
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring the mechanism of formation of [Ce(1,10-phenanthroline)2(NO3)3] by in situ luminescence analysis of 5d–4f electronic transitions†
L. Ruiz Aranaa,
P. Lindenberga,
H. Saida,
M. Radkea,
N. Heidenreichab,
C. S. Cunhaac,
S. Leubnera and
H. Terraschke *a
*a
aInstitut für Anorganische Chemie, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Str. 2, 24118 Kiel, Germany. E-mail: hterraschke@ac.uni-kiel.de
bDESY Photon Science, Notkestr. 85, 22607 Hamburg, Germany
cInstitute of Chemistry, University of São Paulo, Av. Prof. Lineu Prestes 748, 05508-000 São Paulo, SP, Brazil
First published on 15th November 2017
Abstract
This work introduces the application of the in situ luminescence analysis of coordination sensors (ILACS) technique for monitoring the emission of Ce3+ 5d–4f electronic transitions under real reaction conditions during the formation of [Ce(phen)2(NO3)3] (phen = 1,10-phenanthroline). The mechanism of formation indicated by the ILACS data was confirmed by several additional methods, including ex situ and synchrotron-based in situ X-ray diffraction (XRD) analysis, in situ light transmission, and in situ infrared (IR) spectroscopy, among others. Initially, the in situ luminescence measurements presented a broad emission band at 415–700 nm, which was assigned to the Ce3+ ions in ethanolic solution. Upon the addition of the phen solution to the reactor, a gradual shift of the emission band to lower energies (500–900 nm) was observed. This occurs due to the changes in the Ce3+ coordination environment during its incorporation into the solid [Ce(phen)2(NO3)3] complex. In situ IR measurements during the crystallization of [Ce(phen)2(NO3)3] confirmed the kinetics of the crystallization process by detecting changes in the phen and nitrate vibrations at e.g. 842 and 1301 cm−1, respectively. Simultaneous in situ XRD measurements confirmed the induction time of approximately 3 minutes after the addition of the phen solution, previously detected by the in situ luminescence measurements, coinciding with the onset of the [Ce(phen)2(NO3)3] Bragg reflections. In situ monitoring of events occurring during the formation of solid materials is a crucially important step for developing rational synthesis approaches and for tailoring structure-related properties, such as luminescence.
Introduction
The attention given to in situ characterization techniques has increased considerably in the past few years, in particular because of the knowledge they provide about structural changes during formation of solid materials such as nucleation, crystal growth, metal–ligand exchange processes, formation of intermediates, polymorphic phase transitions, and many others.1–3 In situ monitoring of chemical reactions allows, therefore, not only the development of rational synthesis strategies (i.e. improving reaction conditions, increasing product yield or avoiding the formation of side products) but also allows targeted manipulation of synthesis parameters to alter the product structure and, consequently, their structure-related properties.1,4–7 The in situ luminescence analysis of coordination sensors (ILACS) approach8–11 enables monitoring of synthesis processes in real time, under real reaction conditions. Using the spectroscopic sensitivity of lanthanide ions (Ln)12–16 as coordination sensors, it is possible to obtain structural information about changes in the coordination environment of the Ln ions, throughout the entire reaction, from the early stages of the product formation by means of in situ luminescence measurements. A similar approach has been shown by Tiseanu et al.17 to investigate the transition between the amorphous to crystalline phase of ZrO2. Among the advantages of the ILACS strategy is the possibility of carrying out experiments in conventional university laboratories (without the need for synchrotron radiation) and being able to characterize ions, small crystallites, amorphous materials and potential side products present in very low concentrations, ideally complementing other techniques as in situ X-ray diffraction (XRD) analysis.8 Traditionally, Eu3+ and Tb3+ have been used as coordination centers for ILACS measurements, following the behavior of the emission peaks generated by 4f–4f electronic transitions of these ions for monitoring the formation of solid materials.8–10 These 4f–4f transitions are parity-forbidden, causing them to have a low absorption coefficient and sharp absorption bands, which, in turn, generally lead to low emission intensities. Due to shielding effects of the 5s2 and 5p6 subshells on the 4f-orbitals, the position of the emission peaks of the 4f–4f transitions are also almost invariable, being relatively insensitive to changes in coordination environment in comparison to the emission bands assigned to the 5d–4f electronic transitions of lanthanide ions.14–16,18In contrast, 5d–4f electronic transitions, such as those observed for Eu2+ and Ce3+, are parity-allowed, therefore very fast and the respective emission intensities are much higher.18 Additionally, the 5d excited state is not shielded by other outer orbitals, allowing the energy and position of the emission maximum to vary strongly in response to changes in the interplay of the 5d electron and its environment.19 Taken together, these properties make 5d–4f emitters ideal local coordination sensors for detecting changes in covalence (nephelauxetic effect) and crystal field splitting around the cation sites. Since Eu2+ is easily oxidized to its more stable trivalent oxidation state, requiring an inert atmosphere to remain reduced, Ce3+ is the preferred coordination sensor for in situ monitoring of 5d–4f transitions during synthesis in solution under atmospheric conditions. Trivalent cerium ions possess only one optically active electron. In the ground state, this electron is in the 4f-shell (4f1), and, in the excited state configuration, it is in the 5d-shell (5d1). The 4f1-state possesses two energy levels (7F5/2 and 2F7/2) which are separated by ca. 2000 cm−1. The 5d1-state is split by the crystal field into two to five components.20 Emission occurs when the electron transitions from the lowest crystal field component of the 5d1 excited state to the two levels of the 4f1 ground state, yielding in a double-band shaped emission spectrum.20 The spectrum can be particularly well resolved for measurements at low temperature or low doping concentrations.21 A second interesting in situ spectroscopic analysis exploiting the optical properties to monitor complex formation is light transmission spectroscopy, which provides information about the turbidity of the reaction solution as the reaction progresses.2 To perform these measurements, reaction solutions are irradiated with an external light source. An optical fiber submersed in the solution measures the intensity of the light penetrating the solution throughout the progression of the reaction. When solid material begins forming, the turbidity of the solution increases, causing a proportional decrease in the intensity of transmitted light.22 Since this method does not require luminescence properties or the use of specific wavelengths of irradiating light, it is highly versatile and more broadly applicable than in situ luminescence measurements.
Recognition of the potential biological and technical suitability of lanthanide complexes as markers for bioimaging and the production of organic light-emitting diodes (OLEDs)23 has provided additional incentives to investigate complexes containing these ions in their own right. A clear understanding of their structures and the crystallization processes responsible may ultimately allow for monitoring and controlling the structural formation of these materials and for fine-tuning of the structure-related optical properties, relevant for their use in these expanding fields. Among the complexes interesting in this regard is [Ce(phen)2(NO3)3] (phen = 1,10-phenanthroline). [Ce(phen)2(NO3)3] crystallizes as yellow prismatic crystals in the space group C2/c. The cerium central atoms are coordinated by six oxygen atoms of the three nitrate units and four nitrogen atoms of two phen, creating a distorted dicapped square antiprism environment around the metal center.24
This work aims to explore the sensitivity of 5d–4f transitions using the ILACS technique for the monitoring of [Ce(phen)2(NO3)3] formation, confirmed by ex situ and synchrotron-based in situ XRD analysis, in situ infrared (IR) spectroscopy as well as in situ measurements of light transmission, pH values and ion conductivity. While the in situ luminescence measurements provide information about the short-range, in situ XRD concerns the long-range cation order. In situ IR is able to detect the presence of the nitrate and 1,10-phenanthroline ligands in solution or in the complex and in situ measurements of light transmission is able to detect the formation of the solid material by the increase of the solution turbidity. On the other hand, the formation of the complex is detected by in situ pH measurements by the decrease in basicity upon the uptake of phen molecules and by the in situ conductivity measurements by the decrease in conductivity upon the uptake of the ions from the solution. To the best of our knowledge, the optical properties and the mechanism of crystallization of [Ce(phen)2(NO3)3] are reported here for the first time.
Experimental part
Materials
The experiments reported in this work were completed using two distinct experimental setups. Setup I consists of the in situ crystallization cell at the University of Kiel and the Setup II comprises a specially designed mobile cell for performing in situ XRD measurements at synchrotron facilities. For both experimental setups, the preparation of reaction solutions was identical. Initially, 30 mL of an 0.023 M ethanolic solution of Ce(NO3)3·6H2O (99.99%, Alfa Aesar, Germany) was placed in the reactor. Afterwards, 5 mL of a 0.276 M ethanolic solution of 1,10-phenanthroline (99%, Alfa Aesar, Germany) was added to the reactor at a rate of 0.5 mL min−1, under magnetic stirring and at room temperature (Table S1†). In situ characterization methods, explained in detail below, were carried out concomitantly. Following the reaction, the product was centrifuged, dried at 80 °C for 2 h, and subjected to ex situ XRD analysis.In situ measurement assemblies
In order to confirm the measurements obtained from in situ experiments, ex situ XRD analysis were performed on samples removed at e.g. minutes 5, 7, 10, 12, 15, and 20 of the reaction period. As was done with the final product, these samples were centrifuged and dried at 80 °C for 2 hours. The ex situ XRD data was measured in transition geometry on a STOE Stadi-p X-ray powder diffractometer (STOE & Cie GmbH, Darmstadt, Germany) with Cu Kα1 radiation (λ = 1.54056 Å), a Ge monochromator and DECTRIS® MYTHEN 1K detector (DECTRIS, Baden-Daettwil, Switzerland). The reflection spectra were recorded at room temperature with a Varian Techtron Pty UV/Vis/NIR two-channel Cary 5000 spectrometer, using BaSO4 as a reference material.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 (23 keV, 0.53905 Å). The setup used at DESY (Setup II, Fig. S2†) consisted of an adapted glass reaction vessel containing an incurvated cavity in one of its walls to decrease the pathway length of the X-ray beam through the reaction volume. This vessel was placed in reactor holder22 containing an integrated stirring system designed to flexibly fit different beamlines without requiring realignment after each reaction. Additionally, the reactor holder contained two openings for the entrance and exit of X-ray beams and a larger opening arranged at 90° to the path of the X-ray beam for the irradiation of the reactor with a light source for light transmission measurements. The light source for transmission measurements was a UV-LED (365 nm, Sahlmann Photochemical Solutions). Light intensity was measured during the synthesis of the [Ce(phen)2(NO3)3] complex using an optical fiber submersed in the solution and connected to a portable spectrometer (StellarNet Inc., Tampa, FL, USA) equipped with a CCD detector.
26 (23 keV, 0.53905 Å). The setup used at DESY (Setup II, Fig. S2†) consisted of an adapted glass reaction vessel containing an incurvated cavity in one of its walls to decrease the pathway length of the X-ray beam through the reaction volume. This vessel was placed in reactor holder22 containing an integrated stirring system designed to flexibly fit different beamlines without requiring realignment after each reaction. Additionally, the reactor holder contained two openings for the entrance and exit of X-ray beams and a larger opening arranged at 90° to the path of the X-ray beam for the irradiation of the reactor with a light source for light transmission measurements. The light source for transmission measurements was a UV-LED (365 nm, Sahlmann Photochemical Solutions). Light intensity was measured during the synthesis of the [Ce(phen)2(NO3)3] complex using an optical fiber submersed in the solution and connected to a portable spectrometer (StellarNet Inc., Tampa, FL, USA) equipped with a CCD detector.Results and discussion
The emission spectrum of Ce3+ in ethanolic solution consists of a broad band between approximately 415 and 700 nm (λex = 400 nm, Fig. 1, blue curve) with full width at half maximum (FWHM) of 5161 cm−1, which is assigned to the 5d–4f electronic transitions of trivalent cerium ions. The emission spectrum of [Ce(phen)2(NO3)3] presents a broad band between approximately 500 and 900 nm with FWHM of 4006 cm−1, also ascribed to the Ce3+ 5d–4f transitions (λex = 400 nm, Fig. 1, red curve). Using the emission spectra shown in the Fig. 1 (top), the Commission Internationale d'Éclairage (CIE) 1931 color coordinates were calculated, reaching x = 0.4494, y = 0.4339 for the emission of Ce3+ in solution and x = 0.6693, y = 0.3231 for Ce3+ within the [Ce(phen)2(NO3)3] complex (Fig. 1, bottom). Ex situ excitation spectrum of [Ce(phen)2(NO3)3] (λem = 670 nm, Fig. S3†) consists of a broad band with maximum at 395 nm, matching the absorbance range indicated by reflectance spectra of [Ce(phen)2(NO3)3] and the pure 1,10-phenanthroline ligand.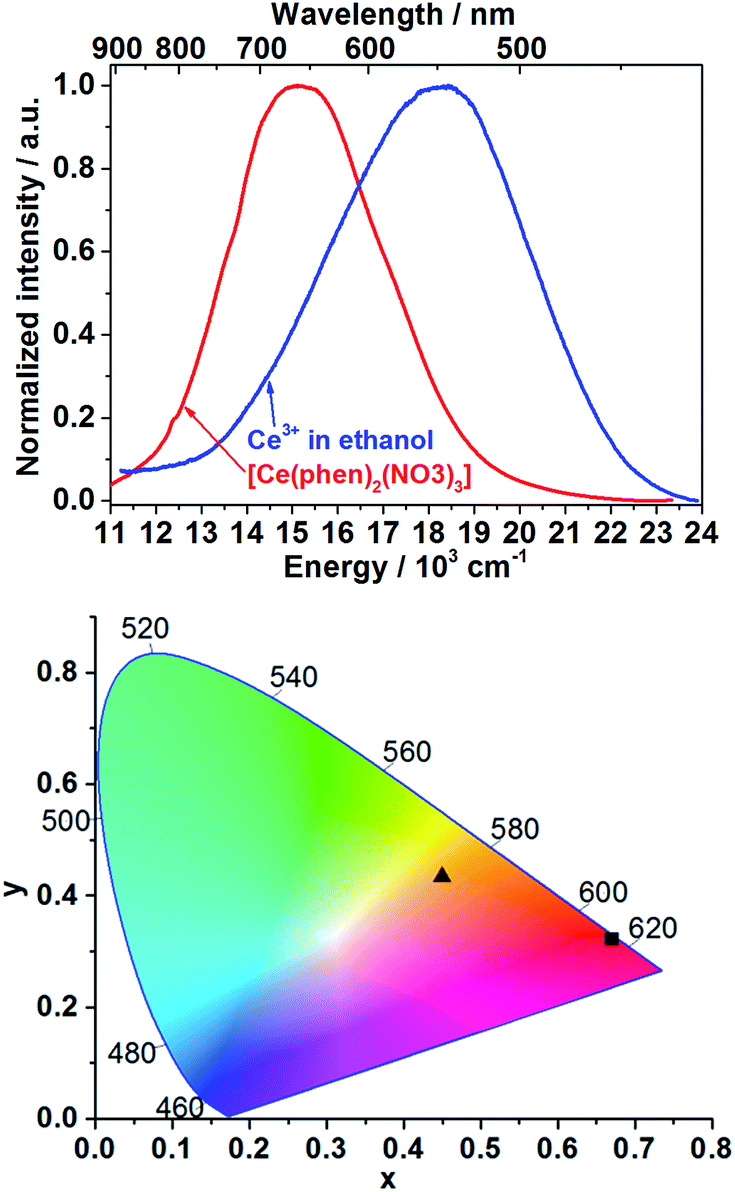 | ||
| Fig. 1 Ex situ emission spectra of Ce3+ in ethanol (blue curve) and after incorporation into the [Ce(phen)2(NO3)3] complex (red curve) at λex = 400 nm (top). CIE 1931 chromaticity diagram plot of the color coordinates of the Ce3+ in ethanolic solution (x = 0.4494, y = 0.4339, ▲) and of [Ce(phen)2(NO3)3] (x = 0.6693, y = 0.3231, ■) (bottom). | ||
Detectable differences in the optical properties of isolated Ce3+ as starting material solved in ethanol and Ce3+ within the [Ce(phen)2(NO3)3] product enables the use of ILACS as a method to monitor the crystallization process. Therefore, the in situ luminescence spectra (λex = 400 nm) were recorded for the duration of crystallization processes of [Ce(phen)2(NO3)3] using Setup I (Fig. S4†). Intensity of emission intensity at 500 nm and at 700 nm were selected for monitoring respectively, the high and low energy bands, to minimize overlap of the precursor and product signals. Fig. 2 and S4† depict the time dependent transformation of the emission band at high energies assigned to Ce3+ in ethanol, before the addition of the ethanolic solution of 1,10-phenanthroline and its gradual conversion to the low-energy emission band assigned to the product complex during the synthesis reaction.
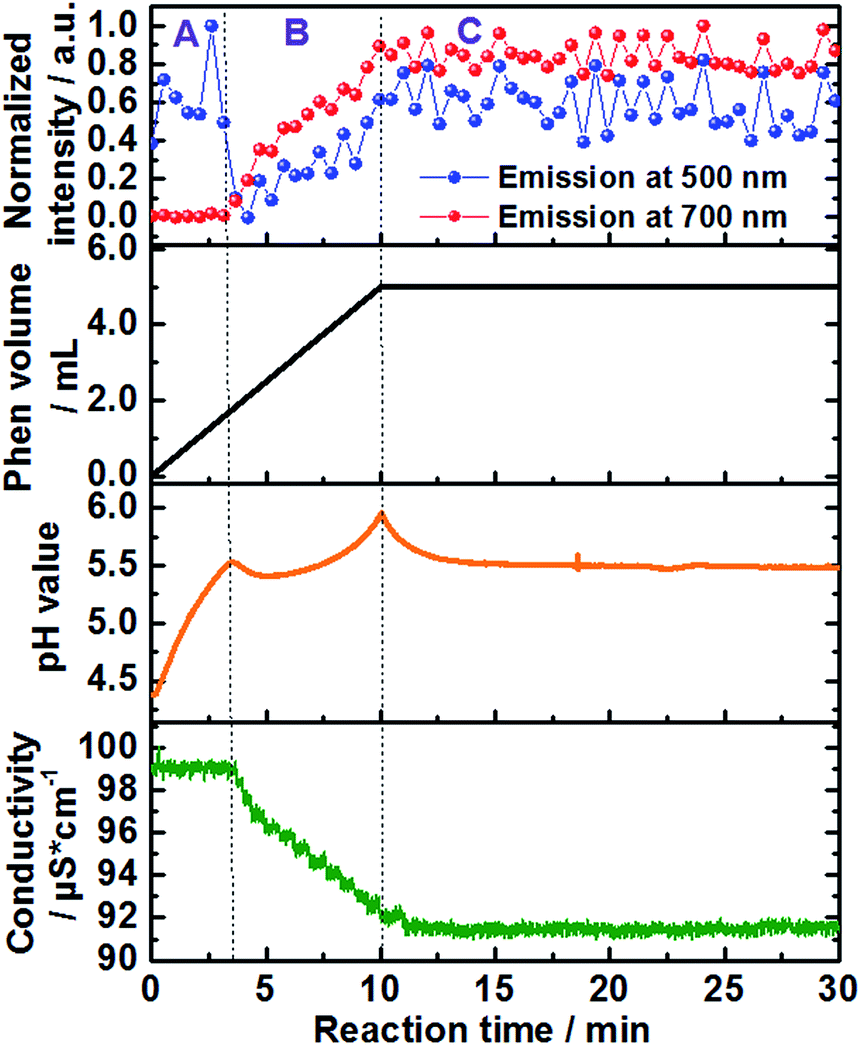 | ||
| Fig. 2 Time-dependence of the normalized emission intensity (λex = 400 nm) at 500 nm (blue curve) and 700 nm (red curve), added volume of the 1,10-phenanthroline solution (back curve) as well as in situ measurements of pH value (orange curve) and ion conductivity (green curve) (experiment 1, Table S1†). | ||
Approximately 3 minutes after initiating addition of the ligand solution (Fig. 2, black curve), the intensity of the high energy band at 500 nm (blue curve) begins decreasing. This corresponds to a simultaneous increase in the intensity of the low-energy emission band of [Ce(phen)2(NO3)3] at 700 nm (Fig. 2, red curve), indicating the formation of the product complex. The shift in the energy of the Ce3+ emission between the starting material and the formed product can be explained by the changes on the coordination environment around the Ce3+ ions during the desolvation and ligand exchange processes, during the uptake of the Ce3+ ions from the solution as well as the consecutive attachment of the phen ligands for the formation of the complex, explained by the nephelauxetic effect. This effect is caused by the coordination of the nitrogen atoms of the phen ligand, enhancing the covalence of the coordination sphere of the trivalent cerium ions in comparison to the ethanolic solvation shell, increasing therefore the crystal field splitting, which leads to the emission band to longer wavelengths.18 This clear shift in the emission spectra assigned to the 5d–4f transitions of Ce3+, showing the transfer between the Ce3+ ions from the solution to the [Ce(phen)2(NO3)3] product offer a significant improvement to the ILACS technique in comparison to the application of Eu3+ ions as coordination sensors. In our previous work,8 for instance, the emission spectra assigned to the 4f–4f electronic transitions of Eu3+ do not shift and the incorporation of Eu3+ ions from the solution into the [Eu(phen)3(NO3)3] complex is detected mostly by changes in the intensity of the emission spectra.
In addition to the comparison of emission intensity at 500 nm and 700 nm, Fig. 2 shows changes in the total added volume of the 1,10-phenanthroline solution, in situ measurements of pH value (orange curve), and ion conductivity (green curve) across the reaction period. In order to better analyze the individual processes involved in the mechanism of formation of the [Ce(phen)2(NO3)3] complex, the reaction was divided in three stages: A, B and C (Fig. 2). Stage A comprises the reaction time between approximately 0 and 3 min, stage B the reaction times from approximately 3 to 10 min, and stage C the reaction time from 10 min onward. During reaction stage A, the pH value increased due to the addition of the basic 1,10-phenanthroline solution. The intensity of the bands at 500 nm, at 700 nm and the ion conductivity do not significantly change. This period may, therefore, be characterized as an induction period, before the formation of the product. At the beginning of stage B, increases in the emission intensity at 700 nm, assigned to the [Ce(phen)2(NO3)3] complex, are accompanied by a rapid decrease in the high energy emission band at 500 nm, assigned to the Ce3+ ions in solution, indicating the begin product formation. The intensity of the emission band at 700 nm continues to climb throughout stage B, reaching a steady state and leveling off in the early part of stage C, indicating no significant complex formation after this period and the completion of the reaction. After its initial drop off, the intensity of the 500 nm emission band begins to climb again, also leveling off in the early part of stage C. The rise in intensity seen at this high-energy band occurs, most probably, due to the slight overlap with the emission band generated by the product and is not from isolated Ce3+ ions (Fig. 1 and S4†). Interestingly, the position of the band assigned to the [Ce(phen)2(NO3)3] complex remains invariable, indicating no significant variation of the coordination environment of Ce3+ between the desolvation of the Ce3+ ions and their incorporation to the product lattice. This indicates that [Ce(phen)2(NO3)3] is directly crystallized, without the formation of reaction intermediates.
The timeline indicated by the ILACS approach was confirmed by concurrent changes in pH value and ion conductivity (Fig. 2). Approximately 3 minutes after addition of the ligand, concomitant with the changes in the emission spectra, there is a transient decrease in pH value as 1,10-phenanthroline molecules are removed from the solution and incorporated to the solid complex. The pH quickly returns to higher values, as additional basic ligand solution is added to the reaction content more quickly than the ligand can be incorporated into product complexes. The pH continues to rise until t ≈ 10 min, at which point no additional ligand is being added, and the pH starts to level off into a steady state, indicating the completion of the reaction. Similarly, at t ≈ 3 min, ion conductivity begins to decrease due to the uptake of free ions out of the solution and into the complex, further confirming the start of the crystallization process. These values continue to decrease until t ≈ 10.5 min, at which point they stabilize.
In order to confirm the formation of the [Ce(phen)2(NO3)3] complex, samples were removed from the reactor at pre-determined times during the crystallization process and subsequently characterized by means of ex situ XRD analysis. As shown in the Fig. S5† (e.g. for experiment 2, Table S1†), for all carried out experiments, the measured XRD patterns matched the theoretically calculated diffraction pattern for [Ce(phen)2(NO3)3] complexes. Moreover, the reflection patterns also indicate a direct pure phase formation of the product, confirming that the high energy emission band observed before crystallization is assigned to Ce3+ ions in the ethanolic solution and not to a crystalline reaction intermediate.
In situ IR spectra further validate mechanism of complex formation. As expected, at t = 0 min, before the addition of the 1,10-phenanthroline solution, the in situ IR spectra show the characteristic bands assigned to the NO3− vibrations at e.g. 816 cm−1, 872 cm−1, 1036 cm−1 and 1300 cm−1 (ref. 27 and 28) within the cerium(III) nitrate solution (Fig. 3 and S6†). Consistent with the data obtained from the all other in situ analyses, there was no change in the intensity of the nitrate IR bands during the first 3 minutes following the addition of the phen solution. After this point, the intensity of the nitrate IR signal begins to increase, indicating the reaction period during crystal growth, and continues to do so until t ≈ 10 min, at which point the intensity stabilizes and remains constant, indicating the end of the reaction. A possible explanation for the observed increase in IR intensity is the crystallization of the [Ce(phen)2(NO3)3] complex on the tip of the sensor, which could cause an increase in the concentration of NO3− ions within the measured area during crystal growth. This hypothesis was confirmed by a similar time-dependent profile of the IR bands at e.g. 842 cm−1, 1420 cm−1 and 1510 cm−1, assigned to the 1,10-phenanthroline vibrations29 (Fig. 3 and S7†). Before the crystallization, the concentration of the 1,10-phenanthroline molecules up to t ≈ 3 min (13.15 × 10−3 mol L−1) is low, generating only slight increases in the intensity of its IR bands (Fig. S7†). Upon crystallization on the sensor tip, the concentration of phen ligand molecules within the measured area increases, corresponding to higher increases in the intensity of the respective IR bands disproportionate to the increases in phen ligand concentration supplied by the continued dosing. In agreement with the in situ measurements of pH value, ion conductivity and luminescence mentioned above, no significant change in the intensity of the IR bands are observed after t ≈ 10 min, also indicating the completion of the crystallization process.
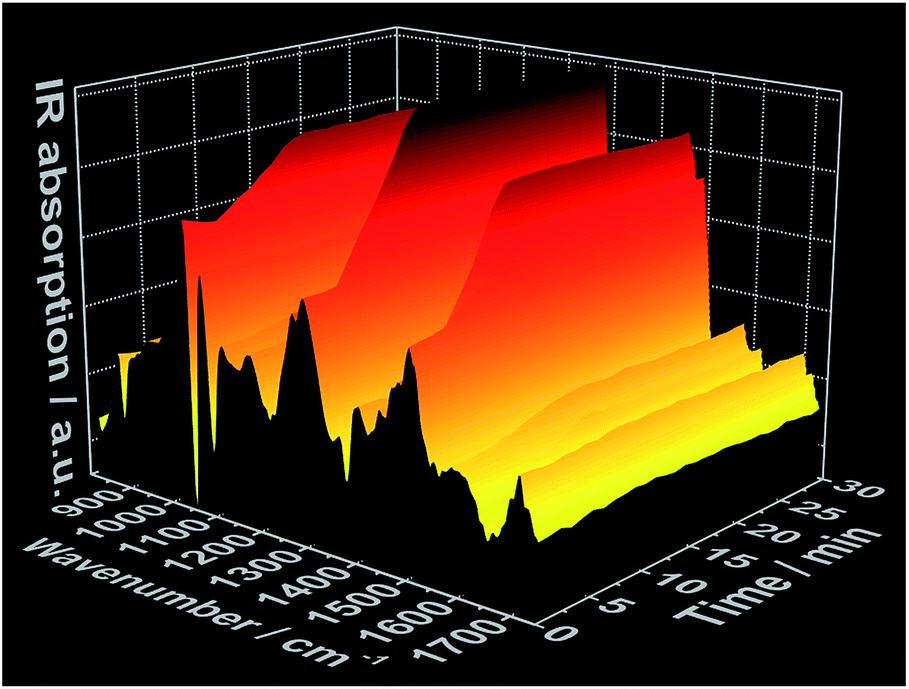 | ||
| Fig. 3 Time-dependent in situ IR spectrum recorded during crystallization of [Ce(phen)2(NO3)3] (experiment 3, Table S1†). | ||
To improve the time resolution of the preliminary data concerning the mechanism of product formation generated by ex situ XRD experiments, synchrotron-based in situ XRD measurements were performed at the beamlines P07B25 (experiment 4, Table S1†) and P0926 (experiment 5, Table S1†) at PETRA III (DESY, Hamburg). Fig. 4 shows, for instance, the time-resolved in situ X-ray diffraction patterns recorded at the P07B beamline during the addition of the 1,10-phenanthroline solution to solved cerium(III) nitrate. Similar to the results outlined in Fig. 2 for in situ luminescence measurements, in situ XRD indicate that [Ce(phen)2(NO3)3] does not immediately crystallize (Fig. S8 and S9†). Instead, the reflections assigned to the [Ce(phen)2(NO3)3] complex first arise after an induction period of approximately 3 minutes. The intensity of these reflections continuously grows across the reaction period, with a greater rate of increase seen during the addition of the 1,10-phenanthroline ligand. Also in accordance with the in situ luminescence results, in situ XRD measurements show no reflections that could be attributed to a reaction intermediate, indicating that the [Ce(phen)2(NO3)3] product is directly crystallized.
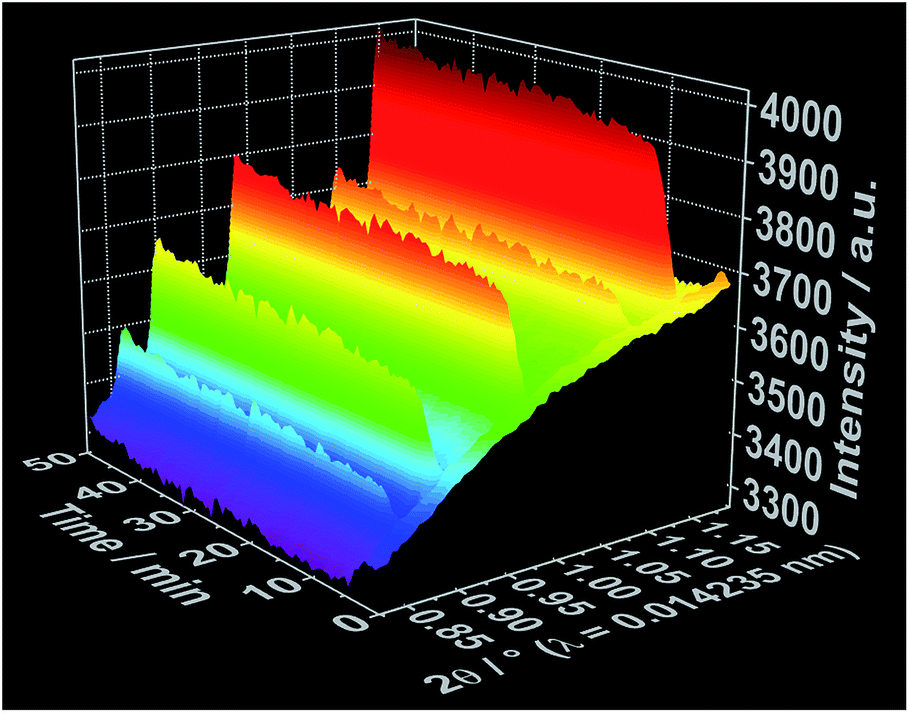 | ||
| Fig. 4 Time-dependent in situ XRD patterns recorded during crystallization of [Ce(phen)2(NO3)3] at the DESY P07B beamline (experiment 4, Table S1†). | ||
As demonstrated in our previous works,22 simultaneous in situ light transmission analysis was carried out simultaneously with in situ XRD analysis at the P09 beamline, allowing observation of phenomena occurring in the solution before the onset of the crystallization process (Fig. 5). The results generated from these experiments showed a strong decrease in light transmission upon the addition of the 1,10-phenanthroline to the cerium(III) nitrate solution which continued to decrease throughout the reaction period, reaching its lowest levels following complete crystallization. Interestingly, the initial decrease in light transmission occurred immediately after the addition of 1,10-phenanthroline, several minutes before the reflections assigned to the crystalline [Ce(phen)2(NO3)3] product could be detected via XRD (Fig. S10–S13†). Three hypotheses may explain this observation: (i) the formation of an amorphous phase before [Ce(phen)2(NO3)3] crystallization increased the turbidity of the solution, (ii) an initial formation of [Ce(phen)2(NO3)3] in solution at concentrations too low to precipitate increased the turbidity of the solution or (iii) 1,10-phenanthroline molecules absorbed irradiating light (at 365 nm), decreasing the measured intensity of the light source.
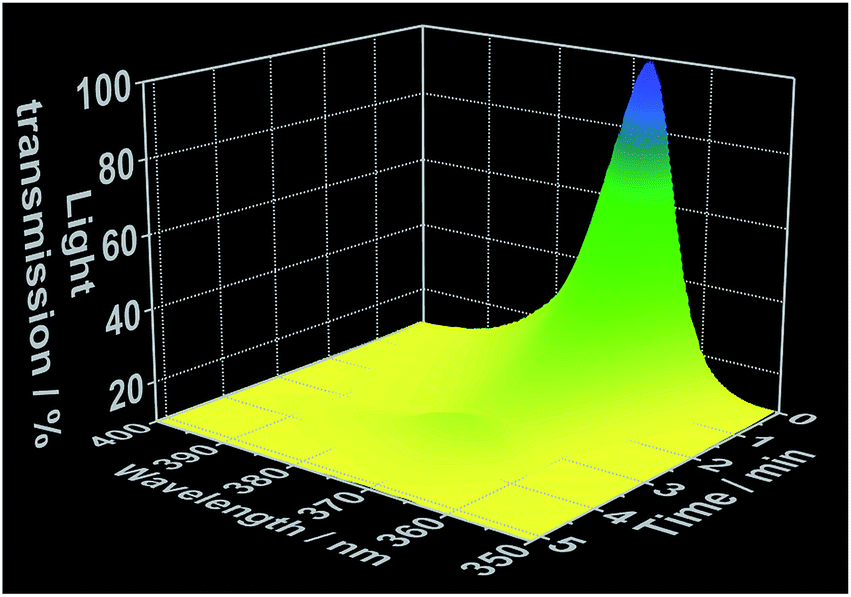 | ||
| Fig. 5 Time-dependent light transmission at 365 nm, measured simultaneously to in situ XRD at the DESY P09 beamline (experiment 5, Table S1†). | ||
We evaluated the viability of each of these hypotheses based on the in situ luminescence measurements shown in Fig. 2 and S3.† If hypothesis (i) were true, there would be a change the coordination environment around the Ce3+ ions during the formation and dissolution of the amorphous phase, inducing shifts on the Ce3+ emission spectra before crystallization. Since this phenomenon was not observed and the typical emission band of Ce3+ in ethanolic solution (Fig. 1) was invariably detected until the rise of the [Ce(phen)2(NO3)3] emission band, hypothesis (i) could be disregarded. Though [Ce(phen)2(NO3)3] is capable of absorbing light at 365 nm (Fig. S3†), making hypothesis (ii) at least theoretically viable, such solution of this complex would induce changes in the ILACS spectra. Since no [Ce(phen)2(NO3)3] emission band was detected prior to crystallization (Fig. 2 and S9†), hypothesis (ii) also could not explain the decrease of the light transmission prior to crystallization (Fig. 5). The existence of a 3–4 minute induction period of in all experiments (Fig. 2, S6, S9 and S11†), eliminates all but hypothesis (iii) as a possible explanation. Therefore, the absorption range of the 1,10-phenanthroline molecules shown by the reflectance spectra in Fig. S3† also overlaps with the spectral range of the irradiated light at 365 nm. Hence, the decrease in light transmission observed immediately following the introduction of the 1,10-phenanthroline solution is caused by absorption of light by the added reactant. After the induction period, the crystallization of the solid material causes the increase of the turbidity of the solution blocking the light transmission, which reaches at this reaction stage its lowest values.
Though it is clear the induction period at the beginning of the crystallization process exists, it is not abundantly clear why the reaction does not immediately take place. We hypothesize the delay is caused by the low pH values of the initial cerium(III) nitrate solution (Fig. 2), which gradually increases during the course of the reaction, eventually reaching the critical pH value of 5.5, at which point the ligand can deprotonate and coordinate the Ce3+. This pH is reached at approximately t = 3–4 min and allows the crystallization process to start.
Conclusions
In conclusion, ILACS was successfully applied to monitor the emission of 5d–4f Ce3+ electronic transitions in order to acquire information about the crystallization mechanism of the [Ce(phen)2(NO3)3] complex. Due to the sensitivity of the emission properties of Ce3+ ions to its coordination environment, it was possible to detect, following an induction time of few minutes, a decrease in the emission band at 415–700 nm (λex = 400 nm) assigned to the Ce3+ in ethanolic solution, indicating the begin of a desolvation process. The crystallization of the [Ce(phen)2(NO3)3] complex could be monitored through measurement of its emission band at 500–900 nm (λex = 400 nm). During the reaction, no additional emission bands besides those assigned to Ce3+ in solution and in the solid product could be observed, indicating that the [Ce(phen)2(NO3)3] complex is directly formed, without the formation of a reaction intermediate. These results were confirmed by the following additional characterization approaches: in situ measurement of the pH value, ion conductivity, infrared spectroscopy, light transmission as well as ex situ and synchrotron based in situ X-ray diffraction analysis, showing the high potential of applying Ce3+ ions as coordination sensors for monitoring crystallization processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. Wolfgang Bensch and Prof. Norbert Stock for providing the equipment needed to perform the experiments. We would also like to thank the German Research Foundation's (DFG) Priority Program 1415 and project TE 1147/1-1, the MATsynCell consortium and the Daimler and Benz Foundation (project 32-11/15) for financial support. Thanks to Milan Köppen for the development of the in situ data helper software used for analyzing the DESY and IR data. Also thanks to Dr Nicole Pienack and the ILACS Group for the enriching discussions. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to also thank Dr Sonia Francoual and Dr Jörg Strempfer for assistance in using the beamline P09 as well as Dr Uta Rütt for the assistance using the beamline P07B.References
- N. Pienack and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 2014 CrossRef CAS PubMed.
- H. Terraschke, M. Rothe and P. Lindenberg, Rev. Anal. Chem., 2017 DOI:10.1515/revac-2017-0003.
- M. Wendt, L. K. Mahnke, N. Heidenreich and W. Bensch, Eur. J. Inorg. Chem., 2016, 34, 5393 CrossRef.
- N. Heidenreich, U. Rütt, M. Köppen, A. K. Inge, S. Beier, A.-C. Dippel, R. Suren and N. Stock, Rev. Sci. Instrum., 2017, 88, 104102 CrossRef CAS PubMed.
- L. Engelke, M. Schäfer, M. Schnur and W. Bensch, Chem. Mater., 2001, 13, 1383 CrossRef CAS.
- M. Schäfer, R. Stähler, W.-R. Kiebach, C. Näther and W. Bensch, Z. Anorg. Allg. Chem., 2004, 630, 1817 Search PubMed.
- C. Schmidt, M. Feyand, A. Rothkirch and N. Stock, J. Solid State Chem., 2012, 188, 44 CrossRef CAS.
- H. Terraschke, L. R. Arana, P. Lindenberg and W. Bensch, Analyst, 2016, 141, 2588 RSC.
- H. Terraschke, M. Rothe, A.-M. Tsirigoni, P. Lindenberg, L. Ruiz Arana, N. Heidenreich, F. Bertram and M. Etter, Inorg. Chem. Front., 2017, 4, 1157 RSC.
- P. Polzin, I. V. Eliani, J. Ströh, M. Braun, N. Ruser, N. Heidenreich, P. Rönfeldt, F. Bertram, M. Suta and H. Terraschke, Phys. Chem. Chem. Phys., 2017 Search PubMed , submitted.
- P. Lindenberg, L. Ruiz Arana, N. Heidenreich and H. Terraschke, Z. Anorg. Allg. Chem., 2016, 642, 1074 Search PubMed.
- H. Terraschke and C. Wickleder, Chem. Rev., 2015, 115, 11352 CrossRef CAS PubMed.
- H. Terraschke, M. Suta, M. Adlung, S. Mammadova, N. Musayeva, R. Jabbarov, M. Nazarov and C. Wickleder, J. Spectrosc., 2015, 2015, 541958 Search PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1 CrossRef CAS.
- W. M. Yen, H. Yamamoto and S. Shionoya, Phosphor Handbook Laser and Optical Science and Technology, CRC Press, Boca Raton, 2006 Search PubMed.
- P. Dorenbos, J. Phys.: Condens. Matter, 2003, 15, 4797 CrossRef CAS.
- C. Tiseanu, V. I. Parvulescu, B. Cojocaru, K. Pemar, M. Sanchez-Dominguez and M. Boutonnet, J. Phys. Chem. C, 2012, 116, 16776 CAS.
- J.-C. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1049 RSC.
- P. Dorenbos, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235110 CrossRef.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer, Berlin, 1994 Search PubMed.
- L. Ning, C. Zhou, W. Chen, Y. Huang, C. Duan, P. Dorenbos, Y. Tao and H. Liang, J. Phys. Chem. C, 2015, 119, 6785 CAS.
- N. Pienack, L. Ruiz Arana, W. Bensch and H. Terraschke, Crystals, 2016, 6, 157 CrossRef.
- K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed.
- Q. Y. Lin and Y. L. Feng, Z. Kristallogr., 2003, 218, 531 CAS.
- N. Shell, A. King, F. Beckmann, T. Fischer, M. Müller and A. Schreyer, Mater. Sci. Forum, 2014, 77, 57 Search PubMed.
- J. Strempfer, S. Francoual, D. Reuther, D. K. Shukla, A. Skaugen, H. Schulte-Schrepping, T. Kracht and H. Franz, Synchrotron Radiat., 2013, 20, 541 CrossRef CAS PubMed.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme, Stuttgart, 2012 Search PubMed.
- M. Gaye, F. B. Tamboura and A. S. Sall, Bull. Chem. Soc. Ethiop., 2003, 17, 27 CrossRef CAS.
- M. M. Campos-Vallette, R. Clavijo, F. Mendizabal, W. Zamudio, R. Baraona and G. Diaz, Vib. Spectrosc., 1996, 12, 37 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07488c |
| This journal is © The Royal Society of Chemistry 2017 |