
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of high DMSO concentration on albumin during freezing and vitrification†
Sampreeti Jena and
Alptekin Aksan *
*
Biostabilization Laboratory, Mechanical Engineering Department, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: aaksan@umn.edu
First published on 11th September 2017
Abstract
IR spectroscopy was used to probe the composition of the freeze concentrated liquid (FCL) during near equilibrium freezing of albumin–dimethyl sulfoxide (DMSO) solutions down to −70 °C. While cooling down to −32 °C, preferential entrapment of DMSO within ice occurred, which was attributed to formation of DMSO–water complexes. These complexes subsequently collapsed on cooling below −32 °C with albumin being preferentially entrapped in ice. Heterogeneity induced in the FCL as a result, was more pronounced in solutions containing lower solute concentrations and thus larger volume fractions of ice. Crystallization of DMSO was not observed on cooling below its eutectic temperature and the FCL vitrified instead at −58 °C, as confirmed by differential scanning calorimetry and low temperature X-ray crystallography. The increase in viscosity of the FCL upon glass transition prevented further compositional changes. Isothermal measurements confirmed that preferential exclusion of either species from ice occurred solely during advance of the ice interfaces and was hence governed by its relative diffusivity. Adverse effects of high DMSO concentrations on the protein were observed between −35 °C and −55 °C and devitrification during thawing proved to be even more detrimental in vitrified samples. Thawing rate had a significant effect on the extent of structural unfolding of the protein in vitrified samples, but not on the ones which were simply frozen. These results will be relevant to vitrification of solutions containing proteins, cells and small tissue systems as such as monocytes, oocytes, ova, embryos, zygotes, vein rings/segments and pancreatic islets.
Introduction
Feasibility of long-term preservation for isolated cells, tissues, and organs with high post-storage viability and function has been a topic of interest for a long time.1–4 Recently rejuvenated interest in this area has mainly been fuelled by the growing demand for vital (e.g. liver, heart and kidneys), and non-vital organs (e.g. ovaries, uterus and eyes) for transplantation.2,3 Although cryopreservation has achieved some success in preserving isolated cells and certain non-vital tissues, attempts at freezing and storing complex organs such as the heart, liver or kidneys have generally resulted in failure.5,6Vitrification, a kinetic phenomenon that occurs during supercooling below a characteristic temperature (called the glass transition temperature), is believed to have the potential to protect and stabilize cells and tissues by entrapping them in a very viscous glass, without exposing them to the detrimental effects of ice.7,8 If cooled sufficiently fast (approximately, at 1 million °C s−1), even pure water can be vitrified, avoiding crystallization. However, reaching a cooling rate this high has not been technically feasible for specimens larger than a couple of micrometres in size.
In research studies conducted for vitrification of tissues and organs, very high concentrations of specialty chemicals (vitrification agents) are incorporated into the solution to ensure that ice crystallization can be inhibited at cooling rates that are technically achievable.8 For example, the vitrification protocols developed for isolated mammalian cells and very small tissues only require cooling rates around 1000–1400 °C min−1, usually attained by immersion in liquid or slush nitrogen.9,10 However, vitrification of large tissues, and organs necessitates achieving vitrification at technically less challenging cooling rates of 0.1–5 °C min−1 (ref. 8 and 11) to also ensure spatial uniformity of temperature history within the sample and to minimize freeze/thaw induced mechanical stresses.12 The main issue is that avoiding ice nucleation during low cooling rates require presence of very high concentrations of vitrification agents. For example, the vitrification solutions (such as VS1, VS4, M22) proposed for kidneys, ovaries, and the liver are composed of glycerol, ethylene glycol, and dimethyl sulfoxide (DMSO) at a total concentration as high as 50% w/w.13–18
During slow cooling following ice nucleation, a growing ice phase excludes the non-crystallizing chemicals in the solution, forming a freeze concentrated liquid (FCL). As the FCL gets more concentrated with decreasing temperature, its glass transition temperature (T′g) increases and the solution eventually vitrifies when its temperature reaches the T′g of the FCL.19,20 Combined with low temperatures and lengthy exposure times, presence of high concentrations of vitrification agents could have very detrimental effects on the biological macromolecules present in the solution, by inducing severe mechanical, thermal, and osmotic stresses and through other specific chemical interactions resulting in protein denaturation, membrane injury, and oxidative damage.21,22 This is believed to be one of the major reasons why highly viable large tissues and functional organs could not be recovered after cryopreservation and vitrification, while partial success could only be claimed for non-vital small tissues such as ovaries,23 blood vessels,24 heart valves,25 and corneas.16,26 Even though kidney and heart have been the most widely researched organs, neither (with the exception of rabbit kidneys17,27) has recovered functionality post-thaw in a reproducible fashion.28
Earlier studies have utilized confocal Raman and IR spectroscopy to study freezing induced micro-heterogeneity in the FCL and specific ice–solute interactions with corresponding changes in protein structure.29–31 Twomey et al.,32 for example, have detected significant changes in the relative ratio of albumin to DMSO in the FCL after slow freezing of aqueous albumin–DMSO solutions.32 However, no study to date has explored the changes in the FCL in situ during slow cooling and vitrification, identifying the effects of kinetic and thermodynamic transitions of the FCL on the macromolecules suspended within.
In this study, we explored the effect of slow freezing on solutions containing high concentrations of DMSO (20–30% w/w) and albumin, by simultaneously quantifying the extent of association of DMSO and albumin with the ice phase using IR spectroscopy and characterizing the thermo-physical state of the FCL (supercooled, vitrified vs. crystalline) in situ at cryogenic temperatures. In parallel experiments, we have characterized the formation and evolution of the amorphous, and crystalline phases in the FCL using Differential Scanning Calorimetry (DSC) and Low Temperature X-ray Diffraction (XRD), respectively. The tertiary and secondary structures of albumin in fresh, vitrified and thawed solutions were quantified using Circular Dichroism Spectroscopy. These studies have revealed that under specific circumstances, achieving vitrification may not necessarily result in the most favourable outcome, which has very important implications in vitrification of large tissues and organs.
Experimental
Materials and methods
Experimental solutions were prepared gravimetrically using DMSO (99.9% purity, Sigma-Aldrich, St. Louis, MO), bovine serum albumin (MW 66.5 kDa, ≥99% purity, Sigma-Aldrich, St. Louis, MO), and ultrapure water (UPW).Fourier transform infrared spectroscopy (FTIR)
Fourier transform infrared (FTIR) spectroscopy was used to study the composition of the freeze concentrated liquid (FCL) in frozen samples. The advantages of FTIR include a higher signal to noise ratio and the ease of in situ spectra collection. The experimental solution (∼100 nL) was sandwiched between two CaF2 windows, sealed with vacuum grease to generate a thin film, ∼2 μm thick. The assembly was then transferred to an infrared microscope attached to the FTIR spectrometer (Thermo-Nicolet Continuum with a mercury cadmium telluride detector, Thermo Electron, Waltham, MA) equipped with a freeze-drying cryostage (FDCS 196, Linkam Scientific Instruments Ltd., UK). Spectra were collected at a resolution of 4 cm−1 and a total of 128 IR scans were averaged for each spectrum to be analyzed in the 4000–930 cm−1 wavenumber range. The IR spectra were analysed using OMNIC (Thermo-Nicolet) software.Aqueous solutions tend to supercool significantly and ice crystallization rarely occurs at the equilibrium freezing temperature. In order to ensure repeatability, a method was devised to control ice nucleation: samples were cooled rapidly at 30 °C min−1 down to the homogenous nucleation temperature (∼−50 °C)33 and then warmed back up to the equilibrium melting temperature at 1 °C min−1 while the sample was continuously visualized through the microscope. When only a few small ice crystals remained in the melting sample, heating was stopped and the sample was re-cooled at a slower rate (0.1–0.3 °C min−1) to avoid concentration variations within the sample due to diffusional limitations and to ensure near-equilibrium freezing. To model popularly used vitrification solutions,13,16 20%, and 30% w/w DMSO solutions containing 6.66 (66.6 mg mL−1) and 10% w/w albumin (100 mg mL−1), respectively, were prepared in ultrapure water. The initial albumin to DMSO mass ratio in all experimental solutions was 0.33 before freezing in accordance with DMSO–protein formulations used in existing studies.32 During slow cooling, IR spectra were collected from 7 different regions within the FCL phase of each sample, while the temperature was held constant at 2 °C intervals between the equilibrium freezing temperature and −70 °C. To explore the effects of cooling rate, 30% DMSO + 10% BSA samples were cooled down at different rates (0.1 °C min−1, 0.3 °C min−1, 0.5 °C min−1 and 0.7 °C min−1) after controlled ice nucleation (as described above). In these experiments, spectra were collected from 7 different regions of the FCL while the temperature was held constant at 5 °C intervals as the samples was cooled down to −53 °C. Note that at each hold temperature, we waited 30 minutes before collecting spectra in the FCL to ensure the sample equilibrated.
To observe temporal changes in the FCL during isothermal hold, time-lapse IR spectra were collected from a fixed region in the FCL for up to 80 minutes: in the first set of experiments, 30% DMSO + 10% BSA solutions cooled at 0.3 °C min−1 (after controlled ice nucleation) were held at different temperatures (−15 °C, −20 °C, −25 °C, and −35 °C) while time-lapse IR spectra were collected. In the second set of experiments, solutions of identical composition were cooled at 0.1 °C min−1, 0.3 °C min−1 and 0.5 °C min−1 (after controlled ice nucleation) and were held at −15 °C while IR spectra were collected. All experiments were conducted in triplicate.
Differential scanning calorimetry (DSC)
A differential scanning calorimeter (Q2000, TA Instruments, New Castle, DE) equipped with a cooling accessory was used in all experiments. The instrument was calibrated using the melting point of tin. Approximately 50 mg of the sample solution was placed in an aluminium pan, which was sealed with a pin-holed lid. Dry nitrogen was used as the purge gas at a flow rate of 50 mL min−1. 30% DMSO + 10% BSA samples were cooled rapidly at 30 °C min−1 down to −60 °C (below the homogenous nucleation temperature), and warmed back up to −13 °C at 1 °C min−1, close to the equilibrium melting temperature (−11 °C). The samples were then cooled very slowly at a rate of 0.3 °C min−1 down to −80 °C and rapidly heated back to room temperature at 10 °C min−1. The DSC data were analyzed using TA Universal Analysis software. Sample solutions were analyzed in duplicate.Powder X-ray diffractometry (XRD)
A powder X-ray diffractometer (D8 ADVANCE; Bruker AXS, Madison, WI) equipped with a variable temperature stage (TTK 450; Anton Paar, Graz-Straßgang, Austria) and a Si strip one-dimensional detector (LynxEye; Bruker AXS, Madison, WI) was used. A 30% DMSO + 10% BSA solution was subjected to a temperature history identical to the one used for DSC analysis. XRD patterns were collected continuously with a dwell time of 900 s during slow cooling at 0.3 °C min−1 down to −80 °C. Solutions were exposed to Cu Kα radiation (40 kV × 40 mA) over an angular range of 5–27° 2θ with a step size of 0.05°. The XRD patterns were analyzed using commercially available software (JADE 2010). Solutions were analyzed in duplicate.UV circular dichroism (CD)
Spectra were recorded using a J-815 circular dichroism spectropolarimeter (JASCO) using a 1 cm path length quartz cuvette over the range of 190–310 nm at ambient temperature. Data were collected every 0.2 nm with a bandwidth of 1 nm, averaged over 8 scans. An integration time of 8 seconds at 50 nm min−1 and a 2 nm slit width were used. The solutions were diluted with ultrapure water to reach a protein concentration of 0.1 mg mL−1. The background spectra of the solvent medium (30% DMSO (w/w) after identical dilution) were subtracted from the final spectra and data was obtained as mean residue ellipticity (Ɵ). To estimate protein secondary structure content, analysis of the relevant CD spectra was carried out using the CDPro software. The basis set 7 of the CDPro software was used. Analysis was performed using the SELCON 3 method.34Separate solutions of composition 30% DMSO + 10% BSA (w/w) were subjected to controlled ice nucleation followed by slow cooling at 0.3 °C min−1 down to −35 °C, −55 °C or −65 °C in the cryostage of the FTIR microscope. The solutions were thawed under ambient conditions (from −35 °C, −55 °C or −55 °C) back up to room temperature. Different samples of the same composition were cooled at the same rate (0.3 °C min−1), but rapidly thawed in the cryo-stage at 20 °C min−1. Protein structure in fresh and thawed solutions were analyzed by CD and compared. Solutions were analyzed in duplicate.
Results and discussion
DMSO is a polar organic compound, and therefore depresses the freezing temperature of water by distorting its hydrogen bonding, inhibiting ice formation.35 Furthermore, it has been shown that DMSO toxicity is directly proportional to the average strength of hydrogen bonding between its polar groups and the intracellular water.17 Therefore, it is crucial to understand how DMSO–water interactions change during freezing, vitrification and thawing of an aqueous protein solution.Concentrations of DMSO and albumin in the FCL at any temperature during cooling were calculated using the baseline corrected areas of the amide II band of albumin (1525–1570 cm−1),36 (ν2 + ν3) combination band of water in the near IR region (4500–5500 cm−1)37,38 and the ν-HCH band (1367–1485 cm−1) of DMSO.39,40 Note that concentration-dependent calibration curves (constructed by varying the amount of albumin in a 30% (w/w) DMSO solution and varying the amount of DMSO in a 30% (w/w) albumin solution) at room temperature are included as ESI data (Fig. S1 and S2†). All the calibration curves constructed were linear in the range analyzed. IR spectra were acquired below −11 °C during near-equilibrium cooling of the 20% DMSO + 6.67% BSA solution and below −13 °C for the 30% DMSO + 10% BSA solution (2 °C below their respective equilibrium freezing temperatures).
The mass ratio of albumin to DMSO (R) in the FCL as a function of temperature during near equilibrium freezing is shown in Fig. 1 for 20% and 30% w/w DSMO solutions containing albumin. Note that at room temperature (before freezing), R was equal to 0.33 in both samples. Distinct changes in the composition of the freeze concentrate (FCL) was observed. While cooling down to approximately −33 °C (Region I in Fig. 1), an increase in R was observed, suggesting preferential entrapment of DMSO in the growing ice phase. This was attributed to association of the DMSO molecules with the surrounding water molecules through hydrogen bonding at ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (ref. 41 and 42) or 1:3 (ref. 35) (DMSO:water), forming clusters. These clusters have significantly reduced diffusivity (by a factor of 30)43,44 and higher affinity for ice (hydrating water molecules reduce the effective hydrophobicity of DMSO) compared to free DMSO molecules and therefore are more likely to be entrapped in an advancing ice interface.
:2 (ref. 41 and 42) or 1:3 (ref. 35) (DMSO:water), forming clusters. These clusters have significantly reduced diffusivity (by a factor of 30)43,44 and higher affinity for ice (hydrating water molecules reduce the effective hydrophobicity of DMSO) compared to free DMSO molecules and therefore are more likely to be entrapped in an advancing ice interface.
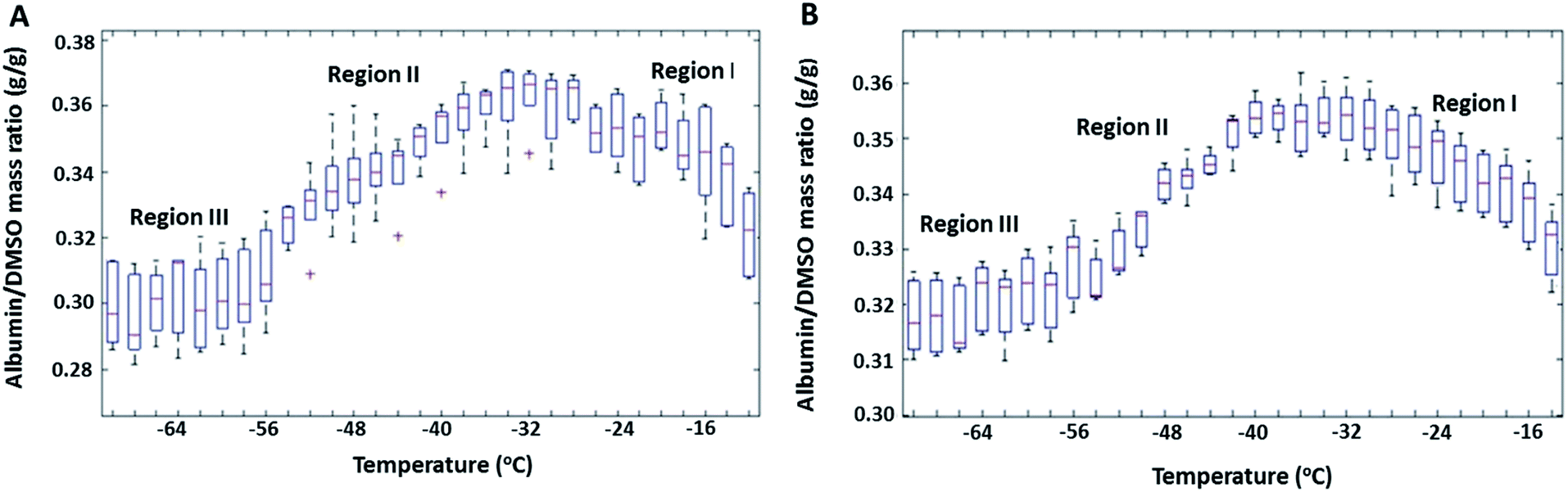 | ||
| Fig. 1 Albumin/DMSO mass ratios (R) measured at 7 different regions of the freeze concentrated liquid as a function of decreasing temperatures during near equilibrium freezing (at 0.3 °C min−1) of (A) 20% DMSO + 6.66% BSA (B) 30% DMSO + 10% BSA solutions. | ||
To gain insight into the hydrogen bonding dynamics in the FCL, the (ν2 + ν3) combination band of water in the NIR region consisting of the δ-OH (i.e., ν2) and the ν-OH (i.e., ν3) bands38,45 was probed. In the presence of DMSO, the band was composed of 2 peaks (Fig. 2A). The higher frequency peak (henceforth referred to as S1) located at 5116 cm−1 was previously attributed to water molecules with one active hydrogen bond (HB), (single HB donor).46 The second peak (henceforth referred to as S*2) located at 4800 cm−1 was close to the location of a peak previously observed in neat water (4920 cm−1). This peak was attributed to the water molecules that have coordination numbers of 3–4, and are simultaneously HB donors and acceptors.46,47 Appearance of the S*2 peak in the (ν2 + ν3) combination band of water confirmed the formation of the DMSO–water clusters in the FCL. Peaks in the neighbourhood of S*2 have been reported in the literature for solutions containing a kosmotrope such as trehalose and are attributed to a new organization in water HB network due to the presence of the kosmotrope.48,49 Location of the S*2 peak at a lower frequency (4800 cm−1) than the S1 peak (5116 cm−1) suggested stronger HB within the DMSO–water clusters.50 In neat water, a high frequency peak has been detected at 5232 cm−1 originating from HB acceptor free water molecules.38,46 This band was entirely absent in solutions containing DMSO, indicating that all water molecules in the FCL were simultaneously HB donors and acceptors.
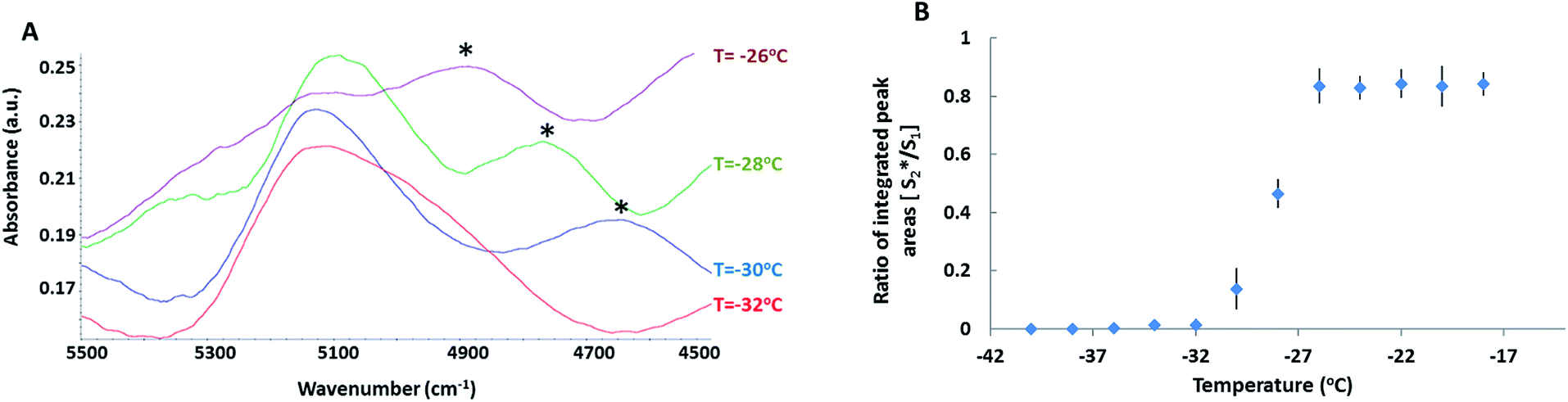 | ||
| Fig. 2 (A) (ν2 + ν3) NIR band of water at a fixed region in the FCL during near equilibrium freezing (at 0.3 °C min−1) of 30% DMSO + 10% BSA solution (* denotes the S*2 peak arising due to DMSO–water cluster formation). (B) Ratio of the integrated areas of S1, and S*2 peaks after deconvolution of the (ν2 + ν3) band at a fixed region of the FCL as a function of temperature during near equilibrium freezing of 30% DMSO + 10% BSA solution (n = 3). | ||
The ratio of the integrated, baseline corrected areas of S*2 and S1 peaks were plotted as a function of decreasing temperature (Fig. 2B). The peak ratio did not change during cooling down to −28 °C. However, below −28 °C, the area ratio decreased rapidly. Below −32 °C, the S*2 peak disappeared completely. Thus DMSO–water clusters collapsed between −28 °C and −32 °C and below −32 °C, water molecules were only engaged in self-association (and possibly with protein molecules). The observed decrease in the population of water–DMSO clusters agreed with previously reported MD simulations, which predicted thermodynamic destabilization of DMSO–water HB complexes in binary solutions when mole fraction of DMSO exceeded 0.5.42,51,52 Another interesting observation was that within this temperature range, the S*2 peak shifted to lower frequencies. This could be caused by the increase in the strength of HB and/or change in the water to DMSO ratio in the HB complexes. MD simulations predict a transition from 1:2 and 1:3 (DMSO:water) HB complexes to 2:1 complexes followed by complete dissociation of all complexes (disordering) with progressively increasing mole fraction of DMSO in the solution.42,51
While cooling from approximately −33 °C down to −59 °C (Region II in Fig. 1), R decreased, suggesting preferential entrapment of albumin in ice. Collapse of DMSO–water clusters below −32 °C resulted in DMSO to exist as free molecules in the FCL. Free DMSO in water is known to have a higher diffusion coefficient than albumin (by a factor of 40).43 Besides, denaturation of albumin, shown to occur between −35 °C and −55 °C by post thaw CD analysis (discussed later), is associated with increased solvation of the unfolded protein.53 High concentrations of unfolded albumin (18% w/w at −32 °C) in the FCL is likely to cause self-association.54 The aforementioned factors lead to a larger hydrodynamic radius and reduced diffusivity of albumin, thereby increasing its tendency to be entrapped in ice, as compared to DMSO. The decrease in R during equilibrium freezing continued down to approximately −59 °C, a temperature below which (Region III in Fig. 1) ice growth was arrested and no further compositional change could be observed in the FCL with cooling.
Albumin and DMSO concentrations in the FCL as determined from the IR spectra were subtracted from concentrations predicted by the DMSO–water phase diagram to calculate the amounts of both species entrapped in the growing ice phase. In the 30% DMSO + 10% BSA solution, 4.69 mg of DMSO per g of water in the FCL was found to be entrapped in ice in Region I, while 1.48 mg of DMSO per g of water in the FCL was found to be entrapped in Region II. On the other hand, the amounts of albumin entrapped within the ice phase in the same regions were calculated to be 2.84 mg (in Region I), and 4.16 mg (in Region II) per g of water in the FCL, respectively. In the 20% DMSO + 6.67% BSA sample, approximately, 7.12 mg, and 1.85 mg of DMSO per g of water in the FCL were entrapped in ice in Regions I and II, respectively. In the same solution, 4.56 mg, and 6.27 mg of albumin per g of water in the FCL were entrapped in ice in Regions I and II. Variation in the R values calculated from 7 spatially different regions in the FCL were calculated as % of the mean R value at each hold temperature and were used as a measure of spatial heterogeneity in the frozen sample. In the 20% DMSO + 6.67% BSA sample, variation was 4–5% in Region I, 2–3% in Region II and 4–5% in Region III. The corresponding values calculated for 30% DMSO + 10% BSA sample were 2–3% in Region I, 0.5–2% in Region II, and 2–3% in Region III. The amounts of albumin and DMSO contained in ice were greater in the 20% DMSO + 6.67% BSA sample than in the 30% DMSO + 10% BSA sample. This was attributed to the larger volume fraction of ice present at any given temperature in the more dilute solution.35,55 Larger ice interface area in contact with the FCL is likely to increase the extent of association of DMSO and albumin with ice. Additionally, FCL of the 20% DMSO + 6.67% BSA solution was more heterogeneous spatially at any given hold temperature than the 30% DMSO + 10% BSA solution due to the reasons mentioned above.
Mole fraction of DMSO in the FCL during equilibrium freezing of binary DMSO–water solutions follows the liquidus line in the phase diagram (Fig. S3†) and thus should have a known and fixed value at a given temperature irrespective of initial DMSO concentration. But the volume of ice is larger in more dilute solutions. Identical trends were observed with decreasing temperature in the 20% DMSO + 6.67% BSA and 30% DMSO + 10% BSA (w/w) solutions suggesting that the nature of preferential association of albumin and DMSO with ice was dictated by the composition of the freeze concentrate (mole fractions of albumin and DMSO). Twomey et al. reported preferential exclusion of albumin during freezing to temperatures below −8 °C and preferential exclusion of DMSO above −8 °C in similar slow freezing studies.32 In those experiments, composition of the FCL were measured only at the end of freezing and compared to solutions at room temperature, as opposed to our method of determining FCL composition at regular temperature intervals throughout freezing.
The effect of cooling rate on FCL composition during slow freezing was also investigated (Fig. 3) in 30% DMSO + 10% BSA (w/w) solutions. Preferential exclusion of either species from ice, reflected by changes in their concentration ratio in the FCL (R), was observed only during slow cooling (rates ≤0.3 °C min−1) (Fig. 3A and B). At faster cooling rates (0.5, and 0.7 °C min−1) (Fig. 3C and D), no significant change was observed in R. It is plausible that faster rate of propagation of the ice interface resulted in non-equilibrium conditions and arbitrary exclusion of solutes from the ice phase, thereby negating the effects of diffusivity and selective interactions of the excluded species with the ice interface.
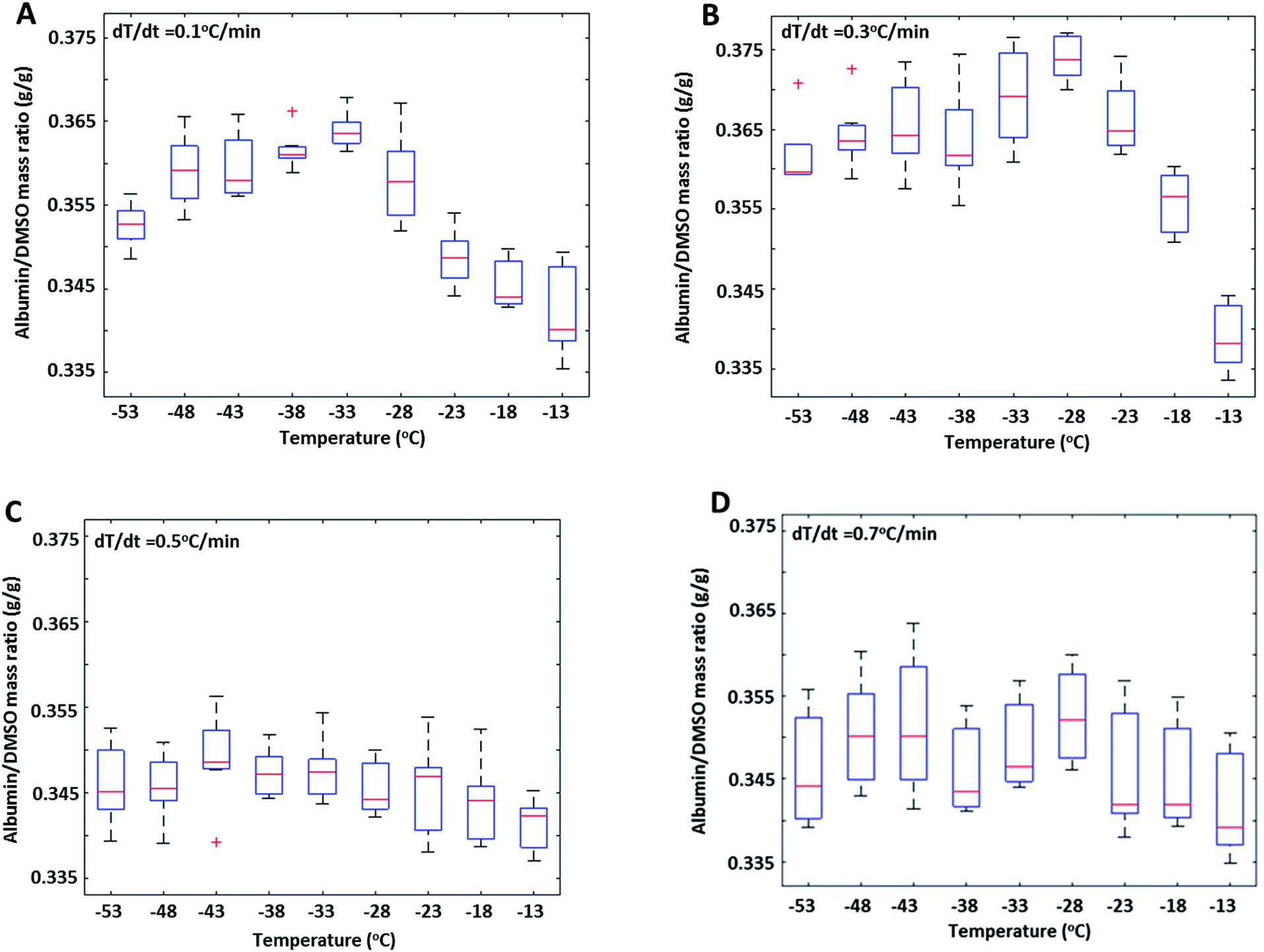 | ||
| Fig. 3 Albumin/DMSO mass ratios (R) measured at 7 different regions of the freeze concentrated liquid as a function of decreasing temperatures during near equilibrium freezing of 30% DMSO + 10% BSA solutions at cooling rates of (A) 0.1 °C min−1 (B) 0.3 °C min−1 (C) 0.5 °C min−1 and, (D) 0.7 °C min−1. | ||
Amide I band of proteins is commonly used to investigate their secondary structures.56,57 However, it could not be used in our studies as it overlaps with the bending peak of water. Far-UV circular dichroism (190–250 nm) is sensitive to the secondary conformation of the protein backbone. On the other hand, near-UV region (250–300 nm) of the CD spectra gives information on the tertiary structure. In the native state, albumin is known to comprise of 50–54% α-helices, 6–8% β-sheets, 14–15% turns and 24–25% random structures.58 The far UV region includes ellipticity contributions arising from several aromatic residues at characteristic wavelengths; phenylalanine and disulphide bonds at 262 nm; tryptophan, close to 300 nm and tyrosine around 280 nm.
Secondary structure content of albumin in freshly prepared (unfrozen) solutions and frozen/thawed solutions that were cooled down to −35 °C, were in good agreement with the native state conformation reported in the literature58 (Tables 1 and 2). There was a slight change in the near UV ellipticity values at 262, 280 and 300 nm, which can be assumed to be negligible. This indicated that BSA retained its native structure at room temperature and during slow freezing down to −35 °C. For mammalian cells, DMSO cytotoxicity has been reported to occur at concentrations as low as 1–5% w/w.59,60 On the other hand, for proteins, detrimental effects of DMSO are protein-dependent and were only observed at very high concentrations (>30% w/w) at room temperature.61,62 Post-thaw CD measurements revealed that BSA in the presence of DMSO was stable during slow freezing down to temperatures as low as −35 °C, despite the DMSO concentration in the FCL having increased to approximately 45% w/w (Fig. S3†). In frozen protein formulations, hydrophobic organic solvents such as DMSO are known to be excluded from the immediate vicinity of the protein molecules (preferential exclusion principle).53 Especially, proteins such as such as BSA and lysozyme with ionic surface groups are tightly bound to a hydrating water layer, preventing direct contact with DMSO.
| Sample | H (r) | H (d) | S (r) | S (d) | Turns | Unordered |
|---|---|---|---|---|---|---|
| a H (r) → regular helix, H (d) → distorted helix, S (r) → regular sheet, S (d) → distorted sheet. Frozen solutions were cooled at 0.3 °C min−1 after controlled nucleation. All samples were analyzed in duplicate. | ||||||
| Fresh | 33.6 ± 0.4 | 18.3 ± 0.3 | 4.9 ± 0.4 | 5.1 ± 0.6 | 15.2 ± 0.5 | 25.2 ± 0.3 |
| Frozen to −35 °C | 33.5 ± 0.8 | 17.9 ± 0.6 | 4.6 ± 0.5 | 5.0 ± 0.7 | 14.6 ± 0.3 | 26.0 ± 0.2 |
| Frozen to −55 °C | 24.1 ± 0.2 | 13.3 ± 0.6 | 4.1 ± 0.7 | 5.2 ± 0.5 | 20.1 ± 0.5 | 32.3 ± 0.4 |
| Frozen to −65 °C | 19.2 ± 0.5 | 11.5 ± 0.8 | 5.2 ± 0.2 | 5.1 ± 0.6 | 23.2 ± 0.4 | 36.3 ± 0.4 |
| Sample | 262 nm | 268 nm | 300 nm |
|---|---|---|---|
| a Frozen solutions were cooled at 0.3 °C min−1 after controlled nucleation. All samples were analyzed in duplicate. | |||
| Fresh | −0.27 ± 0.02 | −0.13 ± 0.03 | 0.22 ± 0.03 |
| Frozen to −35 °C | −0.13 ± 0.02 | −0.14 ± 0.04 | 0.15 ± 0.02 |
| Frozen to −55 °C | 0.12 ± 0.01 | 0.07 ± 0.04 | −0.08 ± 0.03 |
| Frozen to −65 °C | 0.11 ± 0.04 | 0.07 ± 0.02 | −0.07 ± 0.03 |
On the other hand, post-thaw secondary structure of the albumin cooled down to −55 °C revealed a 15% decrease in the fraction of α-helices (H (r) + H (d)), 5% increase in the fraction of turns, and 7% increase in the fraction of random structures (Table 1). There was 22% decrease in the fraction of α-helices, 8% increase in the fraction of turns and 11% increase in the fraction of random structures after thawing the solutions frozen to −65 °C. In solutions frozen to −55 °C and below, the near UV spectra showed an increase in ellipticity at 262 nm and 280 nm, and a decrease at 300 nm (Table 2), indicating increased exposure of aromatic amino acids, tyrosine and tryptophan from the hydrophobic core to the polar medium and presence of an asymmetric environment around disulphide linkages.58,62–64 In summary, irreversible unfolding of the secondary and tertiary structures of albumin was observed in the presence of DMSO when solutions were cooled below −35 °C. Between −35 °C and −55 °C, the corresponding DMSO concentration in the FCL increased from 45% to 57% (w/w). MD simulations have shown that at high DMSO concentrations (50–60% w/w), direct binding of DMSO to protein becomes favourable, possibly due to destabilization of DMSO–water HB complexes.65 Furthermore, there are reports of destabilizing effects of direct hydrophobic interactions with polar organic substances on proteins.66,67 These results suggest that unfolding of BSA during slow freezing occurred between −35 °C and −55 °C, before the FCL vitrified. Secondary unfolding was exacerbated when solutions were cooled below the glass transition temperature (−59 °C) down to −65 °C.
We have also explored the effect of thawing rate on the secondary and the tertiary structure of albumin. Samples that were cooled down to −65 °C and rapidly thawed showed 12% decrease in the fraction of α-helices (compared to 22% decrease in the case of slow thawing) and hence substantially lower structural damage (Table 3). For samples cooled down to −55 °C, there was a 10% decrease in α-helix content when rapidly thawed (compared to 15% decrease in the case of slow thawing). Thus, secondary structure of albumin was more sensitive to thawing induced stresses such as ice recrystallization than DMSO toxicity and temperature induced stresses during cooling. Devitrification induces rapid ice growth and recrystallization due to enhanced mobility and has been identified to be a major challenge for cell and organ vitrification.4,7,9 A rapid heating rate is known to not only delay devitrification during warming but also reduces extent of recrystallization.68 Thus, rapid warming produced the most pronounced effect in samples that were cooled below the glass transition temperature and vitrified. Interestingly, thawing rate did not affect the tertiary structure of albumin at all (Table 4) indicating that tertiary unfolding of albumin occurred during the slow cooling stage itself.
| Sample | H (r) | H (d) | S (r) | S (d) | Turns | Unordered |
|---|---|---|---|---|---|---|
| a H (r) → regular helix, H (d) → distorted helix, S (r) → regular sheet, S (d) → distorted sheet. Frozen solutions were cooled at 0.3 °C min−1 after controlled nucleation. All samples were analyzed in duplicate. | ||||||
| Cooled to −65 °C and slowly thawed | 19.2 ± 0.5 | 11.5 ± 0.8 | 5.2 ± 0.2 | 5.1 ± 0.64 | 23.2 ± 0.4 | 36.3 ± 0.4 |
| Cooled to −65 °C and rapidly thawed | 25.8 ± 0.1 | 14.2 ± 0.3 | 4.8 ± 0.2 | 5.3 ± 0.6 | 21.2 ± 0.4 | 29.3 ± 0.3 |
| Cooled to −55 °C and slowly thawed | 24.1 ± 0.2 | 13.3 ± 0.6 | 4.1 ± 0.7 | 5.2 ± 0.5 | 20.1 ± 0.5 | 32.3 ± 0.4 |
| Cooled to −55 °C and rapidly thawed | 26.9 ± 0.2 | 15.9 ± 0.2 | 4.9 ± 0.5 | 5.1 ± 0.1 | 19.1 ± 0.5 | 28.3 ± 0.4 |
| Sample | 262 nm | 268 nm | 300 nm |
|---|---|---|---|
| Cooled to −65 °C and slowly thawed | 0.11 ± 0.04 | 0.07 ± 0.02 | −0.07 ± 0.03 |
| Cooled to −65 °C and rapidly thawed | 0.10 ± 0.03 | 0.06 ± 0.03 | −0.07 ± 0.01 |
| Cooled to −55 °C and slowly thawed | 0.12 ± 0.01 | 0.07 ± 0.04 | −0.08 ± 0.03 |
| Cooled to −55 °C and rapidly thawed | 0.11 ± 0.04 | 0.08 ± 0.01 | −0.06 ± 0.02 |
The effect of hold temperature (Fig. 4A) and cooling rate (Fig. 4B) on the temporal changes in FCL composition was investigated in 30% DMSO + 10% BSA (w/w) samples during isothermal hold. When the solution was cooled at 0.3 °C min−1 from its equilibrium freezing temperature and then held at −15 °C, R value measured at a fixed region in the FCL continued to increase for 30 minutes and then levelled off. It is possible that continued ice growth and diffusion of species during equilibration, at this relatively high cryogenic temperature led to a gradual increase in R (i.e. preferential exclusion of DMSO), consistent with trends observed during equilibrium freezing. A finite cooling rate would result in some deviation from equilibrium conditions. Thus, when temperature is stabilized, there is a finite time within which the FCL reaches its equilibrium composition corresponding to the hold temperature and ice growth stops.69,70 This finite time period is a combination of the response time of the instrument, thermal equilibration time of the sample as well as the time taken by both species (albumin and DMSO) to diffuse within the FCL. Thus, an apparent spatial heterogeneity may arise in spectra collected due to non-equilibrium concentration and temperature gradients across the sample. This was further confirmed when spectra collected after a hold time of 30 minutes (Fig. 1 and 3) revealed significantly reduced heterogeneity in the FCL than the ones collected immediately at the beginning of the hold period (not shown). Following equilibration, value of R did not change with time, leading us to believe that the extent of association of albumin and DMSO with ice were negligible without ice propagation.
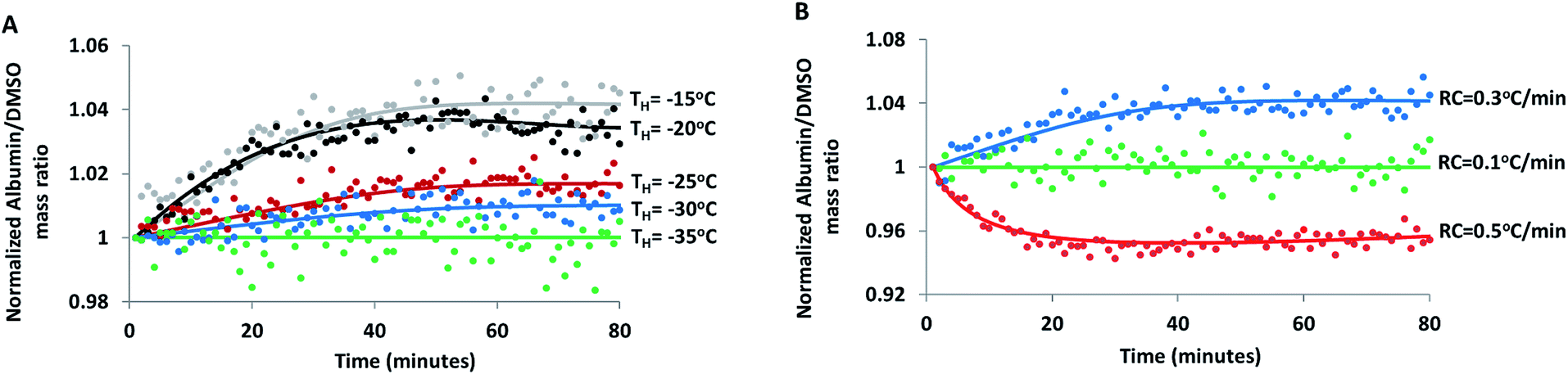 | ||
| Fig. 4 (A) Albumin/DMSO mass ratio (R) measured at a fixed region of the freeze concentrated liquid as a function of time in 30% DMSO + 10% BSA solution cooled at 0.3 °C min−1, held at different temperatures (TH: hold temperature). (B) Albumin/DMSO mass ratio (R) measured at a fixed region of the FCL as a function of time when 30% DMSO + 10% BSA solution is held at −15 °C after cooling at different rates (RC: cooling rate). | ||
The isothermal increase in R was directly correlated to the isothermal storage temperature (Fig. 4A): at hold temperatures of −35 °C and lower, FCL composition did not change with time. From the slope of the liquidus line in the DMSO–water binary phase diagram (Fig. S3†), it is clear that the differential increase in ice volume with differential decrease in temperature (dV/dT) decreases progressively during equilibrium cooling. Temporal changes in composition of the FCL (due to preferential association) at a given hold temperature will be proportional to the increase in ice volume during equilibration. Therefore, the magnitude of increase in R during equilibration was maximum when the hold temperature was −15 °C and progressively decreased with decreasing hold temperature (Fig. 4A).
The temporal evolution of R during isothermal hold at −15 °C was not observed when the sample was cooled very slowly at 0.1 °C min−1 (Fig. 4B), showing that equilibrium conditions prevailed at a very slow cooling rate. On the other hand, with a relatively higher cooling rate of 0.5 °C min−1, time-lapse IR spectra showed a decrease in R during equilibration at the same hold temperature of −15 °C. When cooled at 0.5 °C min−1, the ice volume exceeded the corresponding equilibrium value for the given hold temperature causing it to melt during equilibration, as reflected by the decrease in R.
XRD enabled characterization of the phases crystallizing from the FCL. In the XRD patterns obtained from the frozen 30% DMSO + 10% BSA solution, hexagonal ice was the only crystalline phase detected (Fig. 5A). Note that the peaks at 22.7 (3.93 Å), 24.4 (3.65 Å), 25.8 (3.46 Å) and 33.5 (2.68 Å) 2θ are assigned to hexagonal ice.71 Although formation of the water–DMSO trihydrate eutectic has been reported to occur at −63 ± 1 °C,35 crystallization of DMSO was not detected in the XRD patterns as diffraction peaks. Likewise, in the DSC scans (not shown) no crystallization exotherm was detected during slow freezing, confirming that DMSO remained amorphous.
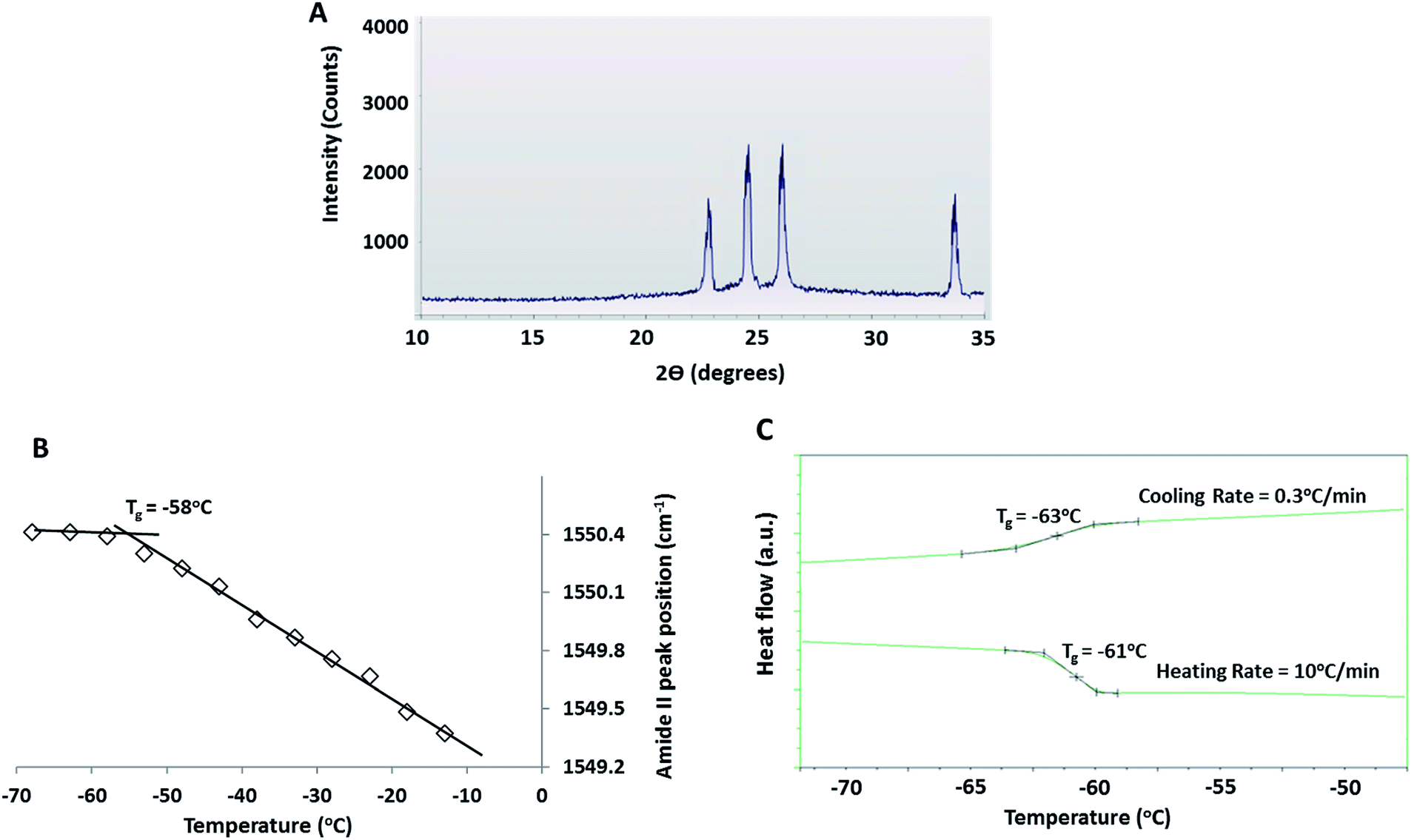 | ||
| Fig. 5 (A) XRD pattern of a frozen 30% DMSO + 10% BSA solution at −70 °C after equilibrium freezing (at 0.3 °C min−1). (B) Amide II peak location in IR spectra taken from 7 different regions of the freeze concentrated liquid during near equilibrium freezing (at 0.3 °C min−1) of 30% DMSO + 10% BSA solution (standard deviation bars were smaller than the marker size and were thus omitted). (C) DSC scans during equilibrium freezing (at 0.3 °C min−1) of a 30% DMSO + 10% BSA solution up to −80 °C and subsequent heating from −80 °C to room temperature at 10 °C min−1. | ||
Position of the protein amide II band in IR spectra gives a measure of flexibility of the protein backbone. Amide II position (in wavenumber) of albumin in the FCL was plotted as a function of temperature during near equilibrium freezing of a 30% DMSO + 10% BSA solution (Fig. 5B). Glass transition of the protein causes stiffening of the protein backbone and is reflected by an abrupt change in the slope of the wavenumber (of amide II) vs. temperature curve at a specific temperature (i.e. the glass transition temperature).49,72 The abrupt change in slope was observed at −58 °C (Fig. 5B). During slow freezing at 0.3 °C min−1, a faint glass transition (small baseline shift) at −63 °C could be detected in the DSC thermograms, indicating vitrification of the freeze concentrate (Fig. 5C). The glass transition can also be detected at −61 °C when the frozen solution was re-heated from −80 °C back to room temperature at 10 °C min−1 (Fig. 5C), indicating devitrification of the previously vitrified FCL. The variations in the glass transition temperatures observed in all these experiments (Fig. 5B and C) was due to glass transition being a kinetic and not thermodynamic transition.73 Note that the pronounced baseline shift associated with glass transition during heating was due to the faster heating rate employed (10 °C min−1).
Below −59 °C (Region III), FCL composition did not further change with decreasing temperature (Fig. 1) and crystallization of DMSO tri-hydrate was ruled out by low temperature XRD and DSC experiments (Fig. 5A and C). The cessation of compositional changes at these temperatures was attributed to increased viscosity of the FCL upon glass transition. The heating/cooling DSC scans (Fig. 5C) and the temperature sensitivity of the amide II peak (Fig. 5B) clearly showed the glass transition of the FCL (measured at −58 °C by FTIR, at −61 °C (during heating) and −63 °C (during cooling) by DSC). Note that the DMSO concentration in the FCL at these temperatures is in the range of 52–55% w/w and the threshold vitrification concentration of aqueous DMSO has been reported to be 49% w/w.4 Presence of large proteins such as BSA and haemoglobin in aqueous sugar solutions has been reported to strengthen hydrogen bonding interactions in the surrounding medium, increasing the glass transition temperature of the medium.74 This explains the higher glass transition temperature recorded for the FCL than the value predicted by the Gordon Taylor equation (∼−85 °C) based on the FCL composition at −63 °C. Additionally, a distinct glass transition has been detected in most hydrated proteins between −73 and −53 °C, which suppresses collective unharmonic motions in the protein molecule.75 Coupling of the protein dynamics with that of that of the amorphous DMSO–water phase has likely increased its glass transition temperature as well.
Conclusions
Vitrification of the FCL at −63 °C inhibited DMSO crystallization and further compositional changes in the FCL during slow freezing of albumin–DMSO solutions down to −70 °C. Continued changes in the relative concentrations of albumin and DMSO in the FCL were observed with decreasing temperature due to preferential association of either species with the growing ice phase. The nature and extent of preferential association were governed by DMSO–water hydrogen bonding interactions, their relative diffusivities and the surface area of ice–FCL interface. When cooled between −35 °C and −55 °C, unfolding of albumin secondary and tertiary structures were observed presumably due to direct DMSO–protein binding. However, when solutions were cooled below the glass transition temperature and thawed, ice recrystallization induced during devitrification proved to be significantly more detrimental to albumin than exposure to DMSO. Adverse effects of ice recrystallization were eliminated with rapid thawing rates (>20 °C min−1). These results implied that while vitrification is desirable for long term stability of the macromolecules, subsequent rapid thawing is a necessity. A vitrified sample therefore yielded significantly lower protein recovery when thawed slowly than a sample which was simply frozen without vitrification. Moreover, adverse effects of high DMSO concentrations mainly observed between −35 °C and −55 °C may be eliminated by increasing the glass transition temperature of the FCL using high Tg chemicals (such as disaccharides) in order to induce vitrification of the FCL above this temperature range. These should minimize the adverse effects of DMSO and re-crystallization, potentially enhancing the viability and functionality of large tissues and organs post-thaw.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by an NSF grant (CBET-1335936) to A. A. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.Notes and references
- G. M. Fahy, B. Wowk and J. Wu, Rejuvenation Res., 2006, 9, 279–291 CrossRef CAS PubMed.
- A. Karow Jr, Organ Preservation for Transplantation, Little, Brown and Company, Boston, 1974, pp. 3–8 Search PubMed.
- T. E. Starzl, J. Surg. Res., 1970, 10, 291–297 CrossRef CAS PubMed.
- B. Best, Vitrification in Cryonics, Online web article, 2011, link: http://www.benbest.com/cryonics/vitrify.html.
- G. M. Fahy, Cryobiology, 1980, 17, 371–388 CrossRef CAS PubMed.
- S. Kubota and R. Lillehei, J. Low Temp. Phys., 1976, 2, 95–105 Search PubMed.
- B. Wowk, Cryobiology, 2010, 60, 11–22 CrossRef CAS PubMed.
- G. M. Fahy and W. F. Rall, Vitrification in Assisted Reproduction, A User's Manual and Trouble-Shooting Guide, Informa Healthcare, London, 2007, pp. 1–20 Search PubMed.
- G. M. Fahy and W. F. Rall, in Vitrification in Assisted Reproduction: A User's Manual and Trouble-Shooting Guide, CRC Press, 2007, pp. 1–20 Search PubMed.
- M. Sansinena, M. V. Santos, N. Zaritzky and J. Chirife, Theriogenology, 2012, 77, 1717–1721 CrossRef CAS PubMed.
- D. E. Pegg, Cryopreservation and freeze-drying protocols, 2007, pp. 39–57 Search PubMed.
- P. S. Steif, M. C. Palastro and Y. Rabin, Cell Preserv. Technol., 2007, 5, 104–115 CrossRef CAS PubMed.
- W. F. Rall and G. M. Fahy, Nature, 1985, 313, 573–575 CrossRef CAS PubMed.
- M. Ghorbani, R. Sadrkhanlou, V. Nejati, A. Ahmadi and G. Tizroo, Vet. Res. Forum, 2012, 3(4), 245–249 Search PubMed.
- E. Alcolak, E. A. Marar, S. C. Mytas, N. Chalvatzas, V. Palapelas, B. Schöpper, K. Diedrich and S. Al-Hasani, Middle East Fertil. Soc. J., 2011, 16, 189–193 CrossRef.
- G. M. Fahy, B. Wowk, J. Wu, J. Phan, C. Rasch, A. Chang and E. Zendejas, Cryobiology, 2004, 48, 157–178 CrossRef CAS PubMed.
- G. M. Fahy, B. Wowk, J. Wu and S. Paynter, Cryobiology, 2004, 48, 22–35 CrossRef CAS PubMed.
- T. Shin, M. Fukushima, R. Sato, K. Nishio, H. Yagi, G. Arai, S. Soh and H. Okada, J. Clin. Oncol., 2015, 33, e20672 Search PubMed.
- L.-M. Her and S. L. Nail, Pharm. Res., 1994, 11, 54–59 CrossRef CAS.
- L.-M. Her, R. P. Jefferis, L. A. Gatlin, B. Braxton and S. L. Nail, Pharm. Res., 1994, 11, 1023–1029 CrossRef CAS.
- G. M. Fahy, T. H. Lilley, H. Linsdell, M. S. J. Douglas and H. T. Meryman, Cryobiology, 1990, 27, 247–268 CrossRef CAS PubMed.
- G. M. Fahy, Cryobiology, 1986, 23, 1–13 CrossRef CAS PubMed.
- B. Courbiere, J. Massardier, B. Salle, C. Mazoyer, J.-F. Guerin and J. Lornage, Fertil. Steril., 2005, 84, 1065–1071 CrossRef PubMed.
- Y. C. Song, B. S. Khirabadi, F. Lightfoot, K. G. Brockbank and M. J. Taylor, Nat. Biotechnol., 2000, 18, 296–299 CrossRef CAS PubMed.
- K. Brockbank and Y. Song, J. Heart Valve Dis., 2004, 13, 297–301 Search PubMed.
- W. J. Armitage, S. C. Hall and C. Routledge, Invest. Ophthalmol. Vis. Sci., 2002, 43, 2160–2164 Search PubMed.
- G. M. Fahy and S. E. Ali, Cryobiology, 1997, 35, 114–131 CrossRef CAS PubMed.
- G. M. Fahy, B. Wowk, R. Pagotan, A. Chang, J. Phan, B. Thomson and L. Phan, Organogenesis, 2009, 5, 167–175 CrossRef PubMed.
- J. Dong, A. Hubel, J. C. Bischof and A. Aksan, J. Phys. Chem. B, 2009, 113, 10081–10087 CrossRef CAS PubMed.
- Y. Lin, J. A. Raymond, J. G. Duman and A. L. DeVries, Cryobiology, 1976, 13, 334–340 CrossRef CAS PubMed.
- J. A. Raymond and A. L. DeVries, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2589–2593 CrossRef CAS.
- A. Twomey, R. Less, K. Kurata, H. Takamatsu and A. Aksan, J. Phys. Chem. B, 2013, 117, 7889–7897 CrossRef CAS PubMed.
- H. Pruppacher, J. Atmos. Sci., 1995, 52, 1924–1933 CrossRef.
- N. Sreerama, S. Y. Venyaminov and R. W. Woody, Protein Sci., 1999, 8, 370–380 CrossRef CAS PubMed.
- D. H. Rasmussen and A. P. MacKenzie, Nature, 1968, 220, 1315–1317 CrossRef CAS PubMed.
- J. Grdadolnik and Y. Maréchal, Biopolymers, 2001, 62, 40–53 CrossRef CAS PubMed.
- V. Fornés and J. Chaussidon, J. Chem. Phys., 1978, 68, 4667–4671 CrossRef.
- M. Takeuchi, G. Martra, S. Coluccia and M. Anpo, J. Phys. Chem. B, 2005, 109, 7387–7391 CrossRef CAS PubMed.
- F. A. Cotton, R. Francis and W. D. Horrocks, J. Phys. Chem., 1960, 64, 1534–1536 CrossRef CAS.
- W. D. Horrocks Jr and F. A. Cotton, Spectrochim. Acta, 1961, 17, 134–147 CrossRef.
- A. Luzar and D. Chandler, J. Chem. Phys., 1993, 98, 8160–8173 CrossRef CAS.
- I. I. Vaisman and M. L. Berkowitz, J. Am. Chem. Soc., 1992, 114, 7889–7896 CrossRef CAS.
- A. Vishnyakov, A. P. Lyubartsev and A. Laaksonen, J. Phys. Chem. A, 2001, 105, 1702–1710 CrossRef CAS.
- K. Yi, Q. F. Li, L. Zhang, N. Li, Y. Zhou, S. K. Ryu and R. G. Jin, J. Eng. Fibers Fabr., 2013, 8, 107–113 CAS.
- W. A. Luck, Structure of water and aqueous solutions, Verlag Chemie, 1974 Search PubMed.
- G. Choppin and M. Violante, J. Chem. Phys., 1972, 56, 5890–5898 CrossRef CAS.
- V. Fornes and J. Chaussidon, J. Chem. Phys., 1978, 68, 4667–4671 CrossRef CAS.
- E. A. Galinski, M. Stein, B. Amendt and M. Kinder, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 1997, 117, 357–365 CrossRef.
- J. Malsam and A. Aksan, J. Phys. Chem. B, 2009, 113, 6792–6799 CrossRef CAS PubMed.
- R. Giangiacomo, Food Chem., 2006, 96, 371–379 CrossRef CAS.
- I. A. Borin and M. S. Skaf, J. Chem. Phys., 1999, 110, 6412–6420 CrossRef CAS.
- C. Nieto-Draghi, J. Bonet Ávalos and B. Rousseau, J. Chem. Phys., 2003, 119, 4782–4789 CrossRef CAS.
- J. F. Carpenter and J. H. Crowe, Cryobiology, 1988, 25, 244–255 CrossRef CAS PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed.
- F. W. Kleinhans and P. Mazur, Cryobiology, 2007, 54, 212–222 CrossRef CAS PubMed.
- W. Gallagher, Course Manual Chem, 2009, vol. 455 Search PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- M. Dockal, D. C. Carter and F. Rüker, J. Biol. Chem., 2000, 275, 3042–3050 CrossRef CAS PubMed.
- C. Yuan, J. Gao, J. Guo, L. Bai, C. Marshall, Z. Cai, L. Wang and M. Xiao, PLoS One, 2014, 9, e107447 Search PubMed.
- G. Da Violante, N. Zerrouk, I. Richard, G. Provot, J. C. Chaumeil and P. Arnaud, Biol. Pharm. Bull., 2002, 25, 1600–1603 CAS.
- P. Huang, A. Dong and W. S. Caughey, J. Pharm. Sci., 1995, 84, 387–392 CrossRef CAS PubMed.
- A. Pabbathi, S. Patra and A. Samanta, ChemPhysChem, 2013, 14, 2441–2449 CrossRef CAS PubMed.
- A. Chakrabartty, T. Kortemme, S. Padmanabhan and R. L. Baldwin, Biochemistry, 1993, 32, 5560–5565 CrossRef CAS PubMed.
- P. C. Kahn, Methods Enzymol., 1979, 61, 339–378 CAS.
- T. Arakawa, Y. Kita and S. N. Timasheff, Biophys. Chem., 2007, 131, 62–70 CrossRef CAS PubMed.
- B. W. Matthews, Hydrophobic Interactions in Proteins, eLS, John Wiley & Sons, Ltd, 2001 Search PubMed.
- P. Privalov, in Protein Structure and Protein Engineering, Springer, 1988, pp. 6–15 Search PubMed.
- S. Jena, J. Horn, R. Suryanarayanan, W. Friess and A. Aksan, Pharm. Res., 2016, 1–17 Search PubMed.
- P. R. Fitzgerald and N. D. Levine, J. Protozool., 1961, 8, 21–27 CrossRef.
- E. Nsabimana, S. Kišidayová, D. Macheboeuf, C. Newbold and J. Jouany, Appl. Environ. Microbiol., 2003, 69, 3826–3832 CrossRef CAS PubMed.
- I. C. f. D. Data, Journal, 2004, PA.
- W. F. Wolkers, A. E. Oliver, F. Tablin and J. H. Crowe, Carbohydr. Res., 2004, 339, 1077–1085 CrossRef CAS PubMed.
- C. A. Angell, J. Phys. Chem. Solids, 1988, 49, 863–871 CrossRef CAS.
- G. Bellavia, S. Giuffrida, G. Cottone, A. Cupane and L. Cordone, J. Phys. Chem. B, 2011, 115, 6340–6346 CrossRef CAS PubMed.
- D. Ringe and G. A. Petsko, Biophys. Chem., 2003, 105, 667–680 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07556a |
| This journal is © The Royal Society of Chemistry 2017 |