. Wang, G. C. Zhang, X. X. Gu and X. L. Mu, Dalton Trans., 2014, 43, 2521 RSC; (b) Q. B. Yuan, S. L. Zhou, X. C. Zhu, Y. Wei, S. W. Wang, X. L. Mu, F. S. Yao, G. C. Zhang and Z. Chen, New J. Chem., 2015, 39, 7626 RSC; (c) X. X. Gu, L. J. Zhang, X. C. Zhu, S. W. Wang, S. L. Zhou, Y. Wei, G. C. Zhang, X. L. Mu, Z. M. Huang, D. J. Hong and F. Zhang, Organometallics, 2015, 34, 4553 CrossRef CAS; (d) X. X. Gu, X. C. Zhu, Y. Wei, S. W. Wang, S. L. Zhou, G. C. Zhang and X. L. Mu, Organometallics, 2014, 33, 2372 CrossRef CAS; (e) F. H. Wang, S. W. Wang, X. C. Zhu, S. L. Zhou, H. Miao, X. X. Gu, Y. Wei and Q. B. Yuan, Organometallics, 2013, 32, 3920 CrossRef CAS; (f) S. L. Zhou, Z. S. Wu, J. W. Rong, S. W. Wang, G. S. Yang, X. C. Zhu and L. J. Zhang, Chem.–Eur. J., 2012, 18, 2653 CrossRef CAS PubMed; (g) Y. J. Wu, S. W. Wang, L. J. Zhang, G. S. Yang, X. C. Zhu, Z. H. Zhou, H. Zhu and S. H. Wu, Eur. J. Org. Chem., 2010, 326 CrossRef; (h) X. C. Zhu, J. X. Fan, Y. J. Wu, S. W. Wang, L. J. Zhang, G. S. Yang, Y. Wei, C. W. Yin, H. Zhu, S. H. Wu and H. T. Zhang, Organometallics, 2009, 28, 3882 CrossRef CAS; (i) L. J. Zhang, H. P. Wu, S. P. Su and S. W. Wang, Chin. J. Chem., 2009, 27, 2061 CrossRef CAS; (j) Y. J. Wu, S. W. Wang, L. J. Zhang, G. S. Yang, X. C. Zhu, C. Liu, C. W. Yin and J. W. Rong, Inorg. Chim. Acta, 2009, 362, 2814 CrossRef CAS; (k) Y. J. Wu, S. W. Wang, X. C. Zhu, G. S. Yang, Y. Wei, L. J. Zhang and H. B. Song, Inorg. Chem., 2008, 47, 5503 CrossRef CAS PubMed; (l) Q. H. Li, S. W. Wang, S. L. Zhou, G. S. Yang, X. C. Zhu and Y. Y. Liu, J. Org. Chem., 2007, 72, 6763 CrossRef CAS PubMed; (m) S. L. Zhou, S. W. Wang, G. S. Yang, Q. H. Li, L. J. Zhang, Z. J. Yao, Z. K. Zhou and H. B. Song, Organometallics, 2007, 26, 3755 CrossRef CAS; (n) L. J. Zhang, S. W. Wang, S. L. Zhou, G. S. Yang and E. H. Sheng, J. Org. Chem., 2006, 71, 3149 CrossRef CAS PubMed; (o) L. J. Zhang, S. W. Wang, E. H. Sheng and S. L. Zhou, Green Chem., 2005, 7, 683 RSC; (p) S. L. Zhou, S. W. Wang, G. S. Yang, X. Y. Liu, E. H. Sheng, K. H. Zhang, L. Cheng and Z. X. Huang, Polyhedron, 2003, 22, 1019 CrossRef CAS; (q) M. Nishiura, F. Guo and Z. M. Hou, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed; (r) Y. D. Lv, J. L. Zhou, X. B. Leng and Y. F. Chen, New J. Chem., 2015, 39, 7582 RSC.
- X. H. He, Y. Z. Yao, X. Luo, J. Zhang, Y. Liu, L. Zhang and Q. Wu, Organometallics, 2003, 22, 4952 CrossRef CAS.
- G. M. Sheldrick, SADABS: Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL 5.10 for Windows NT: Structure Determination Software Programs, Bruker Analytical X-ray Systems, Inc., Madison, WI, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization spectra (1H NMR and 13C NMR) for complexes 1d, 2e and compounds 4a–n. CCDC 1553833–1553836 for 1a–d. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07565k |
| This journal is © The Royal Society of Chemistry 2017 |