
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a series of bis-triazoles as G-quadruplex ligands†
Maysaa M.
Saleh
 ac,
Charles A.
Laughton
a,
Tracey D.
Bradshaw
a and
Christopher J.
Moody
*b
ac,
Charles A.
Laughton
a,
Tracey D.
Bradshaw
a and
Christopher J.
Moody
*b
aSchool of Pharmacy, Centre of Bimolecular Science, University of Nottingham, University Park, Nottingham NG7 2RD, UK
bSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: c.j.moody@nottingham.ac.uk
cFaculty of Pharmacy, Isra University, Queen Alia Airport Road, Amman 11 622, UK
First published on 6th October 2017
Abstract
Maintenance of telomeres – specialized complexes that protect the ends of chromosomes – is provided by the enzyme complex telomerase, which is a key factor that is activated in more than 80% of cancer cells, but absent in most normal cells. Targeting telomere maintenance mechanisms could potentially halt tumour growth across a broad spectrum of cancer types. Telomeric ends of chromosomes consist of noncoding repeat sequences of guanine-rich DNA. These G-rich ends can fold into structures called G-quadruplexes. Stabilization of G-quadruplexes by small binding molecules called G4 ligands can prevent telomerase enzyme from maintaining telomere integrity in cancer cells. G-quadruplexes can exist in other parts of the genome too, especially within promoter sequences of oncogenes, and also be interesting drug targets. Here, we describe the development of a new series of novel bis-triazoles, designed to stabilize G-quadruplex structures selectively as G4 ligands. FRET assays showed two compounds to be moderately effective G4 binders, with particular affinity for the quadruplex formed by the Hsp90a promoter sequence, and good selectivity for G-quadruplex DNA vs. duplex DNA. However, CD spectroscopy failed to provide any information about the folding topology of the human telomeric G-quadruplex resulting from its interaction with one of the ligands. All the new ligands showed potent cell growth inhibitory properties against human colon and pancreatic cancer cell lines, as evidenced by the MTT assay; notably, they were more potent against cancer cells than in fetal lung fibroblasts. Docking studies were performed to rationalize the affinity of these ligands for binding to the telomeric parallel G-quadruplex DNA.
Introduction
In eukaryotes, the ends of chromosomes are protected by telomeres, the specialized protein–DNA complexes that prevent end-to-end fusions, recombination and damage to the genome.1 In humans, telomeres consist of the sequence (5′-GGGTTA-3′), which is referred to as the “telomeric repeat”.2 These repeating sequences are predominantly double-stranded. However there is an overhanging single-stranded G-rich 3′-end, known as 3′-overhang or G-strand, which plays an important structural and functional role.3 Each chromosome end contains approximately 1000 to 2000 telomeric repeats.2 In human somatic cells, telomere length decreases with each cell division event until it reaches a critical level, at which point cell division ceases, preventing loss of vital chromosomal genetic information.4,5 Telomeres are replenished by an enzyme called telomerase, a “ribonucleoprotein complex” that functions as a reverse transcriptase to add multiple copies of (5′-GGGTTA-3′) – in all vertebrates – to the end of the G-strand of the telomere. Human telomerase consists of two main components: a protein called hTERT (human telomerase reverse transcriptase) and an RNA (3′-CAAUCCCAAUC-5′) known as hTR (human telomerase RNA) or hTERC (human telomerase RNA component). The protein hTERT uses hTERC to add a six-nucleotide repeating sequence (5′-GGGTTA-3′) to the 3′ G-strand of chromosomes.6,7 A number of studies has revealed that telomerase activity is almost absent in normal somatic cells. However, a low level of telomerase activity has been discovered in mitotically active cells, including skin, lymphocytes, and endometrium because telomerase activity in these proliferating cells is regulated by growth. But in >80% cancers, tumour cells possess short telomeres and high levels of telomerase activity.3 For this reason, most telomere-related antitumour strategies target the telomerase enzyme by which telomere maintenance is provided; the inhibition of telomerase activity results in the reduction of telomere length, and ultimately delayed cell death and abrogation of tumorigenicity in vivo.6The telomeric DNA G-strand folds to form G-quadruplexes, also known as G4-DNA, which are four stranded structures formed by nucleic acid sequences which are rich in guanine (Fig. 1). Each structure contains several stacked square arrangements of guanines called G-quartets.8 Each G-quartet is composed of four guanines held together by a cyclic arrangement of eight hydrogen bonds. The presence of a central monovalent cation (typically potassium) helps to maintain the stability of the structure.6,9,10
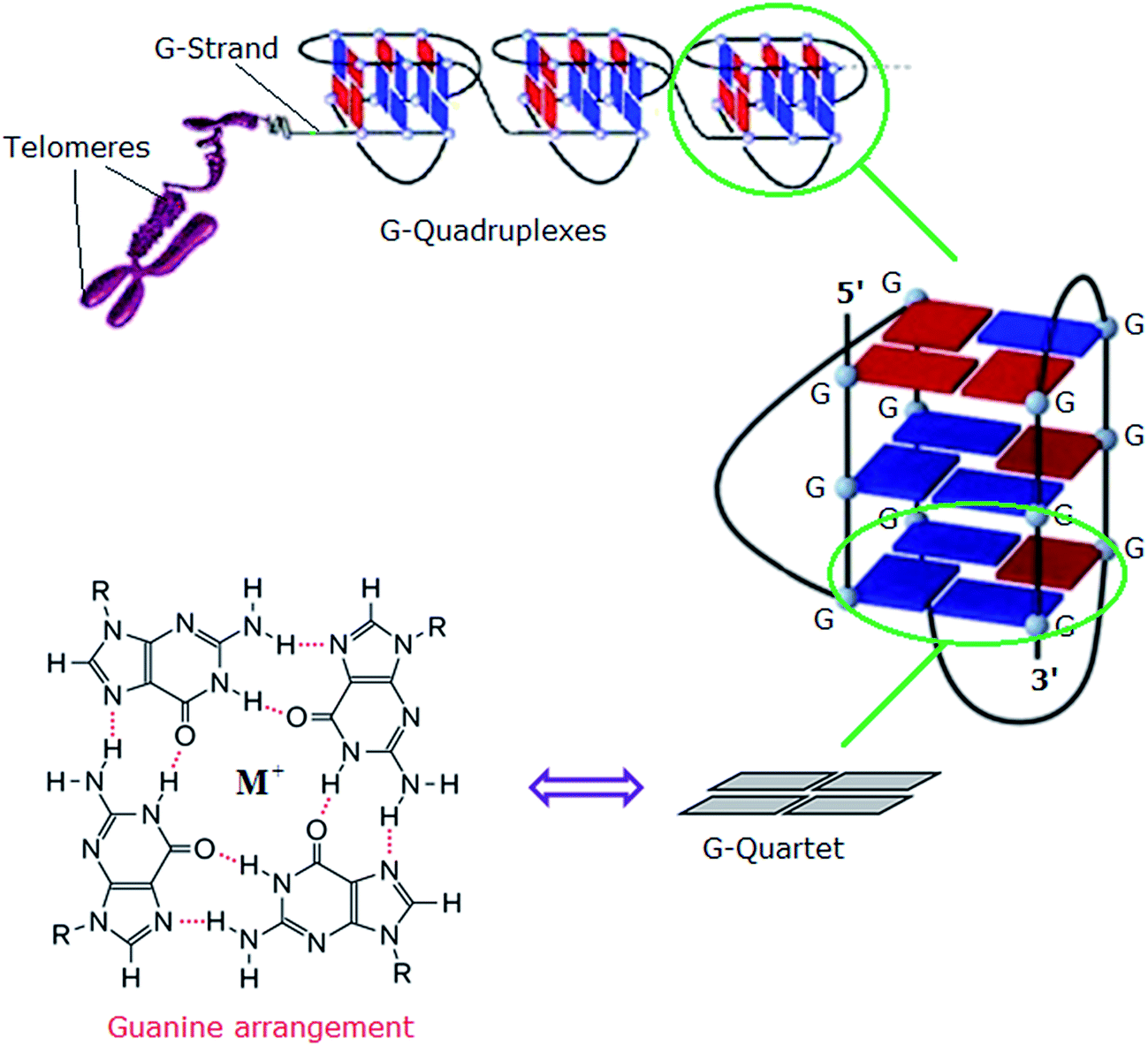 | ||
| Fig. 1 Left: structure of a G-quartet involving four coplanar guanines with a central monovalent cation (guanine arrangement). Right: an intramolecular G-quadruplex. Blue and red boxes represent guanine bases in the anti and syn conformations, respectively.6 Adapted from Y. Xu, T. Ishizuka, K. Kurabayashi, M. Komiyama, Angew. Chem., Int. Ed. 2009, 48, 7833–7836. Copyright Wiley-VCH Verlag GmbH & Co. KGaA.9 Reproduced with permission. And L.-P. Bai, M. Hagihara, K. Nakatani, Z.-H. Jiang, Nature Sci. Rep. DOI: 10.1038/srep06767, 2014.10 Reproduced with permission from Nature Publishing Group. | ||
Two principal categories of G-quadruplex nucleic acids have received attention as potential therapeutic targets: (i) those capable of being formed at the single-stranded G-overhang of telomeric DNA, for which appropriate small molecule ligands can induce G-quadruplex formation, stabilize its structure, inhibit telomerase activity and telomere elongation, and selectively target telomere maintenance, (ii) those within genomes, especially within promoter sequences of genes involved in cellular proliferation and oncogenes such as c-myc, c-kit, and Hsp90. It has been suggested that ligands that bind selectively with promoter G-quadruplexes could in principle down-regulate the expression of these genes and thus have an anticancer effect.11,12 The G-quadruplex stabilization occurs, in most cases, via π–π staking and electronic interactions resulting in binding of the ligand on the G-quartet of the external face of the G-quadruplex (external stacking).13 A wide range of small molecules such as BRACO-19,14 telomestatin,15 RHPS4,16 MMQ3,17 Mn(III)-porphyrin,18 BMSG-SH-3,12 and TRZ,1,19 have been investigated as G-quadruplex binding and stabilizing ligands (Fig. 2).
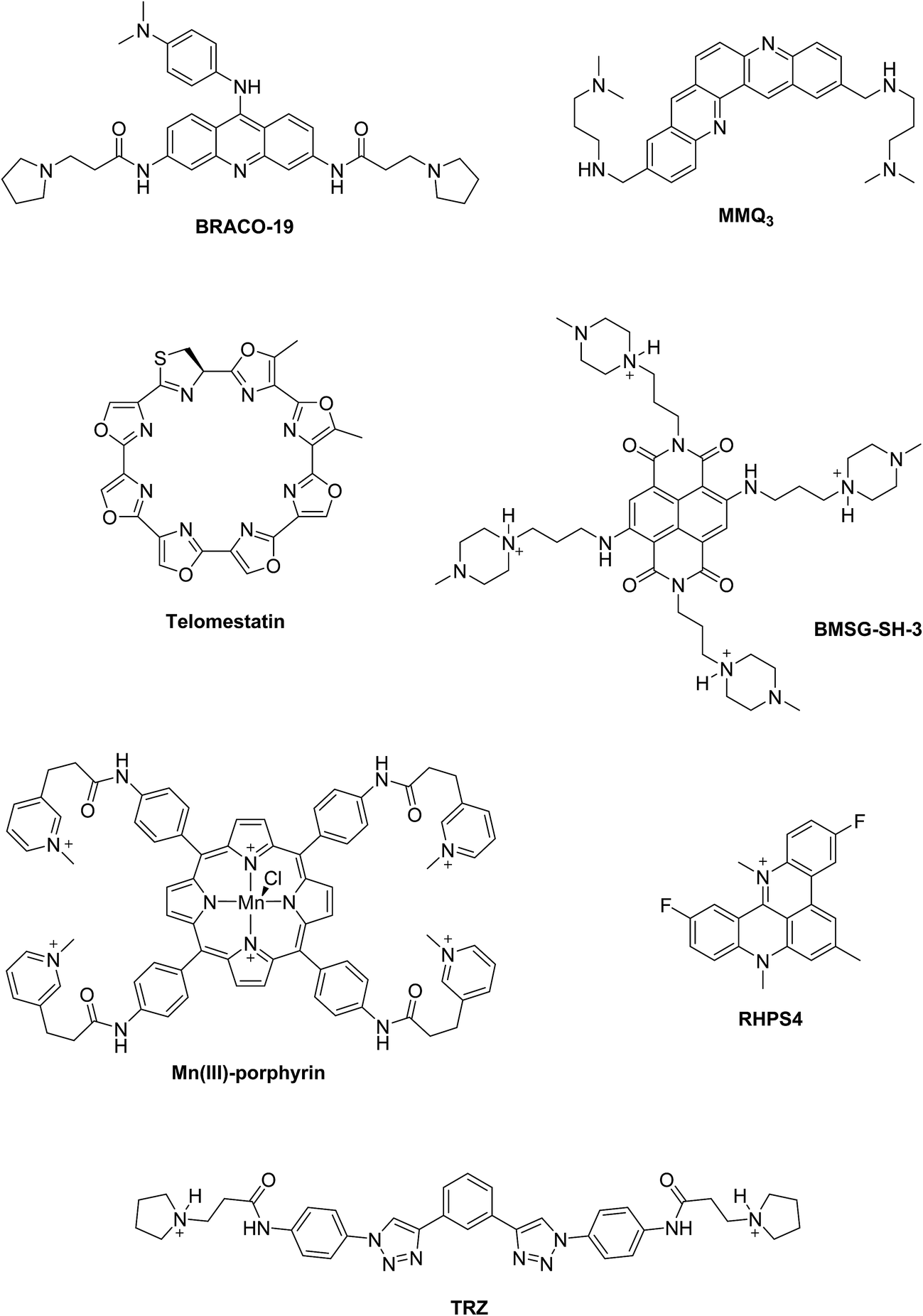 | ||
| Fig. 2 Examples of G-quadruplex binding and stabilizing ligands reported in the literature. | ||
An efficient G-quadruplex ligand should feature; (i) a large aromatic flat surface, much larger than that of a duplex binder to improve the aromatic–aromatic overlap and provide selectivity, (ii) short alkyl chain substituents normally terminating in an amino group that is fully cationic at physiological pH. The positively charged amino groups improve the water-solubility of the ligand and create electrostatic interactions between the ligand and the G-quadruplex–DNA scaffold.11,13
We report here the design, synthesis, biophysical and biological evaluations, and molecular modeling studies of a new series of bis-triazoles, based upon a benzo[2,3-d, 5,6-d]ditriazolo-4,8-dione scaffold, which were developed to stabilize G-quadruplex structures selectively, as G4 ligands and experimental antitumour agents. We selected this scaffold for a number of reasons: firstly it features a novel planar G-quadruplex stacking moiety; secondly synthetic routes to the final compounds are highly convergent, quick, and flexible; and thirdly the symmetrical nature of the ligands potentially simplifies the analysis of their interactions with DNA.
Results and discussion
Synthetic chemistry
A new series of bis-triazoles were designed to display both hydrophobic and hydrophilic characteristics to bind to and stabilize G-quadruplex structures, as G4 ligands. The design included an aromatic core, represented by the bis-(benzyltriazolo)benzoquinone moiety, that promotes π-stacking with the G4 platform, and two protonatable side arms (amino groups) around the aromatic core. The positively charged amino groups are expected to improve the hydrophilicity of the ligands and create electrostatic interactions with the G4-grooves.The synthesis of the hydrophobic aromatic core required a three-step process, as shown in Scheme 1. Heating the commercially available p-nitrobenzyl bromide 1 with sodium azide afforded p-nitrobenzyl azide 2 in yields of 82%.20 1,3-Dipolar cycloaddition reaction of 2 with benzoquinone 3 in the ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under reflux condition in ethyl acetate yielded 66% of bis-triazole 4.21–23 Bis-triazole core 5 was obtained in good yield (92%) by palladium catalyzed hydrogenation of 4 with hydrogen. 13C NMR spectra for bis-triazoles 4 and 5 showed one peak for one carbonyl carbon which confirmed the symmetrical trans-regioisomer rather than cis-isomer that would show two different carbonyl signals.
:1 under reflux condition in ethyl acetate yielded 66% of bis-triazole 4.21–23 Bis-triazole core 5 was obtained in good yield (92%) by palladium catalyzed hydrogenation of 4 with hydrogen. 13C NMR spectra for bis-triazoles 4 and 5 showed one peak for one carbonyl carbon which confirmed the symmetrical trans-regioisomer rather than cis-isomer that would show two different carbonyl signals.
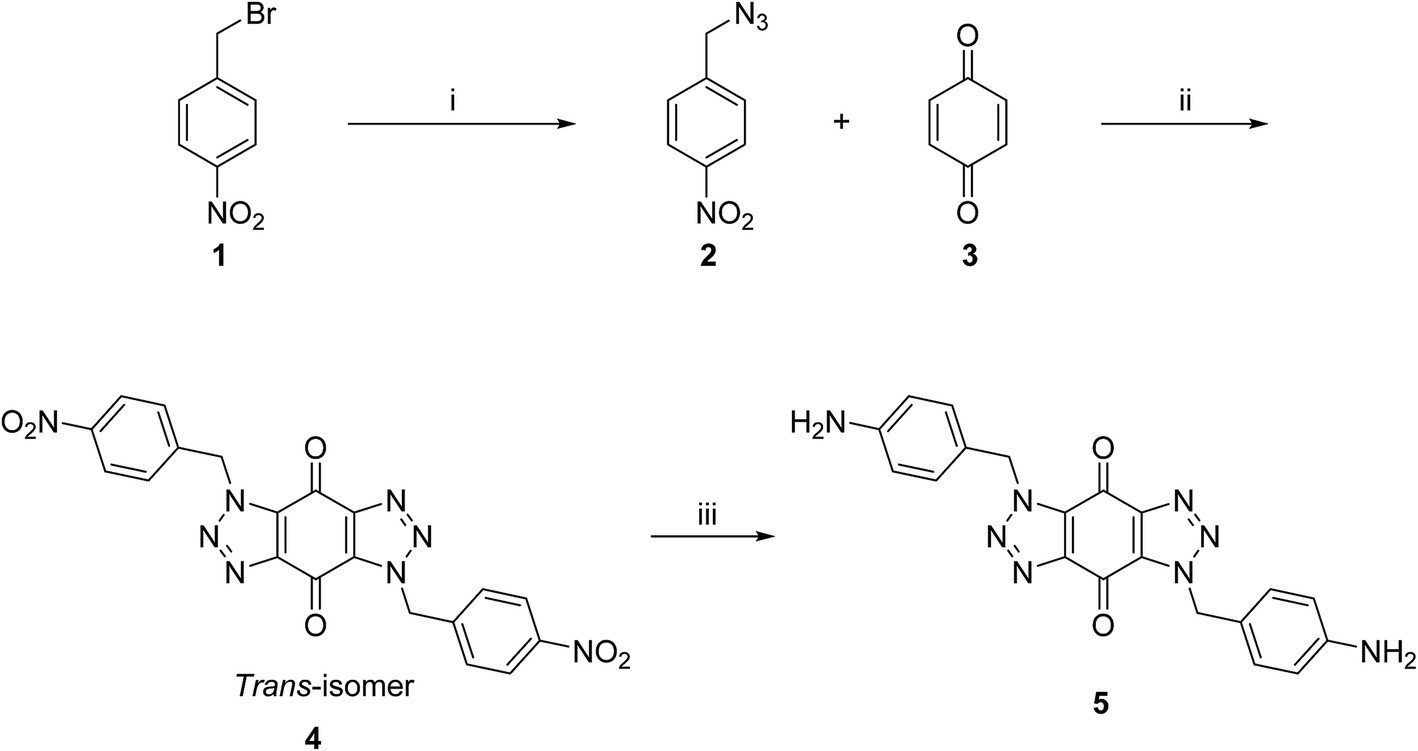 | ||
| Scheme 1 Synthetic route for the aromatic core of bis-triazole 5. Reagents and conditions: (i) NaN3, acetone, reflux, 24 h, 82%; (ii) ethyl acetate, reflux, 48 h, 66%; (iii) H2, Pd/C, DMF, rt, 4 h, 93%. | ||
A three-step route was followed to obtain the side chains 24–33 (Scheme 2). The halo esters 6a–d were heated with secondary amines (diethylamine, morpholine and piperidine) to give the corresponding amino esters 7–13 in yields of 55–94%,24 while the morpholine derivative 10 was commercially available. Conversion into the corresponding amino acids 14–23 was achieved by treating esters 7–13 with potassium hydroxide. Acids 14, 19 and 20 were available commercially. Treating acids 14–23 with oxalyl chloride and a catalytic amount of DMF under an inert atmosphere produced the side chain acid chlorides 24–33, which were used immediately for the next step.
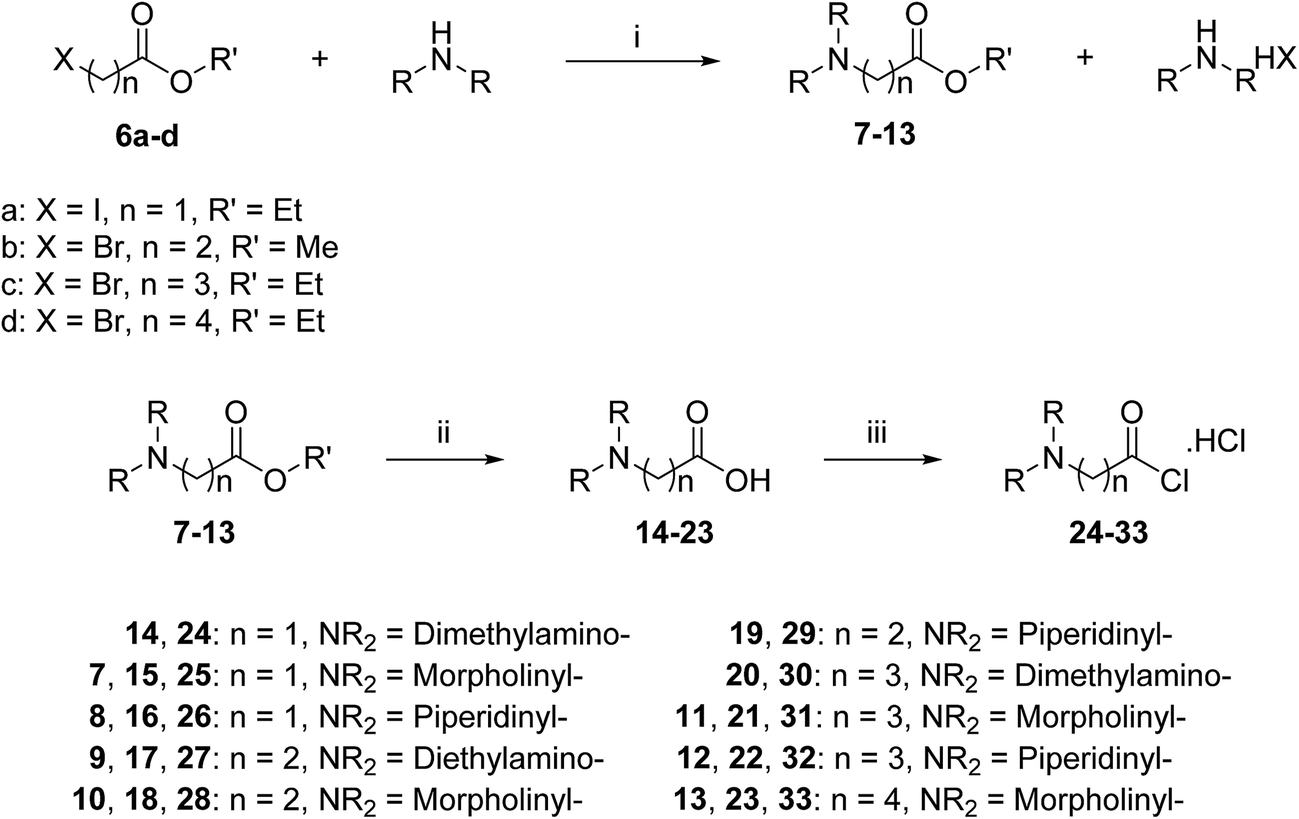 | ||
| Scheme 2 General synthetic route for side chains 24–33. Reagents and conditions: (i) acetonitrile, reflux, 2–8 h, 55–94%; (ii) (a) KOH, H2O/ethanol or methanol, rt, 3–6 h, (b) 0.25 M HCl; (iii) oxalyl chloride, DMF, CH2Cl2, rt, 2–20 h. | ||
The acid chlorides 24–33 were coupled successively to the hydrophobic aromatic core 5 in the presence of pyridine as a catalyst and under inert atmosphere to give novel compounds 34–43 in yields of 38–90%. Treating amines 34–43 with hydrogen chloride in methanol solution, converted them into the corresponding hydrochloride salts 44–53 in good yields (65–95%), as shown in Scheme 3. The salts were purified either by trituration in hot methanol or recrystallization from methanol–light petroleum (1:1).
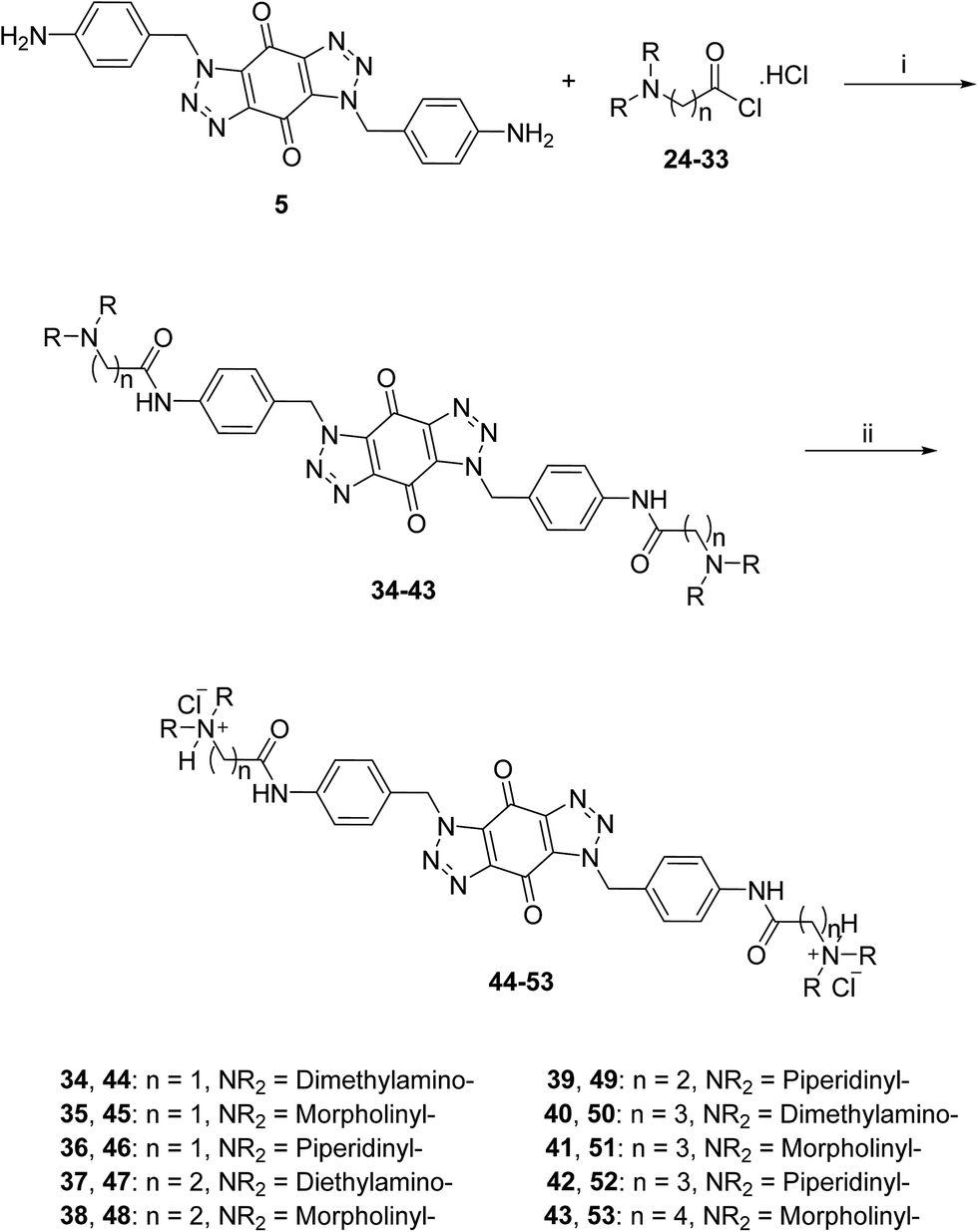 | ||
| Scheme 3 General synthetic route for the target ligands 44–53. Reagents and conditions: (i) (a) pyridine, DMF, rt, 16 h, (b) NaHCO3(aq), 38–90%; (ii) 0.5 M HCl/CH3OH, rt, 4 h, 65–95%. | ||
A mechanism is proposed in Scheme 4 for the formation of bis-triazole 4. The first step is the 1,3-dipolar cycloaddition reaction of azide 2 with the activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of p-benzoquinone 3 to produce the mono-triazoline intermediate A which is tautomerized and oxidized by the excess of 3 to form the mono-triazole adduct C derivative. A second 1,3-dipolar cycloaddition reaction and oxidation takes place in the same way to produce the trans-bis-triazole 4.21,22 The TLC analysis of the reaction mixture filtrates indicted the presence of excess organic azides and large amounts of hydroquinone which supports the proposal that excess benzoquinone plays the role of the required oxidant.
C of p-benzoquinone 3 to produce the mono-triazoline intermediate A which is tautomerized and oxidized by the excess of 3 to form the mono-triazole adduct C derivative. A second 1,3-dipolar cycloaddition reaction and oxidation takes place in the same way to produce the trans-bis-triazole 4.21,22 The TLC analysis of the reaction mixture filtrates indicted the presence of excess organic azides and large amounts of hydroquinone which supports the proposal that excess benzoquinone plays the role of the required oxidant.
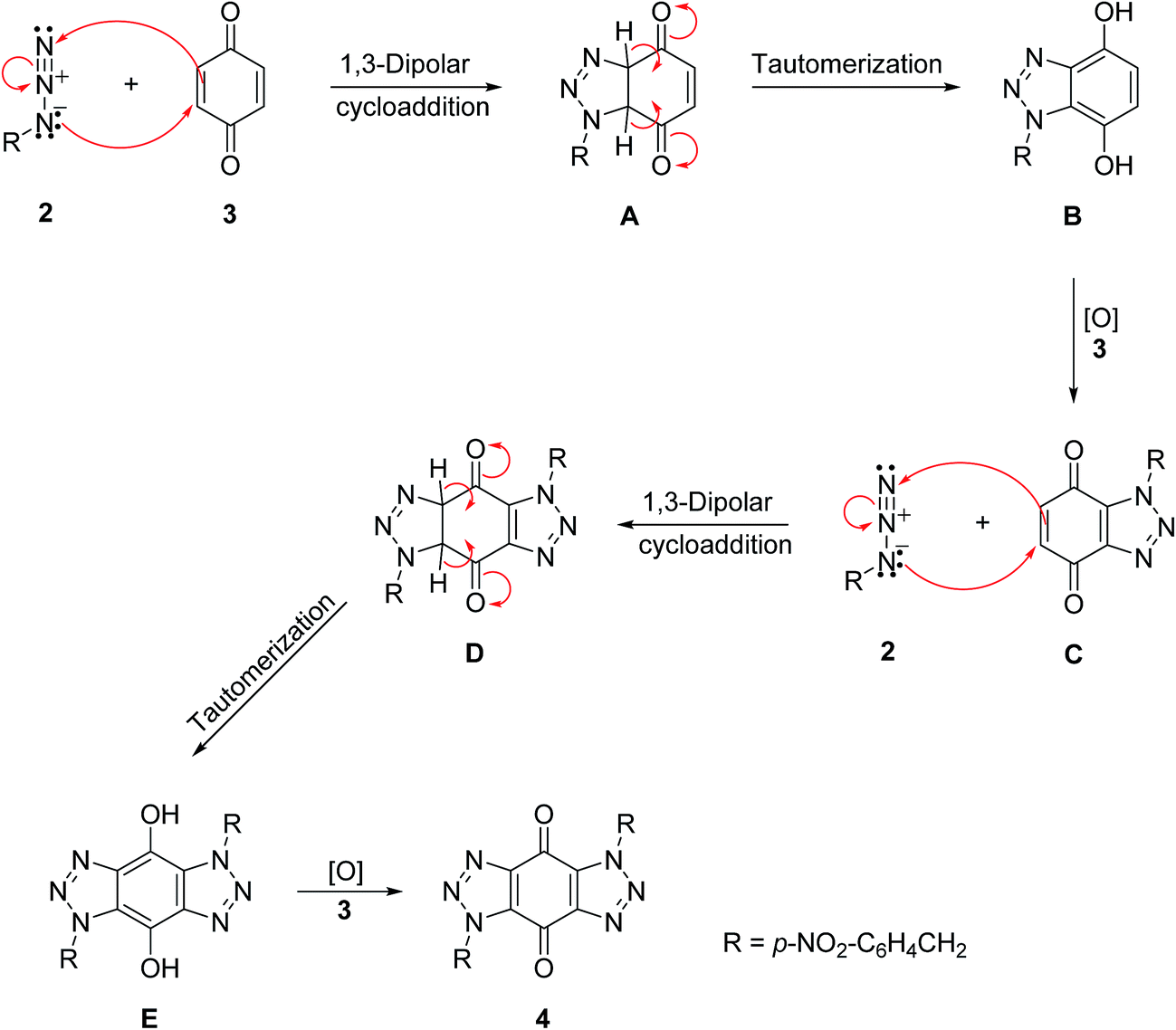 | ||
| Scheme 4 A mechanism for the formation of bis-triazole 4.21,22 | ||
Binding to DNA
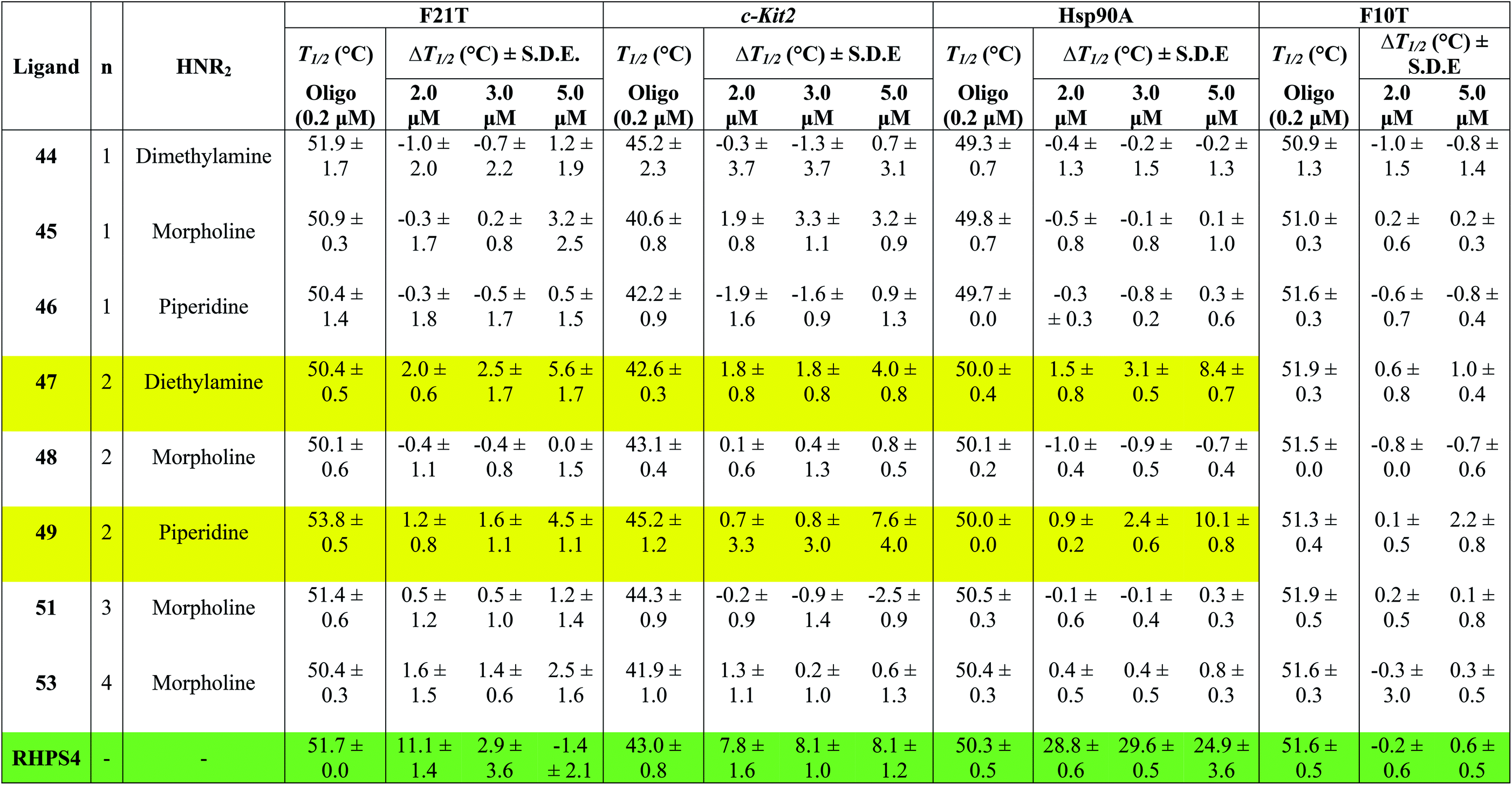
Table 1 shows the effect of three different concentrations of compounds 44–49, 51, 53 and the known G4 binder RHPS4 (ref. 16) (Fig. 2) on the melting temperature (ΔT1/2) of the labeled oligomers F21T, c-kit2, Hsp90a and F10T, respectively.
|
The data show that ligands 44–49, 51 and 53 show a moderate ability to stabilize G-quadruplexes. Though most values are appreciably smaller than those for RHPS4, they are still significant in the sense that they allow us to evaluate relative affinities and trends. The ligands with aminoacetyl 44–46 (n = 1) are poor G-quadruplex stabilizers, the reason might be that the short side-chains impede effective positioning of the protonated amine in the sugar-phosphate backbone loops, or simply do not provide sufficient surface area for the ligand to bind to the G-quadruplex. The morpholino ligands 45, 48, 51 and 53 showed no significant G-quadruplex stabilization, which might result from reduced electrostatic interactions in the backbone and grooves by the uncharged morpholine moiety. For example, the presence of diethylamino group (ligand 47) and piperidino group (ligand 49) in place of morpholino group of the inactive ligand 48, significantly improved the biochemical properties and the binding affinity of 47 and 49 towards the three types of G-quadruplexes.
The results indicated that ligands 47 and 49 induce significant stabilization (ΔT1/2 > 3.50 °C) of the three types of G-quadruplex structures at 5.0 μM concentration, with particular affinity for the quadruplex formed by the Hsp90a promoter sequence. Therefore, the study reveals the ligands 47 and 49 to be the best binders of the series towards telomeric G4-DNA, and promoter sequences G4-DNA of c-Kit2 and Hsp90a, respectively, over the concentration range examined. Good selectivity for G-quadruplex DNA vs. duplex DNA was displayed by the ten ligands over the concentration range used. None of them were observed to significantly increase the melting temperature of duplex forming strand F10T (ds), suggesting a very low stability for any duplex–ligand complex.
According to the FRET assay results, we find that an increase in length of the side chain by one –CH2– unit results in a large increase in the ability of the compounds to stabilize G-quadruplex DNA structure. Also, the nature of the amine substituent at the terminus of the side chain greatly affects the ability of the compounds to stabilize quadruplex DNA.
In a number of cases (e.g.47 and 49), there is a noticeable jump in ΔT1/2 from 3 μM to 5 μM. One possible explanation for this is that these ligands show cooperative binding to the DNA. This would not be unusual for quadruplex binders – e.g. cooperativity in binding of RHPS4 to the c-kit DNA quadruplex has been detected by mass spectroscopy.19
There are a few examples of negative ΔT1/2 values but on the whole they are within experimental error of zero. However a negative ΔT1/2 value is of course quite possible, if the ligand stabilizes some unfolded form of the DNA more strongly than it stabilizes the quadruplex fold.
When comparing the melting temperatures of the eight ligands 44–49, 51 and 53 with those of the known G-quadruplex binder RHPS4, we notice that all the new ligands are less effective at G-quadruplex stabilization. The key difference between the bis-triazoles series 44–53 and the G4 binders RHPS4 (ref. 16), trisubstituted acridine BRACO-19 (ref. 14) and tetrasubstituted naphthalene diimide BMSG-SH-3 (ref. 12) (Fig. 2) is the presence of the two sp3 methylene groups within the aromatic region that might perturb the planarity of the hydrophobic aromatic core, and thus affect the π–π staking of the ligands on to the terminal G-quartet. As well as the presence of four hydrophilic arms terminating in protonated piperazine groups and three terminating in protonated pyrrolidine in BMSG-SH-3 and BRACO-19, respectively, increases their electronic interaction with the G4 grooves, in comparison to the bis-triazoles 47 and 49.
:DNA ratio of at least 5:1 was required to observe significant quadruplex stabilization, stock solutions of bis-triazole 47 and the known binder RHPS4 were added to the 22 base human telomeric G4 oligonucleotide AGGG(TTAGGG)3 (h-Tel) in potassium chloride buffer medium to generate solutions of (3.3 μM) h-Tel with (16.5 μM) ligand 47 and RHPS4 of the ratio 1:5 of DNA to ligand, (1.0 μM) of h-Tel with (15.0 μM) of ligand 47 and RHPS4 of the ratio 1:15 of DNA to ligand, and (1.0 μM) of h-Tel with (25.0 μM) of ligand 47 and RHPS4 of the ratio 1:25 of DNA to ligand.
The CD spectrum (Fig. 3) of the native h-Tel (red line) shows a strong positive band at 290 nm, with a small minimum at 235 nm and a number of additional broad positive bands apparent at 250 nm and 275 nm. This spectrum is likely to correspond to a mixture of parallel and anti-parallel folds possibly including hybrid forms as well.8,19,31,32 The CD spectra A, B and C show that the G-quadruplex topology of h-Tel is retained on binding to ligand 47, with minor changes in the spectra (green line) being apparent, indicating that 47 does not induce major changes in telomeric topology on binding. In contrast, binding of RHPS4 (blue line) induces major changes in the CD spectra that are interpreted as almost complete conversion to the anti-parallel-stranded topology. These results suggest that, compared to RHPS4, 47 is a relatively promiscuous G-quadruplex ligand, and/or that CD spectroscopy is not sensitive enough to clearly resolve the topological dependence in the relatively weak binding affinity of ligand 47 – compared to RHPS4 – towards the human telomeric G4 structure, (shown by the FRET melting assay). Because ligand 47, our best telomeric G4 stabilizer, proved not to have a detectable influence on the topology of the quadruplex (parallel versus antiparallel form), we did not consider it of value to investigate other, structurally related but lower affinity, ligands such as 49.
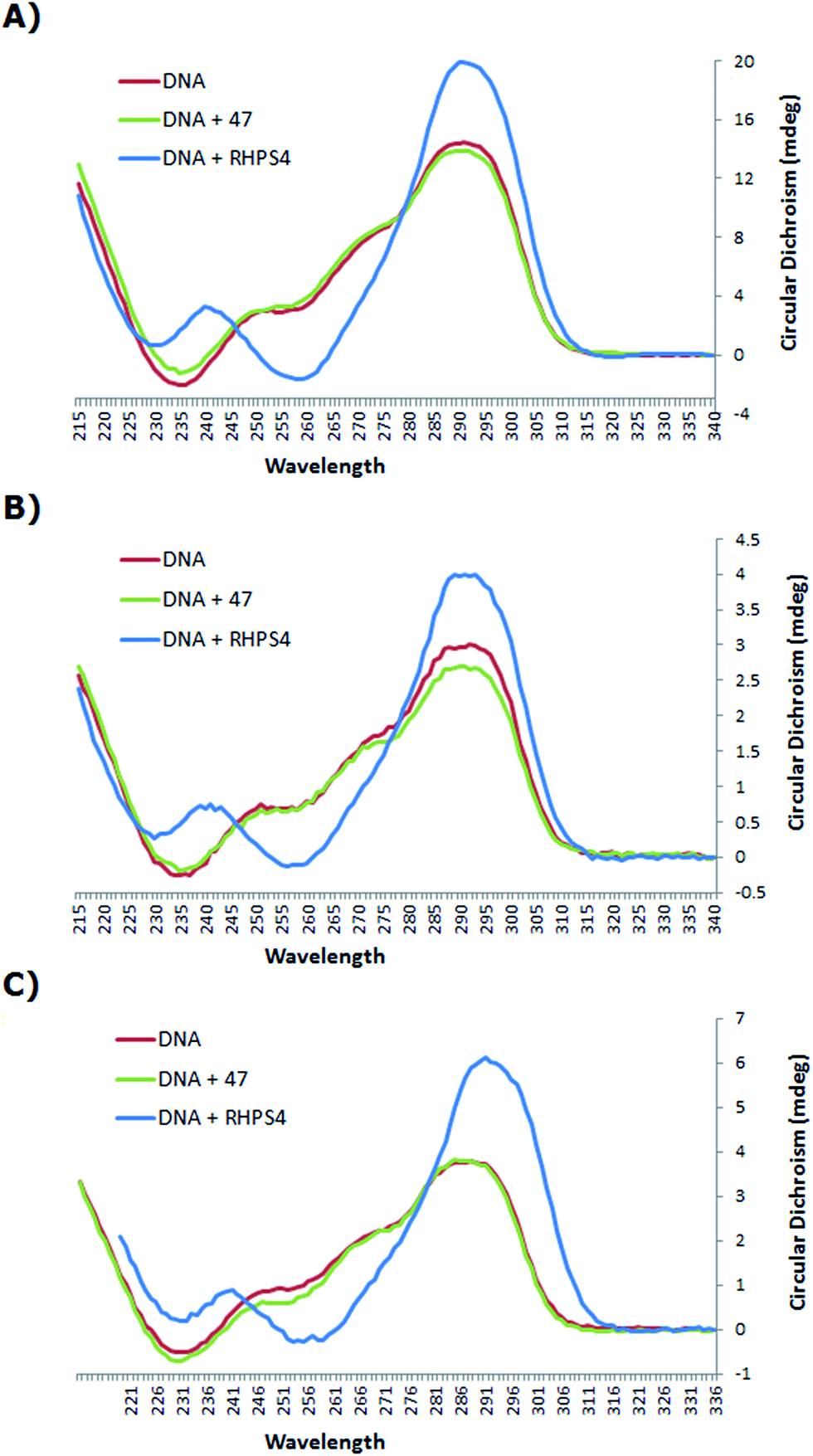 | ||
| Fig. 3 CD spectra of h-Tel and the complexes with ligand 47 and RHPS4 in potassium chloride buffered to pH = 7.0, at 25 °C, (A) 3.3 μM of h-Tel with 16.5 μM of ligand 47 and RHPS4 (of the ratio 1:5 of DNA to ligand); (B) 1.0 μM of h-Tel with 15.0 μM of ligand 47 and RHPS4 (of the ratio 1:15 of DNA to ligand); (C) 1.0 μM of h-Tel with 25.0 μM of ligand 47 and RHPS4 (of the ratio 1:25 of DNA to ligand). | ||
In analysing the docking data, we concentrated on high-scoring poses that were conserved across all members of the congeneric set of ligands, in order to investigate and explore the binding interactions between the ligands and G4-DNA.
The study showed that all the ligands should have affinity for both 3′- and 5′-faces of the human telomeric parallel standard G-quadruplex DNA structure. Fig. 4 shows the VMD top views of the 3′-face of the telomeric parallel G-quadruplex DNA template, and the set of ligands 44–53 bound to the 3′-side of G4-DNA in the best pose for their common core. Despite differences in the sidechains, the majority of the ligands bound in a very similar fashion with only minor variations in some intermolecular interactions between models. Furthermore, all the ligands were oriented and posed with the 3′-face in fashions that agree with the accepted requirements for the stabilization of G-quadruplex structures; the ligands' hydrophobic aromatic cores are positioned over the external G-quartet and their hydrophilic amino side arms are directed into the grooves and TTA loops of the telomeric G4-DNA, as shown in the VMD top views (Fig. 4B).
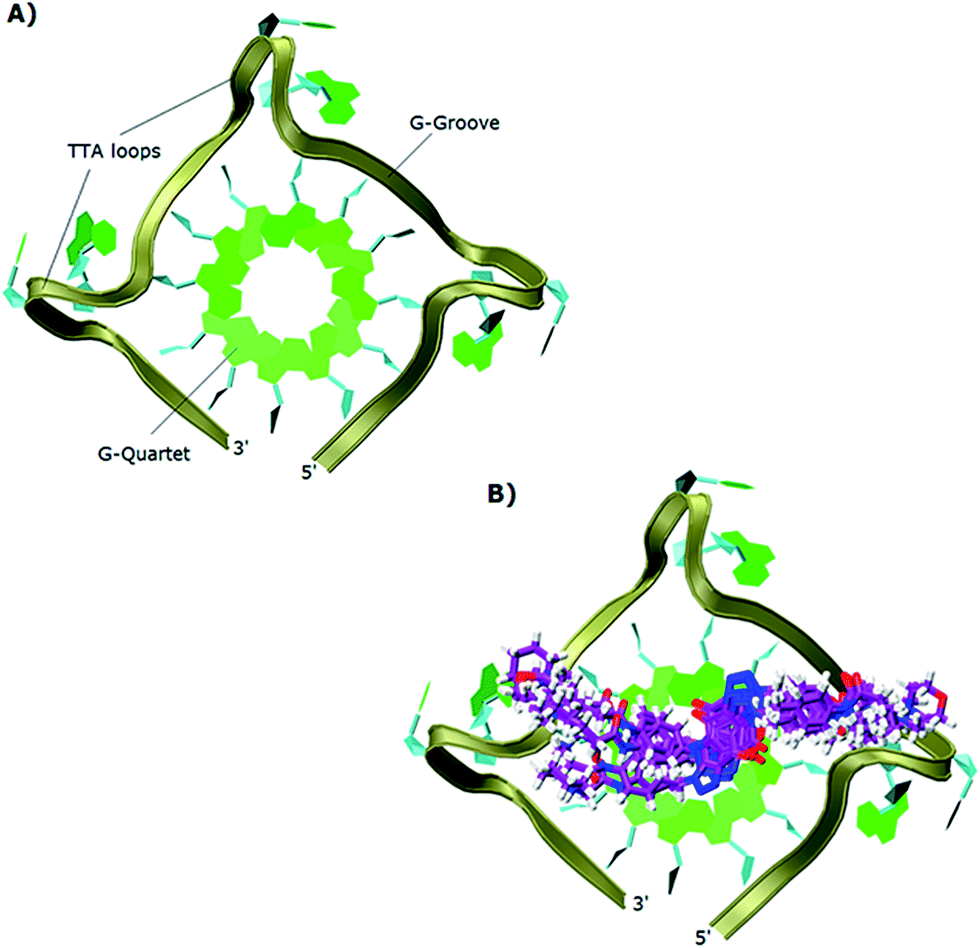 | ||
| Fig. 4 Top views in VMD visualizing software of: (A) 3′-side template of the telomeric parallel G4-DNA; (B) the common pose for ligands 44–53 with the 3′-side of G4-DNA. | ||
In order to gain insight into the nature of interactions between synthesized bis-triazoles 44–53 and the 3′-side of the telomeric parallel G-quadruplex, the side view of the common pose for ligand 47 (the best binder of the series) with 3′-face was studied closely. The side view in VMD (Fig. 5) shows that the aromatic core of ligand 47 does not stack perfectly over the external guanine tetrad, resulting in weak π–π interactions between the two. This explains and rationalizes the moderate binding affinity the ligands revealed by the FRET melting data (ΔT1/2). The main reason for this ineffective stacking is the presence of the two sp3 methylene groups within the aromatic region which perturb the planarity of the hydrophobic aromatic core. The side view also shows the amide nitrogen and the tertiary amine functionality at the ends of the side chains both contribute towards stabilization of the complex by forming hydrogen bonds to phosphate groups of one groove and two TTA loops. Furthermore, the presence of a positive charge on the protonated tertiary amino group would enhance the electrostatic interactions with the negatively charged phosphate groups in the loops.
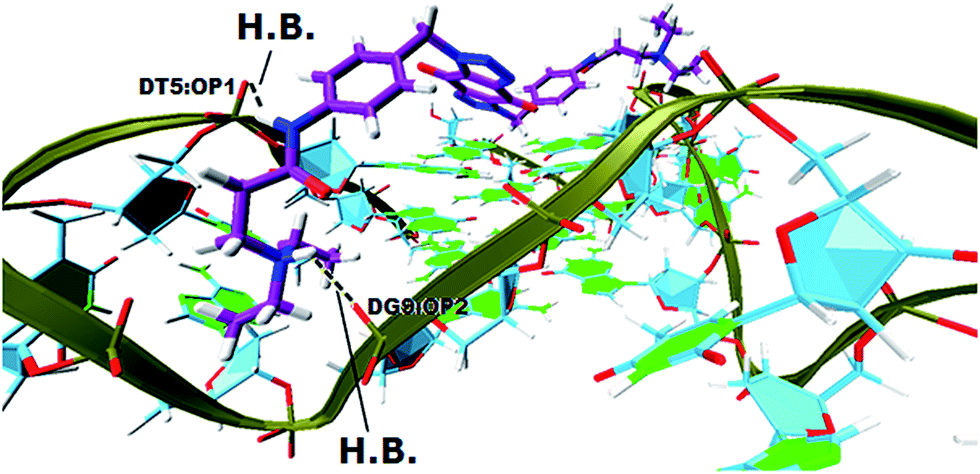 | ||
| Fig. 5 A side view of ligand 47 with the 3′-side G4 template (hydrogen bonds in yellow lines). | ||
Similar results were obtained from docking the bis-triazoles 44–53 to the 5′-side of G4-DNA template: a high-scoring “consensus” pose is found in which the benzoquinone core stacks appreciably, but sub-optimally, with the G-quadruplex face and the side chains orient to find favourable electrostatic binding opportunities in the sugar–phosphate grooves and loops.
000 deaths globally in 2008.38 Pancreatic carcinoma is particularly resistant to chemotherapy, often diagnosed with metastatic disease and has an appalling five year survival rate (<5%).39 Thus, the development of new therapies for such malignant diseases represents a current unmet medical need.
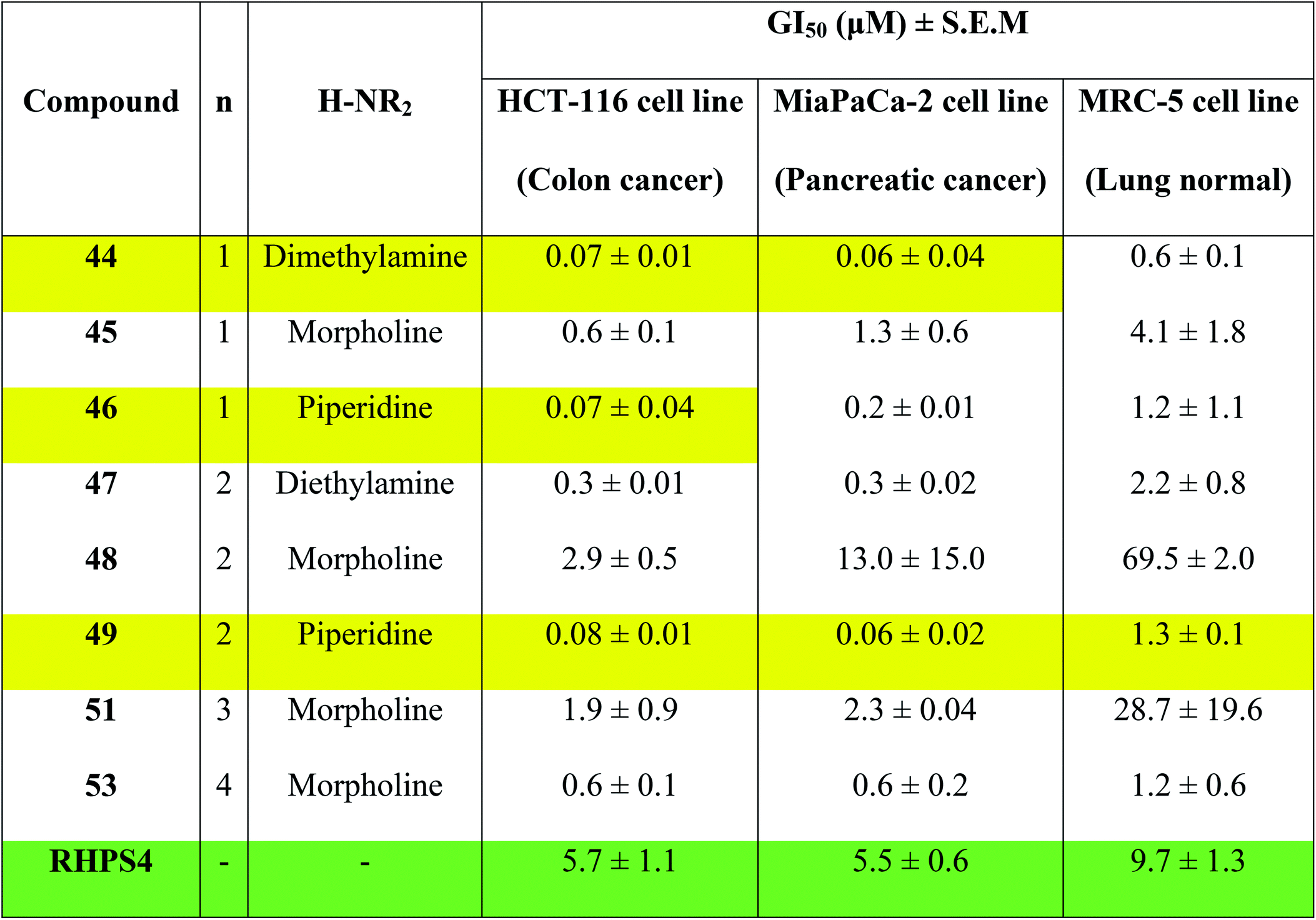
Concentrations at which cell growth is inhibited by 50% (GI50) after 72 h exposure of cells to bis-triazoles 44–53 and RHPS4 were calculated from dose–response curves and are presented in Table 2. RHPS4 structure is shown in Fig. 2.
|
As can be seen from Table 2, most of the compounds 44–49, 51, 53 and RHPS4 show potent cellular growth inhibitory activity against both carcinoma cell lines in the concentration range (0.01–100 μM) used. The highest potency against human cancer cell lines is shown by the ligand 44 (a non G4 binder) with GI50 values of 70 nM for HCT-116 and 60 nM for MiaPaCa-2, ligand 46 (a non G4 binder) which has a GI50 value of 70 nM for HCT-116, and our best c-Kit2 and Hsp90a G4 binder 49 which shows GI50 values of 80 nM for HCT-116 and 60 nM for MiaPaCa-2. The three compounds show almost identical activity with uniform growth inhibitory responses by the two cancer cell lines HCT-116 and MiaPaCa-2 (except 46). We see that ligand 47, the potent telomeric G4 stabilizer, has relatively lower anti-proliferative activity against both human cancer cell lines. This strongly suggests that the synthesized bis-triazoles 44–49, 51, 53 perturb molecular targets other than the telomere, and mechanisms of action in addition to G-quadruplex binding may contribute to antitumour activity.
The results show that the eight synthesized ligands have higher potencies against the human cancer cell lines HCT-116 and MiaPaCa-2 than in normal human lung MRC-5 fibroblasts, inferring good selectivity in their cellular growth inhibitory activity for human cancer cells over human normal cells. These results suggest 44–53 have promise as new therapeutic anticancer agents.
In particular, the results indicate that compound 49 is >16-fold more active against cancer human cell lines HCT-116 and MiaPaCa-2 than against normal human MRC-5 cells. Compared to RHPS4, 49 is both more potent against the two tumour cell lines (GI50 values ≤ 0.08 mM) and more selective for them over MRC-5.
Conclusions
In conclusion, the design, synthesis, biophysical and biological evaluation of a new series of bis-triazoles 44–53 as potential G-quadruplex stabilizers have been described. The target ligands can be obtained through an efficient, convergent, synthetic route in moderate to good yields. In FRET assays two ligands show significant binding to the three types of G-quadruplex structures with selectivity towards the promoter sequence of the Hsp90a oncogene. The synthesized bis-triazoles 44–49, 51 and 53 are selective towards G4-DNA vs. duplex-DNA. The planarity of the aromatic core, number of the hydrophilic side arms and the nature of the amino group at their termini greatly affect the ability of the compounds to stabilize the G-quadruplex. Circular dichroism-based studies of the topology of the human telomeric G-quadruplex obtained from its interaction with ligand 47 suggest that both parallel and anti-parallel folds might be present. In MTT assays, most of the synthesized ligands show potent cell growth inhibitory activity against human carcinoma (colon and pancreatic) cell lines. Notably they are more potent towards human cancer cells than embryonic lung fibroblasts. The weak correlation between FRET and MTT assay results suggests that the synthesized ligands may have additional mechanisms of action besides targeting G-quadruplexes.Docking studies give us a clear idea about the nature of the ligand–quadruplex DNA interactions and rationalize the moderate binding affinity the ligands show in the FRET experiments, suggesting that the hydrophobic core is not planar enough for optimal π–π stacking interactions between the aromatic portion of the ligands and the external G-quartet. This information can guide the optimization of ligands 44–53, for increased binding affinity and selectivity toward G-quadruplex structures.
Experimental section
FRET analysis
All oligonucleotides and their fluorescent conjugates were purchased from Sigma. DNA was initially dissolved as a stock 100 μM solutions in purified water; further dilutions were carried out in the relevant buffer. The ability of the compounds to stabilize G-quadruplex DNA was investigated using a fluorescence resonance energy transfer (FRET) assay modified to be used as a high-throughput screen in a 96-well format. The dual labeled oligonucleotides F21T (5′-FAM-GGGTTAGGGTTAGGGTTAGGG-TAMRA-3′), c-kit2 (5′-FAM-CCCGGGCGGGCGCGAGGGAGGGGAGG-TAMRA-3′) and Hsp90a (5′-FAM-GGGCCAAAGGGAAGGGGTGGG-TAMRA-3′); donor fluorophore FAM: 6-carboxyfluorescein; acceptor fluorophore TAMRA: 6-carboxy-N,N,N′,N′-tetramethylrhodamine used as the FRET probes, were diluted from stock to the correct concentration (0.2 μM) in a 60 mM potassium cacodylate buffer (pH 7.4) and then annealed by heating to 95 °C for 10 min, followed by cooling to room temperature in the heating block for 3.5 h. An analogous protocol was used in the preparation of the labeled hairpin DNA for experiments assessing the duplex-stabilizing interactions of the compound. The oligonucleotide used (F10T) was purchased as the dual-labeled probe forming the palindromic decamer sequence d(TATAGCTATA) with T6 between two sequences to make the hairpin loop (5′-FAM-TATAGCTATATTTTTTTATAGCTATA-TAMRA-3′) and annealed under the same conditions as the other oligonucleotides. Compounds were stored at −20 °C as 1 mM stock solutions in a mixture of hydrochloric acid (75% 1 mM HCl and 25% purified water) and DMSO (9:1), after which 60 mM potassium cacodylate buffer (pH 7.4) was used in all subsequent steps. All experimental values were determined in triplicate. 96-well polypropylene plates (Agilent Technologies) were prepared by aliquoting 50 μL of the annealed DNA into each well, followed by 50 μL of compound solutions.1,25 Measurements were made on a real-time PCR apparatus40 (MX3005P, Stratagene) with excitation at 450–495 nm and detection at 515–545 nm. Fluorescence readings were taken at intervals of 0.5 °C over the range 25–99 °C for 148 cycles, with a constant temperature being maintained for three minutes at 25 °C and 30 seconds prior to each reading to ensure a stable value. Final analysis of the data was carried out using a Microsoft Excel 2010. Esds in ΔT1/2 are ±4.0 °C. In brief, for the determination of the melting temperature, the raw data from the fluorescence detector was analysed by two ways: (i) normalizing the fluorescein emission between 0 and 1, then calculating the temperature for which the normalized emission equals to 0.5 which is defined as T1/2, and the normalized melting curve was plotted as normalized fluorescein emission vs. compound concentration; (ii) the fluorescein emission data were first smoothed using 8-point running average, subsequently the first derivative of the smoothed data was calculated, then the temperature that matches the maximum of the first derivative is determined which is defined as Tmax. The first derivative melting curve was plotted as d(fluorescein emission)/dT vs. compound concentration.26
CD spectroscopy
CD and UV spectra of h-Tel oligonucleotides were recorded on an Applied Photophysics Chirascan plus spectrophotometer interfaced with Chirascan software. The optical system consisted of a 150 W ozone producing xenon lamp, circular light polarizer and end-mounted photomultiplier. Temperature regulation was performed using a Poly Science water bath and Quantum Northwest TC125 controller. Calibration was performed prior to these experiments with D-camphorsulfonic acid. Data were recorded in a 1 cm path length quartz cuvette containing 3 mL samples. Bandwidth was set to 1.0 nm for entrance and exit, and data was recorded with steps of 1 nm over the wavelength range 340–215 nm. The number of samples was set to 100000 for CD and 10000 for UV with adaptive sampling enabled. Each reported curve is the average of 3 repeat scans.
The oligonucleotide 5′-AGGGTTAGGGTTAGGGTTAGGG-3′ (h-Tel) was supplied lyophilized and purified by desalting by Sigma. Dilution of the lyophilized sample was performed using 100 mM potassium chloride and 10 mM potassium phosphate buffer solution at pH 7.0. The concentration of DNA stock solution was determined using molar extinction coefficient 228500 mol−1 cm−1 at a UV maximum of 260 nm. Compounds to be tested for interaction with h-Tel were dissolved in methanol to make a 2.0 mM solution. Solutions of 1.0 and 3.3 μM h-Tel in potassium chloride buffer were annealed by heating to 95 °C for 10 minutes before being allowed to slowly cool to ambient temperature over 4 hours, with or without the addition of a ligand at ratios of 1:5, 1:15 and 1:25 DNA to ligand concentration. Analysis of data was performed using Microsoft Excel. Baseline data was collected using buffer solution in the same cuvette and subtracted from sample spectra, after which the data was normalized to zero at 340 nm.28,41
MTT assay
The antiproliferative activity of experimental agents was evaluated by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay36 on 3 different human cell lines: colon (HCT-116) and pancreatic (MiaPaCa-2) carcinoma, and embryonic lung fibroblasts (MRC-5). Cell lines were sourced from the American Type Culture Collection (ATCC) and stored in liquid nitrogen. Low passage numbers (≤20) were used in all experiments. Lonza MycoAlert™ mycoplasma detection kits were used (according to manufacturer's instructions) ensuring cultures were contamination-free. The carcinoma cell lines were maintained in RPMI-1640 (Sigma-Aldrich (UK), CN: R8758) medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS). MRC-5 fibroblasts were maintained in Minimum Essential Medium (MEM) Eagle (Sigma-Aldrich (UK), CN: M0275) supplemented with 10% (v/v) FBS, 1% sodium carbonate (7.5%), 1% non-essential amino acids (0.1 mM), 1% HEPES (1 M), 1% L-glutamate (200 mM) and 1% penicillin. The cells were seeded into 96-well plates at density of 3 × 103 per well (180 μL per well) and allowed to adhere for 24 h at 37 °C/5% CO2. Agent top stock solutions (10 mM in DMSO) were then freshly made. Serial dilutions were prepared in the same media as mentioned above for addition to carcinoma and normal cells. Control wells received vehicle alone (20 μL per well). Final test agent concentrations in the wells were; 0.01, 0.05, 0.1, 0.5, 1, 5, 10, 50 and 100 μM. The final concentration of DMSO in the wells never exceeded 1%. Vehicle control assays were performed (0.0001–1% DMSO). Experimental plates were incubated for a further 72 h period at 37 °C/5% CO2. Cell viability was recorded at the time of agent addition (T0) and after 72 h exposure: following addition of MTT solution (2 mg mL−1 in PBS: 50 μL per well), experimental plates were incubated for 3 h to allow reduction of MTT by viable cells to insoluble dark purple formazan crystals. The supernatant in each well was then aspirated, and cellular formazan was solubilized by addition of DMSO (150 μL per well). Absorbance was read at a wavelength of 550 nm by using an Anthos Labtec systems plate reader. The measured intensity is proportional to metabolic activity, which correlates with viable cell numbers. Estimated GI50 values (test agent concentrations that inhibit cell growth by 50%) were calculated.39Molecular docking studies
The automated docking studies were carried out using “Glide docking” tools of Glide software, which uses the ChemScore function of Eldridge et al.42 to calculate Glide docking scores. The programme ran 10000 docking trials per ligand, and wrote out at most 50 poses for each ligand.
Chemistry
Details of all synthetic procedures are included in the ESI.†PDB ID code
The PDB ID code for the published telomeric parallel G-quadruplex DNA structure is 1KF1.Conflicts of interest
There are no conflicts to declare.Abbreviations
| CD | Circular dichroism |
| c-Kit2 | Labelled G-quadruplex forming oligomer of the promoter sequence c-kit2 oncogene |
| FAM | 6-(Carboxylfluorescein) |
| FRET | Fluorescence resonance energy transfer |
| F10T | Hairpin double helix forming labeled oligomer |
| F21T | Labeled G-quadruplex forming oligomer of human telomeric sequence |
| GI50 | Concentration inhibiting 50% cell growth |
| Glide | Grid-based ligand docking with energetic |
| HCT-116 | Human colorectal carcinoma cell line |
| Hsp90a | Labeled G-quadruplex forming oligomer of the promoter sequence Hsp90a oncogene |
| MiaPaCa-2 | Human pancreatic carcinoma cell line; MRC-15 embryonic lung fibroblast cell line |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| OD | Optical density |
| TAMRA | 6-Carboxytetramethylrhodamine |
| VMD | Visual molecular dynamics |
Acknowledgements
We thank the Islamic Development Bank (IDB) in Jeddah/KSA for financial support. We are grateful to Prof. Steven Neidle and Dr Cristina Ciancimino from University College of London (UCL) for their help in using the FRET assay, Dr Abigaill Emtage from University of Nottingham/Malaysia Campus for her advice in using Glide program, and Prof. Mutasem Taha from Jordan University for his guidance and scientific advice about targeting telomere and telomerase.References
- A. D. Moorhouse, S. Haider, M. Gunaratnam, D. Munnur, S. Neidle and J. E. Moses, Mol. BioSyst., 2008, 4, 629–642 RSC.
- N. J. Elwood, Cancer Control, 2004, 11, 77–85 CrossRef.
- L. K. Wai, Medsc. Gen. Med., 2004, 6, 19–29 Search PubMed.
- A. M. Olovinkov, Dokl. Akad. Nauk SSSR, 1971, 201, 1496–1499 Search PubMed.
- A. M. Olovnikov, J. Theor. Biol., 1973, 41, 181–190 CrossRef CAS.
- A. De Cian, L. Lacroix, C. Douarre, N. Temime-Smaali, C. Trentesaux, J.-F. Riou and J.-L. Mergny, Biochimie, 2008, 90, 131–155 CrossRef CAS.
- R. Pruzan, K. Pongracz, K. Gietzen, G. Wallweber and S. Gryaznov, Nucleic Acids Res., 2002, 30, 559–568 CrossRef CAS.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS.
- Y. Xu, T. Ishizuka, K. Kurabayashi and M. Komiyama, Angew. Chem., Int. Ed., 2009, 48, 7833–7836 CrossRef CAS.
- L.-P. Bai, M. Hagihara, K. Nakatani and Z.-H. Jiang, Sci. Rep., 2014, 4(6767), 1–10 Search PubMed.
- N. H. Campbell, M. Patel, A. B. Tofa, R. Ghosh, G. N. Parkinson and S. Neidle, Biochemistry, 2009, 48, 1675–1680 CrossRef CAS.
- M. Gunaratnam, M. de la Fuente, S. M. Hampe, A. K. Todd, A. P. Reszka, A. Schätzlein and S. Neidle, Bioorg. Med. Chem., 2011, 19, 7151–7157 CrossRef CAS PubMed.
- D. Monchaud and M.-P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 RSC.
- M. J. B. Moore, C. M. Schultes, J. Cuesta, F. Cuenca, M. Gunaratnam, F. A. Tanious, W. D. Wilson and S. Neidle, J. Med. Chem., 2006, 49, 582–599 CrossRef CAS.
- K. Shin-ya, K. Wierzba, K.-i. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Seto, J. Am. Chem. Soc., 2001, 123, 1262–1263 CrossRef CAS PubMed.
- E. Gavathiotis, R. A. Heald, M. F. G. Stevens and M. S. Searle, J. Mol. Biol., 2003, 334, 25–36 CrossRef CAS PubMed.
- J. L. Mergny, L. Lacroix, M. P. Teulade-Fichou, C. Hounsou, L. Guittat, M. Hoarau, P. B. Arimondo, J. P. Vigneron, J. M. Lehn, J. F. Riou, T. Garestier and C. Helene, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3062–3067 CrossRef CAS PubMed.
- I. M. Dixon, F. Lopez, A. M. Tejera, J.-P. Esteve, M. A. Blasco, G. Pratviel and B. Meunier, J. Am. Chem. Soc., 2007, 129, 1502–1503 CrossRef CAS.
- T. P. Garner, H. E. L. Williams, K. I. Gluszyk, S. Roe, N. J. Oldham, M. F. G. Stevens, J. E. Moses and M. S. Searle, Org. Biomol. Chem., 2009, 7, 4194–4200 RSC.
- X.-L. Wang, K. Wan and C.-H. Zhou, Eur. J. Med. Chem., 2010, 45, 4631–4639 CrossRef CAS.
- S. T. Abu-Orabi, M. Saleh, L. Al-Momani, I. Jibril and Y. Yousef, Jordan J. Chem., 2006, 1, 109–120 CAS.
- M. Saleh, Master, Yarmouk University, 2000.
- C. Marminon, J. Gentili, R. Baret and P. Nebois, Tetrahedron, 2007, 63, 735–739 CrossRef CAS.
- H. Devarajan-Ketha and K. B. Sloan, Bioorg. Med. Chem. Lett., 2011, 21, 4078–4082 CrossRef CAS.
- D. Renciuk, J. Zhou, L. Beaurepaire, A. Guedin, A. M. Bourdoncle and J.-L. Mergny, Methods, 2012, 57, 122–128 CrossRef CAS.
- A. D. Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M.-P. Teulade-Fichou, L. Lacroix and J.-L. Mergny, Methods, 2007, 42, 183–195 CrossRef PubMed.
- J.-L. Mergny, Biochemistry, 1999, 38, 1573–1581 CrossRef CAS PubMed.
- R. W. Woody, Methods Enzymol., 1995, 246, 34–71 CAS.
- W. C. Johnson, Protein Struct. Funct. Genet., 1990, 7, 205–214 CrossRef CAS.
- G. D. Fasman, Circular dichroism and conformational analysis of biomolecules, Springer, 1996 Search PubMed.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Nucleic Acids Res., 2006, 34, 2723–2735 CrossRef CAS PubMed.
- A. T. Phan, V. Kuryavyi, K. N. Luu and D. J. Patel, Nucleic Acids Res., 2006, 35, 6517–6525 CrossRef.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- L. L. C. Schrodinger, The Glide 2.5 calculations used First Discovery, version 2.5021, New York, June 2003 Search PubMed.
- G. N. Parkinson, R. Ghosh and S. Neidle, Biochemistry, 2007, 46, 2390–2397 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- F. Denizot and R. Lang, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS.
- K. R. Bauer, M. Brown, R. D. Cress, C. A. Parise and V. Caggiano, Cancer, 2007, 109, 1721–1728 CrossRef.
- M. Cini, T. D. Bradshaw, W. Lewis and S. Woodward, Eur. J. Org. Chem., 2013, 3997–4007 CrossRef CAS.
- R. A. J. Darby, M. Sollogoub, C. McKeen, L. Brown, A. Risitano, N. Brown, C. Barton, T. Brown and K. R. Fox, Nucleic Acids Res., 2002, 30, e39 CrossRef.
- A. Patel, W. Lewis, M. S. Searle, M. F. G. Stevens and C. J. Moody, Tetrahedron, 2015, 71, 7339–7343 CrossRef CAS.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, J. Comput.-Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemistry experimental protocols, copies of CD spectra, copies of LC-MS data for the tested compounds 44–53, 1H and 13C NMR spectra for compounds 34–53. See DOI: 10.1039/c7ra07257k |
| This journal is © The Royal Society of Chemistry 2017 |