DOI:
10.1039/C7RA07432H
(Paper)
RSC Adv., 2017,
7, 38805-38811
The combination of glass dust and glass fiber as fluxing agents for ceramifiable silicone rubber composites
Received
5th July 2017
, Accepted 3rd August 2017
First published on 9th August 2017
Abstract
Ceramifiable composites based on silicone rubber (SR) filled with mica powder, glass dust (GD) and short glass fiber (SGF) were successfully prepared for high-temperature applications. The effect of the GD/SGF phr ratio in the fluxing agents on the ceramifiable and mechanical properties of the silicone rubber composites was studied. The results demonstrated that the composites showed excellent properties when the ratio of GD/SGF was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the corresponding tensile strength was 6.5 MPa. The elongation at break of the composites reached 245.4% and the flexural strength of sintered samples produced at 1000 °C was 16.2 MPa. Thermogravimetric analysis (TGA) results indicated that the char residue was 57.5% at 700 °C, which was much higher than the virgin SR residue. The ceramization process at different sintering temperatures was consequently studied through infrared spectroscopy (IR), X-ray diffraction (XRD) and scanning electron microscopy (SEM). The linear shrinkage and flexural strength of sintered samples increased with increasing sintering temperature from 600 to 1000 °C. The FTIR and XRD tests indicated that the fluorophlogopite mica structure was destroyed and a new cristobalite crystalline phase was formed at elevated temperature. The SEM revealed that a coherent and dense microstructure of sintered samples was formed with increased temperatures, which contributed to the flexural strength improvement.
:1 and the corresponding tensile strength was 6.5 MPa. The elongation at break of the composites reached 245.4% and the flexural strength of sintered samples produced at 1000 °C was 16.2 MPa. Thermogravimetric analysis (TGA) results indicated that the char residue was 57.5% at 700 °C, which was much higher than the virgin SR residue. The ceramization process at different sintering temperatures was consequently studied through infrared spectroscopy (IR), X-ray diffraction (XRD) and scanning electron microscopy (SEM). The linear shrinkage and flexural strength of sintered samples increased with increasing sintering temperature from 600 to 1000 °C. The FTIR and XRD tests indicated that the fluorophlogopite mica structure was destroyed and a new cristobalite crystalline phase was formed at elevated temperature. The SEM revealed that a coherent and dense microstructure of sintered samples was formed with increased temperatures, which contributed to the flexural strength improvement.
1. Introduction
Compared to the polyolefin materials, silicone rubber (SR) has been extensively used in various domains including spaceflight, automobiles and thermal protection systems, due to its corresponding excellent performance in chemical stability, abrasion resistance and electrical insulation.1–4 Silicone rubber could be transformed into amorphous silica ash after pyrolysis and it is quite loose without any strength, leading to the bad flame retardancy of silicone rubber.5 Therefore, it is critical to improve the ablation resistance properties of silicone rubber at high temperature, to fulfill the increasing requirements in special applications such as in fire resistant wires and cables. Ceramifiable polymer composites are a new type of fire-resistant material, which could be used extensively throughout the buildings and transportation industries. The ceramifiable polymer composites can be converted into ceramics after pyrolysis and the formed ceramics can protect the inner copper wires from melting in the presence of fire and ensure the integrity of the electric circuit for 30 min when subjected to fire or elevated temperature.6,7
Generally, the ceramifiable silicone rubber composites are comprised of the silicone rubber matrix, the refractory fillers (montmorillonite, talc, mica), the fluxing agents (glass frits, zinc borate, APP) and other additives.8–11 The refractory fillers can provide self-supporting skeleton structures and retain the original shape of the materials following the silicone rubber decomposition.12 In addition, these fillers can improve the quality of the silica residues through a eutectic reaction at elevated temperature. Fluorophlogopite mica (KMg3(AlSi3O10)F2) is a kind of ceramifiable additive, which could be obtained by the high temperature melting crystallization of chemical raw materials. Differently from the natural mica, the synthetic fluorophlogopite mica exhibits excellent electrical insulation, good chemical stability and improved high temperature resistance.13–15 In addition, the fluxing agents including zinc borate, APP and glass dust with low softening point temperature were incorporated into the silicone rubber to promote the low temperature ceramification and improve the strength of the ceramic residues.16–18 At present, a high number of researchers have been conducted on the addition of glass frits with low softening point temperatures in the ceramizable polymer composites. Mansouri et al. prepared the ceramizable silicone rubber composites using glass frits with different softening point temperature as fluxing agents at a low temperature (600–1000 °C) and the sintering mechanisms were originally proposed simultaneously.19–21 Li et al. investigated the ceramifiable ethylene–vinyl acetate composites with glass frits with low melting temperature and it was discovered that the mass ratio of glass frits and mica powder had a marked effect on the mechanical properties of the residues formed at high temperatures.22
The short glass fibers (SGF) are commonly utilized as functional fillers in thermoplastic composites to increase the mechanical properties. In addition, the SGF has a low conductivity and excellent heat insulation with a high softening point temperature, which could be utilized as an ablative material.23–25 A high number of research works were conducted on the application of glass dust (GD) into silicone rubber composites, whereas the study of the GD with low softening point temperature and the SGF with high softening point temperature combination as fluxing agents has not been reported in ceramifiable silicone rubber composites.
In the present work, the ceramifiable silicone rubber composites based on the GD and SGF combination as fluxing agents were formulated for optimal comprehensive properties (mechanical and ceramifiable properties) to be reached. The effect of GD/SGF phr ratio on the linear shrinkage, the mechanical strength of sintered samples at high temperature and the mechanical properties, along with the thermal stability of the composites was systematically investigated. The self-supporting property of silicone rubber composites and linear shrinkage and flexural strength of the sintered samples were tested at various temperatures. Moreover, the X-ray diffraction (XRD), scanning electron microscopy and (SEM) and Fourier-transform infrared spectroscopy (FT-IR) were used to investigate the ceramization process at different sintering temperature.
2. Experimental
2.1. Materials
High-temperature vulcanizing (HTV) silicone rubber (R401/70S) was purchased from Wacker Chemie Company and 2,5-dimethyl-2,5-bis(tert butyl peroxy) hexane (DBPMH) as vulcanizing agent was received from Sinopharm Chemical Reagent Co., Ltd., China. The short glass fiber (SGF) with the softening temperature of 930 °C and the fluorophlogopite were supplied by the Lingshou Jiali Processing Factory, China. The glass dust (GD) with the softening temperature of 430 °C was supplied by Donggu New Materials Co., Ltd., China. Table 1 presents the chemical compositions of fluorophlogopite and glass dust.
Table 1 Chemical compositions of fluorophlogopite and glass dust (wt%)
| Oxide |
Fluorophlogopite |
Glass dust |
| SiO2 |
33.43 |
1.42 |
| MgO |
28.96 |
26.02 |
| K2O |
11.80 |
22.65 |
| P2O5 |
0.15 |
28.99 |
| CaO |
0.52 |
0.37 |
| Al2O3 |
10.67 |
9.47 |
| F |
14.19 |
9.71 |
| Others |
0.28 |
1.37 |
2.2. Sample preparation
All the inorganic fillers were dried at 80 °C for 12 h in an oven. The matrix and fillers were mixed in a two-roll open mill to obtain homogeneity at room temperature for 15 min. And then the vulcanizing agent DBPMH was incorporated into the composites. The resulting mixtures were then vulcanized at 180 °C and a pressure of 15 MPa for 10 min. The post-cured was conducted in drying cabinet at 200 °C for 2 h. The compounding formulas of prepared samples are given in Table 2.
Table 2 Formulations of ceramifiable silicone rubber compositesa
| Composition (phr) |
Silicone rubber |
Mica |
Glass dust |
Glass fiber |
DBPMH |
| Parts per hundreds of rubber. |
| SR0 |
100 |
— |
— |
— |
0.8 |
| SR1 |
100 |
40 |
20 |
— |
0.8 |
| SR2 |
100 |
40 |
10 |
10 |
0.8 |
| SR3 |
100 |
40 |
13.3 |
6.7 |
0.8 |
| SR4 |
100 |
40 |
15 |
5 |
0.8 |
| SR5 |
100 |
40 |
— |
20 |
0.8 |
2.3. Sintering
Heating of the flat sheet samples to the various temperatures (T = 600–1000 °C) was executed in a muffle furnace in air atmosphere at a heating rate of 10 °C min−1. All samples were kept at target temperature for 30 min and then naturally cooled to room temperature.
2.4. Characterizations
2.4.1. X-ray fluorescence (XRF). Chemical compositions of fluorophlogopite mica and glass dust were studied with a Shimadzu XRF-1800 spectrometer by X-ray fluorescence technique.
2.4.2. Self-supporting property. The specimen of dimensions 50 mm × 5 mm × 2 mm were placed on a rectangular piece of refractory. Furthermore, the length of the samples was perpendicular to the width of the brick, and 20% of the length extended from the width of the brick.26 Then, the specimens were sintered at various temperatures for 30 min and testing the sintered samples if it could sustain its own weight.
2.4.3. Mechanical property test. A computer-controlled electronic tensile testing machine (CMT2203) was used to evaluate the mechanical properties of the samples, according to the GB/T 528-1998. The cross-head speed for mechanical tests was 500 mm min−1 and the results of five specimens were averaged.
2.4.4. Flexural strength measurements. The Sans universal testing machine CMT4204 was used to perform three-point bend tests in conformity to the GB/T 9596-2006 standard. The cross-head speed was maintained at 0.5 mm min−1 and at least five specimens were averaged.
2.4.5. Linear shrinkage. The linear shrinkage of the sintered samples were measured using the following equation:
| Linear shrinkage (%) = (L1−L2)/L1 × 100% |
where L1 is the dimensional length before sintering, L2 is the dimensional length after sintering.
2.4.6. Apparent porosity. Apparent porosity of the samples sintered at different temperatures was performed using the Archimedes' displacement method.
2.4.7. Thermogravimetric analysis (TGA). Thermogravimetric analysis (TGA) was carried out on a TA Instruments STA409PC under nitrogen atmosphere. The samples (approximately 10 mg) were heated in the temperature range of 25–800 °C at a heating rate of 10 °C min−1.
2.4.8. X-ray diffraction (XRD). The phase composition of the sintered samples was conducted by X-ray diffraction (XRD) with a D/max 2550 VB/PC X-ray diffractometer (Rigaku Corporation, Japan).
2.4.9. SEM. The microstructure of the sintered samples after sintering was characterized by a scanning electron microscope (SEM S3400N HITACHI Instruments) at an accelerating voltage of 5 kV. The cross-section of the samples was sputtered with a thin layer of gold.
2.4.10. FTIR. Fourier-transform infrared spectroscopy (FTIR) of sintered samples sintered at differents temperatures was recorded on a Nicolet 6700 spectrometer with the range of 4000–400 cm−1.
3. Results and discussion
3.1. Mechanical properties of ceramifying silicone rubber composites
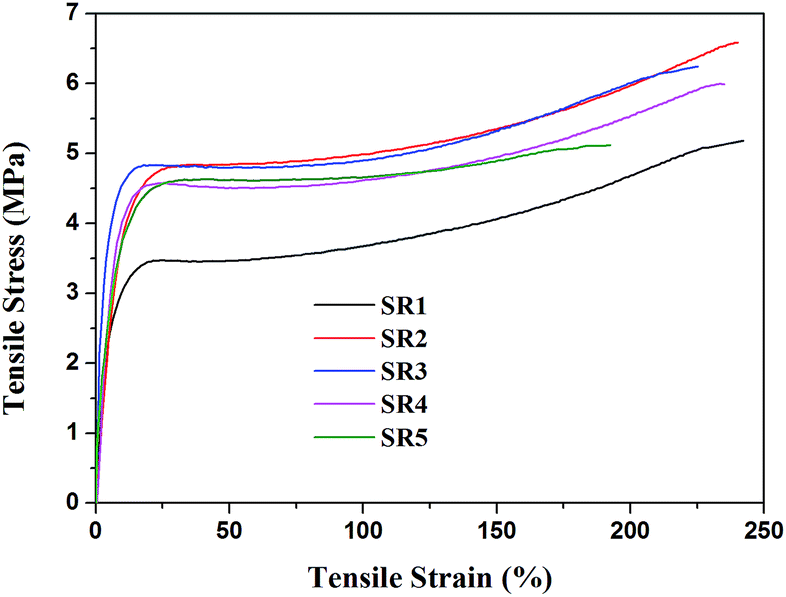
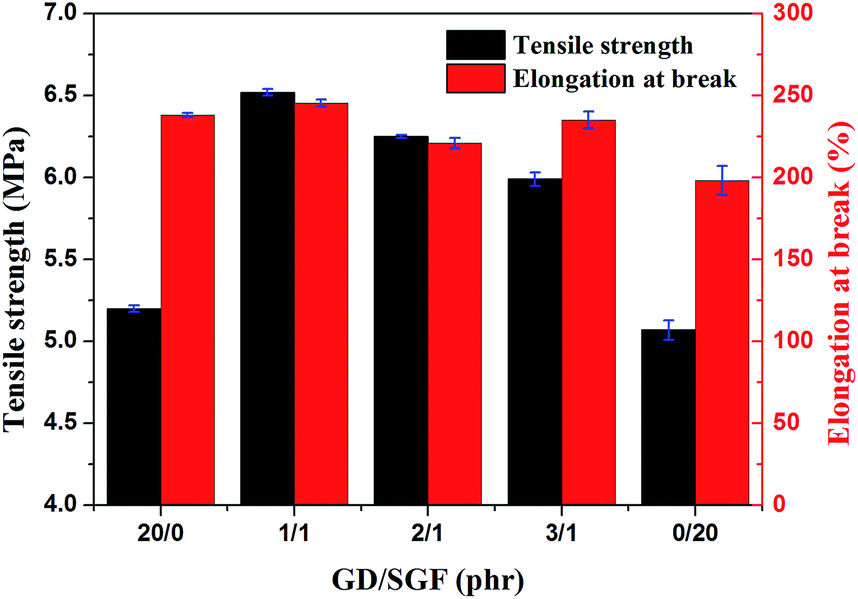
The stress–strain curves obtained from tensile test are presented in Fig. 1. The mechanical properties (tensile strength and elongation at the break) of all the composites are illustrated in Fig. 2. It could be clearly seen from stress–strain curves (Fig. 1) that all the silicone rubber composites showed the typical tensile behavior of elastic rubber. The tensile strength and elongation at the break of the virgin silicone rubber (SR0) was 10.3 MPa and 450%, respectively, which was reported in our previous paper. It was seen that the tensile strength and elongation at the break of all the SR composites were lower than the virgin silicone rubber. This result was attributed to the fact that the R401/70S we purchased was a high-strength silicone rubber with fumed SiO2 and the sample SR0 had reached the optimal mechanical balance.27 Consequently, the additions of inorganic fillers in the high-strength silicone rubber would break this balance and reduce the mechanical properties of the silicone rubber composites. It could be found that the sample SR5 exhibited the lowest mechanical strength and elongation at the break among all silicone rubber composites. This occurred because the poor compatibility between the glass fiber and the silicone rubber matrix, leading to the significantly decrease in the mechanical strength of ceramifiable silicone rubber.28 In addition, it could be observed that the tensile strength of silicone rubber composite decreased from 6.5 to 5.9 MPa with the ratio of GD/SGF increased from 1:1 to 3:1 and the sample SR2 exhibited the best mechanical properties of all the samples.
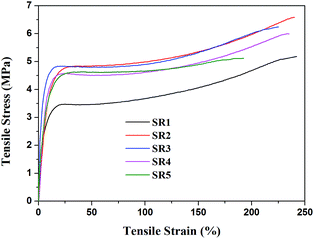 |
| | Fig. 1 Stress–strain curves of ceramifying silicone rubber composites. | |
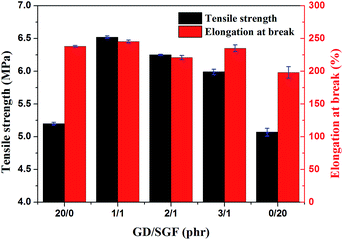 |
| | Fig. 2 The effect of GD/SGF phr ratio on the mechanical properties of ceramifying silicone rubber composites. | |
3.2. Linear shrinkage and flexural strength of the sintered samples
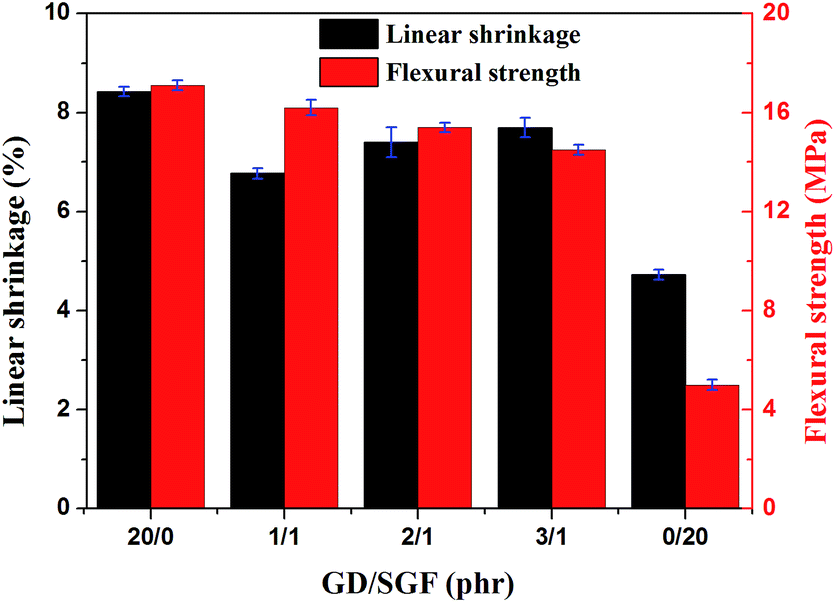
The dimensional stability of the silicone rubber composites at elevate temperature could be evaluated by the linear shrinkage measurement of the sintered samples after sintering at 1000 °C for 30 min. The effect of GD/SGF phr ratio on the dimensional stability and flexural strength of the sintered samples at 1000 °C is illustrated in Fig. 3. It could be observed that the sample SR1 following sintering displayed the maximum linear shrinkage, while the sintered sample of sample SR5 presented the minimal linear shrinkage at 1000 °C. It was indicated that the sample SR5 had good dimensional stability, whereas the sample SR1 presented the worst thermal stability at high temperature. In addition, the increased concentration of GD in the fluxing agents led to the increase in linear shrinkage of the sintered samples and the minimum linear shrinkage was reached when the phr ratio of GD/SGF was 1:1 among the samples SR2, SR3 and SR4. Flexural strength is a prerequisite for to be applied in fireproofing applications. It should be noted that the sintered samples of sample SR5 displayed the minimum flexural strength (5 MPa). Although the sample SR5 exhibited the best dimensional stability at high temperature among all specimens, the mechanical strength of the residues of sample SR5 was still poor, which could not meet the practical application. In addition, it could be seen that the residues of sample SR2 exhibited the highest flexural strength (16.2 MPa) and increased concentration of GD in the fluxing agents led to the flexural strength reduction of the sintered samples.
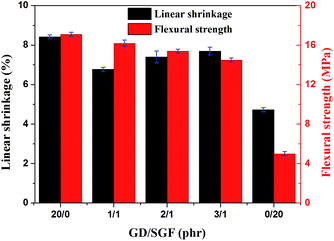 |
| | Fig. 3 The effect of GD/SGF phr ratio on linear shrinkage and flexural strength of the sintered samples. | |
3.3. Thermogravimetric analysis
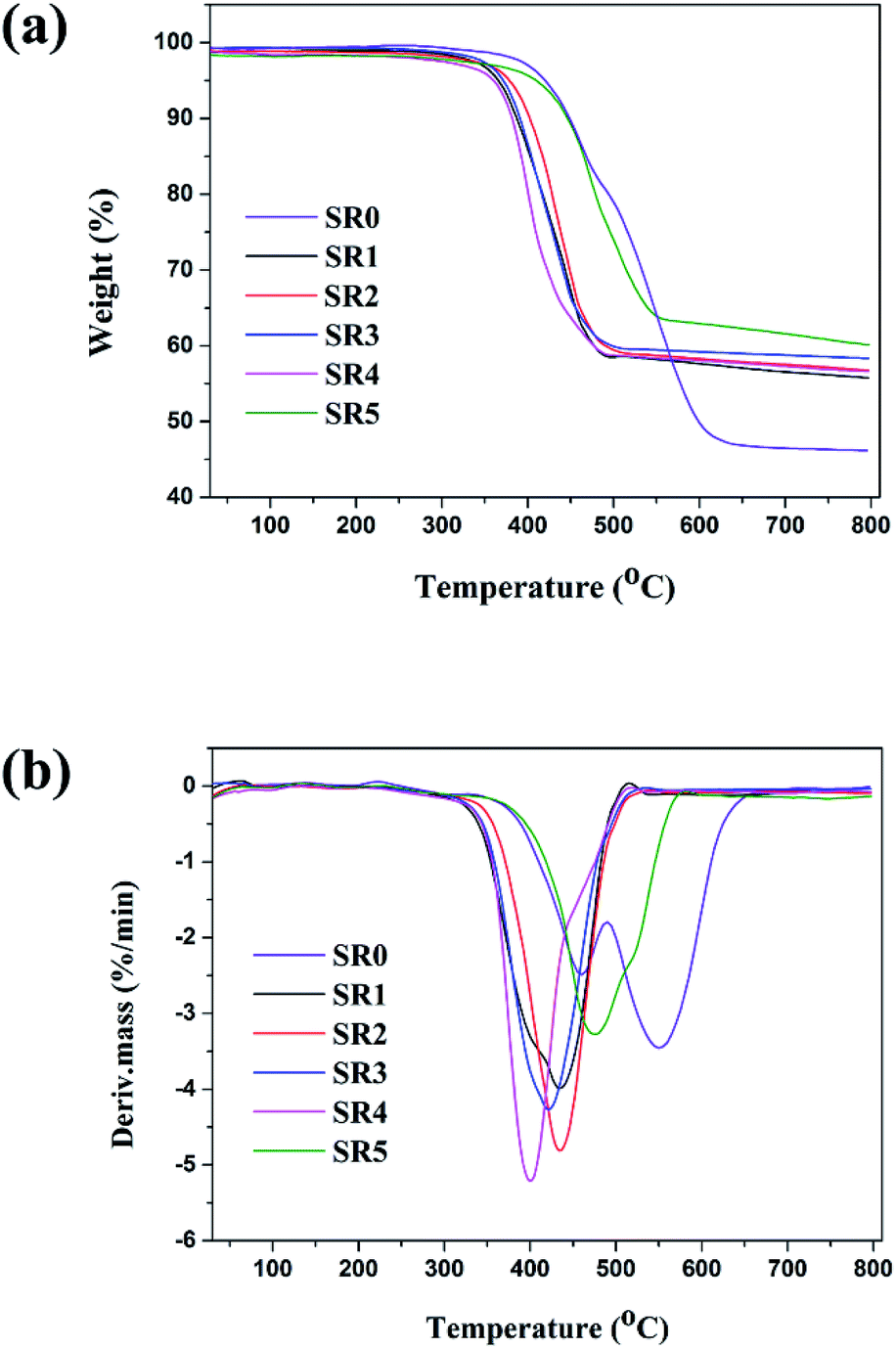
TGA and DTG curves of SR and ceramifiable silicone rubber composites under N2 atmosphere are presented in Fig. 4 and the detailed data for the specimen are listed in Table 3. It could be seen that the thermal degradation of the virgin SR consisted of two main steps and a 46.5% residue that remained at 700 °C. The first decomposition step occurred at 410.9 °C with a maximum weight loss at 461 °C. The second decomposition step occurred from 514.6 to 597.8 °C with a maximum weight loss at 549 °C. Compared to the pure SR, the onset degradation temperature of the ceramifiable silicone rubber composites decreased. This could be attributed to the presence of metal oxide in glass dust and other fillers, which would accelerate the thermal degradation of these composites.29,30 Compared with the sample SR0, the ceramifiable SR composites exhibited an enhanced thermal behavior at temperatures ranging from 550 to 700 °C, producing higher amounts of residues. Among all silicone rubber composites, the sample SR5 exhibited the highest onset degradation temperature (Tonset = 424.6 °C), while the sample SR1 displayed the lowest onset degradation temperature (Tonset = 369.8). Compared with the sample SR1, the thermal stability of the SR2 was improved significantly and the char residue increased from 56.6% to 57.5%. As the increased concentration of GD in the fluxing agent compounds, the onset decomposition temperature (Tonset) decreased from 390.6 °C to 371.1 °C. This behavior occurred possibly due to the higher thermal stability of glass fiber compared to the glass dust at high temperature, resulting in the sample SR2 having higher thermal stability.31 From the analysis above, it was clear that the sample SR2 (GD/SGF = 1:1) exhibited the excellent mechanical and ceramifiable properties of the silicone rubber composites. Therefore, the optimal formula (sample SR2) would be selected to further study the ceramization process of silicone rubber composites at different sintering temperatures in the following sections.
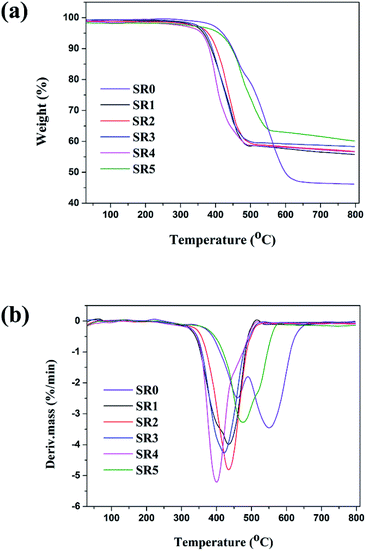 |
| | Fig. 4 Thermal stability of ceramifying silicone rubber composites. | |
Table 3 TGA data of silicone rubber composites in nitrogen atmosphere
| Sample |
Tonset (°C) |
Tmax1a (°C) |
Tmax2 (°C) |
Residues (wt%) at 700 °C |
| Tmax: the temperature at the maximum mass loss rate. |
| SR0 |
410.9 |
461.0 |
549 |
46.5 |
| SR1 |
369.8 |
436.5 |
— |
56.6 |
| SR2 |
390.6 |
436.7 |
— |
57.5 |
| SR3 |
372.6 |
421.8 |
— |
58.8 |
| SR4 |
371.1 |
401.9 |
— |
57.3 |
| SR5 |
424.6 |
475.6 |
— |
61.6 |
3.4. Ceramization process of silicone rubber composites at different temperatures
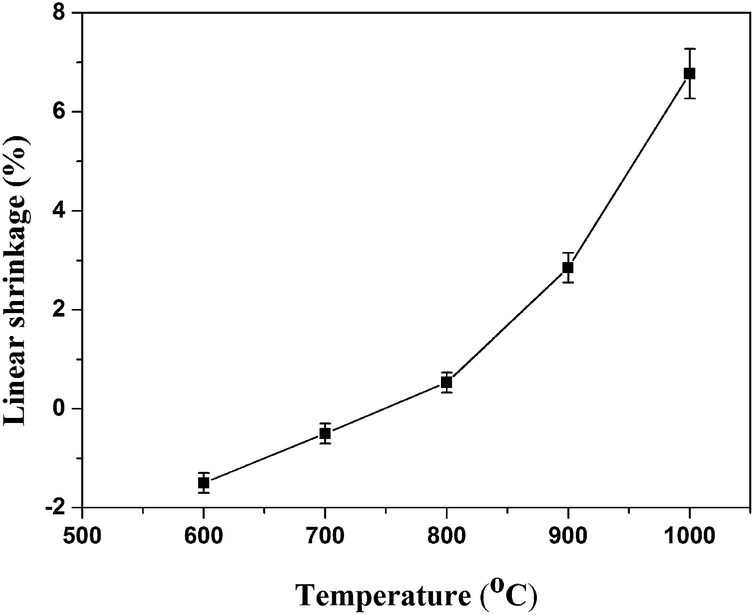
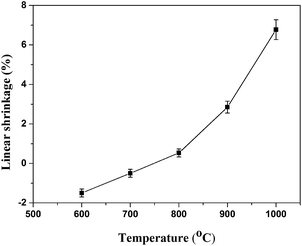
3.4.1. Linear shrinkage and self-supporting property of sintered samples. The linear shrinkage of sintered samples after sintering at different temperatures is illustrated in Fig. 5. It could be observed that the linear shrinkage of sintered samples increased from −1.5% to 6.77% at the temperature range from 600 to 1000 °C. The value of linear shrinkage was negative when the sintering temperature at 600 and 700 °C, indicating that the decomposition of silicone rubber matrix causing the expansion and a high number of holes remained in the residue. The similar results could be found in Guo's research.18 It should be noted that the linear shrinkage of sintered samples changed from negative to positive with increasing sintering temperature from 700 to 800 °C, indicating a degree of sintering in the sintered samples had occurred. The value of linear shrinkage reached a maximum when the sintering temperature was 1000 °C, indicating that more liquid phase at elevated temperatures was produced to fill the holes and voids, leading to the linear shrinkage increasements.
 |
| | Fig. 5 The linear shrinkage of the sintered samples. | |
Fig. 6 presents the effects of sintering temperature on the self-supporting properties of the silicone rubber composites. It could be clearly observed that the self-supporting was performed well for the weight of the residues to be supported in the range of temperatures from 600 °C to 1000 °C.
 |
| | Fig. 6 Self-supporting of ceramifying silicone rubber composites. | |
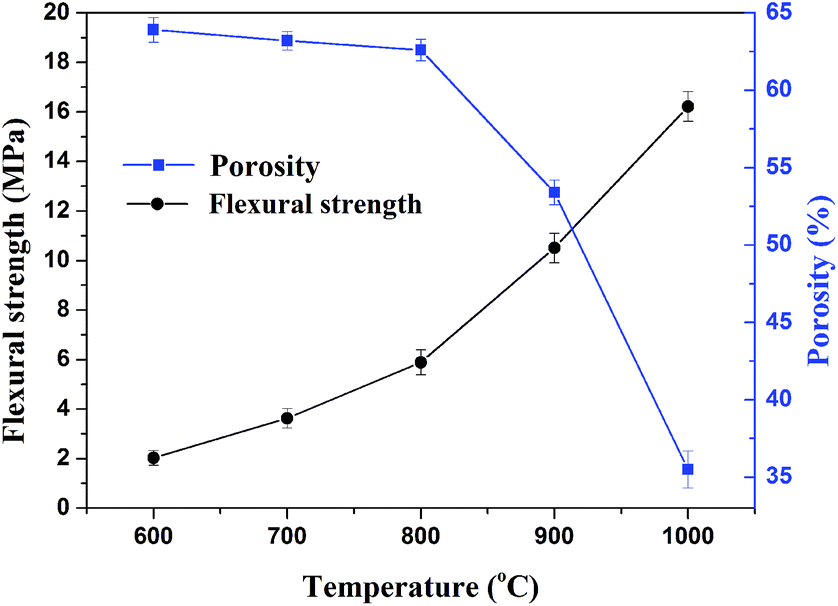
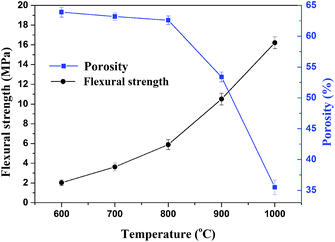
3.4.2. Flexural strength and apparent porosity. The flexural strength and apparent porosity of the sintered samples under different sintering temperatures for 30 min are presented in Fig. 7. Apparently, the apparent porosity of sintered samples decreased and the flexural strength of sintered samples increased with increasing sintering temperature from 600 to 1000 °C. Below 800 °C, the apparent porosity gradually varied with the sintering temperature, whereas beyond 800 °C, the porosity suddenly decreased due to a glassy phase increase into the certain pores, forming a more compact structure. When the sintering temperature was 1000 °C, the flexural strength of sintered samples presented the maximal value of 16.2 MPa and the apparent porosity was reduced to 35.5%. The prepared porosity ceramics with excellent mechanical strength was predicted to provide satisfactory barrier properties for both heat and mass flow.9
 |
| | Fig. 7 The flexural strength and apparent porosity of the sintered samples. | |
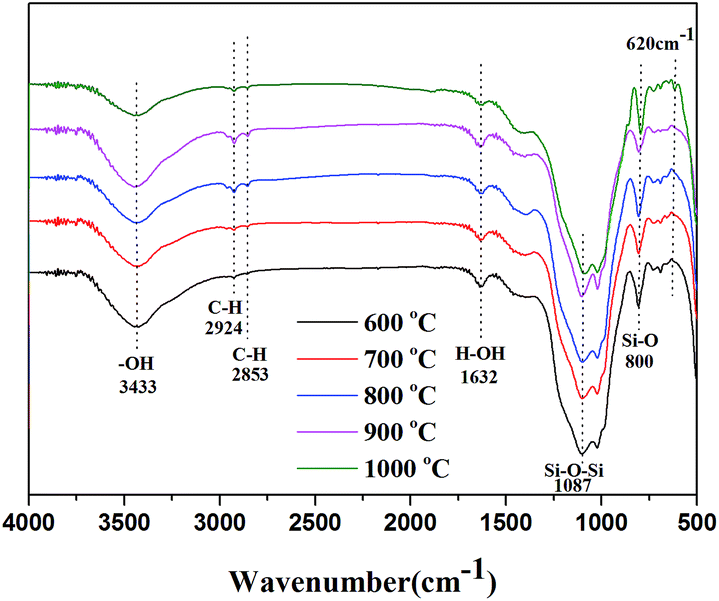
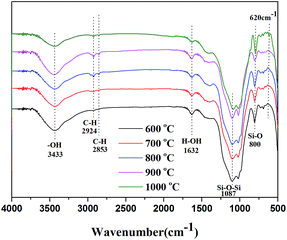
3.4.3. FT-IR analysis of sintered samples. Fig. 8 shows the FTIR spectra of the sintered samples after sintering at various temperatures for 30 min. The peaks at 3445 cm−1 was assigned to the stretching vibration of hydroxyl group and the absorption peak at 1637 cm−1 was attributed to the existence of moisture in the sintered samples. For the all sintered samples, the peaks at 1087 cm−1 (Si–O–Si asymmetric stretching) and 800 (Si–O bending vibration) cm−1 were attributable to the quartz or amorphous silica.32,33 The peaks at about 2924, 2853 cm−1 were assigned to the stretching vibration of C–H in Si–CH3.26,34 Additionally, a new characteristic peak at 620 cm−1 for sintered samples sintered at 1000 °C was appeared, which was attributed to the characteristic peak of cristobalite.35,36 It indicated that the quartz or amorphous silica had been transformed into the cristobalite at high temperature and the crystal structure of cristobalite could be further verified by XRD test.
 |
| | Fig. 8 FT-IR spectra of the sintered samples. | |
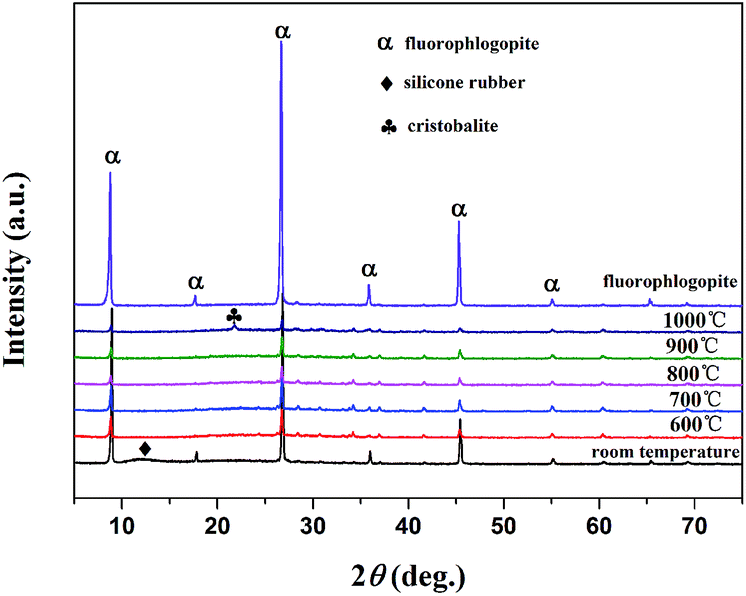
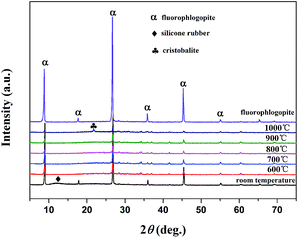
3.4.4. XRD analysis. The XRD patterns of sintered samples sintered at different temperatures of 600 °C, 700 °C, 800 °C, 900 °C and 1000 °C are illustrated in Fig. 9. As displayed in Fig. 9, it could be observed that the mainly crystalline phases of the silicone rubber composites at room temperature was fluorophlogopite mica (JCPDS file no. 16-0344) and the silicone rubber (2θ = 12.4°). The X-ray pattern for the silicone rubber composites at room temperature displayed the peaks at 2θ = 12.4°, corresponding to the amorphous structure peak of the silicone rubber. The XRD diffraction peaks of the silicone rubber composites disappeared after sintering at 600 °C, indicating the silicone rubber decomposition had taken place. It should be noted that the characteristic peaks of the sintered samples sintered at 600 °C were in accordance with the peaks of the fluorophlogopite mica, indicating that the fluorophlogopite mica was the main crystalline phase in the residue. It could also be observed that the XRD spectra were significantly similar and the characteristic peak of the fluorophlogopite mica became weaker as the sintering temperature increased from 600 °C to 700 °C, 800 °C and 900 °C. Following heating to 1000 °C, a new peak appeared at 2θ = 21.7° except the peaks of the fluorophlogopite mica, which was attributed to the cristobalite crystalline phase appearance.37 The fluorophlogopite mica disassembly and the new cristobalite crystals appearance demonstrated that the eutectic mixture must have formed during sintering.
 |
| | Fig. 9 XRD patterns of the sintered samples. | |
3.4.5. SEM analysis. Fig. 10 illustrates the cross-section morphology of sintered samples prepared at different sintering temperatures for 30 min. As shown in Fig. 10(a), the interface between the SGF and the GD was clear and a loose structure was formed due to the degradation of silicone rubber, which resulted in the weak mechanical strength of sintered samples. Fig. 10(b) and (c) showed the interfacial adhesion between the SGF and the solidified phase was slightly enhanced with limited binding. Compared to the microstructure of the sintered samples sintered at 800 °C in Fig. 10(c), the interface between the SGF and the solidified phase became more blurry, and a dense structure was found with the sintering temperature increased to 900 °C, leading to a higher flexural strength (10.5 MPa). As shown in Fig. 10(e), the short glass fibers were disappeared completely and a continuous and porous microstructure was formed at 1000 °C, which caused the improvement of flexural strength of sintered samples.
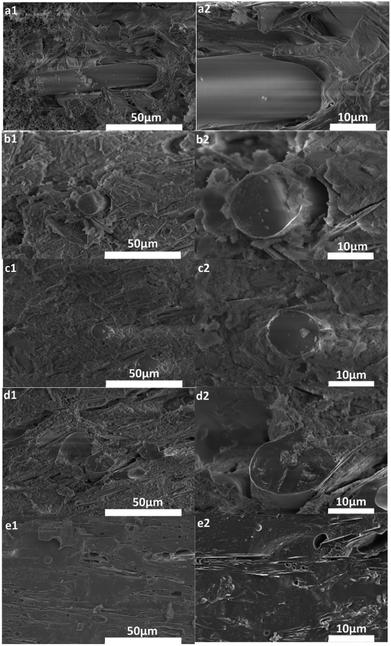 |
| | Fig. 10 Cross-sectional images of sintered samples after sintering at different temperatures: 600 °C (a1, a2), 700 °C (b1, b2), 800 °C (c1, c2), 900 °C (d1, d2), 1000 °C (e1, e2). | |
4. Conclusions
In our work, the ceramifiable silicone rubber composites were successfully prepared for high-temperature applications. The effect of GD/SGF phr ratio on the mechanical and ceramifiable properties of the composites was studied. When the phr ratio of GD/SGF was 1:1, the corresponding tensile strength was 6.5 MPa and the elongation at the break was 245.4%, and its linear shrinkage of sintered samples was 6.7%, which exhibited the optimum condition. TGA revealed that the onset degradation temperature of the composites was 390.6 °C and the char residues was 57.5%. The ceramization process of the composites at various sintering temperatures was consequently investigated. The flexural strength of the sintered samples at high temperature was 16.2 MPa and a relatively low apparent porosity of 35.5% was achieved. The XRD displayed the decrease of the fluorophlogopite mica phase along with the new cristobalite crystals production, indicating that the eutectic reaction had occurred at high temperatures. The SEM results indicated that the dense and continuous microstructure was formed with the increasing of sintering temperature.
Acknowledgements
This work was supported by the Scientific and Technological Achievements Transformation Program of Jiangsu Province (SBA2014010034), and the Ningbo Industrial Major Projects (201601ZD-A01026).
References
- S. M. Shang, L. Gan, M. C. W. Yuen, S. X. Jiang and N. M. Luo, Composites, Part A, 2014, 66, 135–141 CrossRef CAS.
- D. Kronlund, M. Lindén and J. H. Smått, Constr. Build. Mater., 2016, 124, 1051–1058 CrossRef CAS.
- L. Liu, M. Tian, W. Zhang, L. Q. Zhang and J. E. Mark, Polymer, 2007, 48, 3201–3212 CrossRef CAS.
- Y. Z. Song, J. H. Yu, L. H. Yu, F. E. Alam, W. Dai, C. Y. Li and N. Jiang, Mater. Des., 2015, 88, 950–957 CrossRef CAS.
- E. Delebecq, S. Hamdani-Devarennes, J. Raeke, J. M. Lopez-Cuesta and F. Ganachaud, ACS Appl. Mater. Interfaces, 2011, 3, 869–880 CAS.
- M. Imiela, R. Anyszka, D. M. Bielinski, Z. Pędzich, M. Zarzecka-Napierała and M. Szumera, J. Therm. Anal. Calorim., 2016, 124, 197–203 CrossRef CAS.
- J. Dul, G. Parys, Z. Pędzich, D. M. Bielinski and R. Anyszka, Int. J. Adv. Mater. Manuf. Charact., 2012, 1, 29–35 Search PubMed.
- R. Anyszka, D. M. Bieliński, Z. Pędzich and M. Szumera, J. Therm. Anal. Calorim., 2015, 119, 111–121 CrossRef CAS.
- S. Hu, F. Chen, J. G. Li, Q. Shen, Z. X. Huang and L. M. Zhang, Polym. Degrad. Stab., 2016, 126, 196–203 CrossRef CAS.
- L. G. Hanu, G. P. Simon and Y. B. Cheng, Polym. Degrad. Stab., 2006, 91, 1373–1379 CrossRef CAS.
- Z. Al-Hassany, A. Genovese and R. A. Shanks, eXPRESS Polym. Lett., 2010, 4, 79–93 CrossRef CAS.
- X. P. Zhang, Y. Y. Guan, Y. Xie and D. Qiu, RSC Adv., 2016, 6, 7970–7976 RSC.
- Q. S. Wang, J. Li, Y. Lei, Y. Wen, Z. X. Wang, X. Y. Zhan, F. Wang, F. M. Wang, Y. Huang, K. Xu and J. He, Adv. Mater., 2016, 28, 3596–3601 CrossRef CAS PubMed.
- M. Garai, N. Sasmal, A. R. Molla, S. P. Singh, A. Tarafder and B. Karmakar, J. Mater. Sci., 2014, 49, 1612–1623 CrossRef CAS.
- M. Garai and B. Karmakar, J. Alloys Compd., 2016, 678, 360–369 CrossRef CAS.
- M. Imiela, R. Anyszka, D. M. Bielinski, Z. Pedzich, M. Zarzecka and M. Szumera, J. Therm. Anal. Calorim., 2016, 124, 197–203 CrossRef CAS.
- J. C. Zhou, X. Chen, Y. Y. Wang and B. X. Zhao, Mater. Lett., 2016, 169, 197–199 CrossRef CAS.
- J. H. Guo, Y. Zhang, H. J. Li and X. Zhang, J. Alloys Compd., 2016, 678, 499–505 CrossRef CAS.
- L. G. Hanu, G. P. Simon and Y. B. Cheng, Mater. Sci. Eng., A, 2005, 398, 180–187 CrossRef.
- J. Mansouri, R. P. Burford and Y. B. Cheng, Mater. Sci. Eng., A, 2006, 425, 7–14 CrossRef.
- J. Mansouri, C. A. Wood, K. Roberts, Y. B. Cheng and R. P. Burford, J. Mater. Sci., 2007, 42, 6046–6055 CrossRef CAS.
- Y. M. Li, C. Deng and Y. Z. Wang, Compos. Sci. Technol., 2016, 132, 116–122 CrossRef CAS.
- M. I. Martín, F. A. López, F. J. Alguacil and M. Romero, Ceram. Int., 2014, 40, 2769–2776 CrossRef.
- Y. Li, X. D. Cheng, W. Cao, L. L. Gong, R. F. Zhang and H. P. Zhang, Constr. Build. Mater., 2016, 112, 933–939 CrossRef CAS.
- H. W. Di, C. Deng, R. M. Li, L. P. Dong and Y. Z. Wang, RSC Adv., 2015, 5, 51248–51257 RSC.
- X. H. Gong, T. Y. Wu, J. Ma, D. Zhao, Y. C. Shen and T. W. Wang, J. Alloys Compd., 2017, 706, 322–329 CrossRef CAS.
- D. Z. Chen, J. R. Nie, S. P. Yi, W. b. Wu, Y. L. Zhong, J. Liao and C. Huang, Polym. Degrad. Stab., 2010, 95, 618–626 CrossRef CAS.
- D. M. Laura, H. Keskkula, J. W. Barlow and D. R. Paul, Polymer, 2002, 43, 4673–4687 CrossRef CAS.
- J. H. Wang, C. T. Ji, Y. T. Yan, D. Zhao and L. Y. Shi, Polym. Degrad. Stab., 2015, 121, 149–156 CrossRef CAS.
- J. M. Guo, W. Gao, Y. Wang, D. Liang, H. J. Li and X. Zhang, Polym. Degrad. Stab., 2017, 136, 71–79 CrossRef CAS.
- S. H. Lee and S. Q. Wang, Composites, Part A, 2006, 37, 80–91 CrossRef CAS.
- L. Fiocco, B. Michielsen and E. Bernardo, J. Eur. Ceram. Soc., 2016, 36, 3211–3218 CrossRef CAS.
- J. R. Jones, L. M. Ehrenfried and L. L. Hench, Biomaterials, 2006, 27, 964–973 CrossRef CAS PubMed.
- K. Jeevajothi, D. Crossiya and R. Subasri, Ceram. Int., 2012, 38, 2971–2976 CrossRef CAS.
- S. Ansari, J. M. Varghese and K. R. Dayas, Polym. Adv. Technol., 2009, 20, 459–465 CrossRef CAS.
- S. H. Xue, H. Xie, H. Ping, Q. C. Li, B. L. Su and Z. Y. Fu, RSC Adv., 2015, 5, 71844–71848 RSC.
- X. J. Jiao, T. Q. Li, Y. M. Li, Z. W. Zhang, F. Z. Dai and Z. H. Feng, Ceram. Int., 2017, 43, 8208–8213 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab