DOI:
10.1039/C7RA06873E
(Paper)
RSC Adv., 2017,
7, 44867-44873
Alkylthiol-enabled Se powder dissolving for phosphine-free synthesis of highly emissive, large-sized and spherical Mn-doped ZnSeS nanocrystals†
Received
20th June 2017
, Accepted 13th September 2017
First published on 19th September 2017
Abstract
The enhanced dissolution of Se without organo-phosphines is a key issue in the synthesis of oil-soluble selenide nanocrystals. In this article, the dissolution of Se powder was greatly promoted by addition of 1-dodecanethiol at 200–220 °C. This Se precursor was of high reactivity and was suitable for the preparation of Se semiconductor nanocrystals. With this greener Se precursor, approximately spherical and large-sized Mn:ZnSeS quantum dots (QDs) could be prepared after multiple injections of the Zn precursor, and the size of Mn:ZnSeS can reach over 8.5 nm after the tenth injection of the Zn precursor. The preparation of this novel selenium precursor is simple and convenient, and provides a meaningful method for the synthesis and application of other selenium nanocrystals.
1. Introduction
Semiconductor quantum dots (QDs) have been extensively studied because of their flexible solution synthesis, excellent optical properties and promising applications, such as biological labeling, solid-state lighting, LEDs, etc.1–7 However, although a semiconductor QD material has high photoluminescence (PL) quantum yield (QY), with CdSe QDs as a representative, it is a great threat to human health because of its highly toxic heavy metal element (Cd), which has greatly limited the application in many fields.8 So far, colloidal semiconductor QDs have become an important research area in chemistry and materials science because of their size-dependent properties and promising applications.9
Wide band gap semiconductor nanocrystals, such as zinc chalcogenide nanocrystals doped with transition metal, can solve the above concern, which can maintain the advantages of intrinsic nanocrystals and possess other unique properties, such as larger Stokes shift, luminescence under high temperature, better photochemical stability.10–13 Mn(Cu)-doped ZnSe QDs (d-dots) without heavy-metal element are the most typical representative, which have been widely studied.12,14–17 Although people have been able to synthesize the high-quality doped metal selenium nanocrystals, but its synthetic chemistry is still in its infancy mainly because of the lack of high reactivity, stable, greener precursor.18 At present, the most synthetic methods of selenide nanocrystals use the organo-phosphines as the ligands to dissolve selenium powder.12,19 Since 2005, many chemists tried to find other substitutes to replace the Se–organophosphine precursor, including dissolving selenium powder with 1-octadecene (ODE) or olive oil under high temperature,20–22 using SeO2 as selenium source,23 recently dissolving selenium powder by sodium borohydride and thiols.18 In 2005, Zou and co-workers reported that Se powder was first reduced to H2Se and the long chain alkane was oxidized to alkene.24 The resulting H2Se gas was used as the selenium source to form the CdSe QDs. In 2010, we reported that Se powder could react with fatty amine at high temperature, such as oleylamine, and the prepared Mn:ZnSeS QDs possessed pure doping emission and luminous efficiency of 40–60%.14 However, in systematic study, the activity of selenium precursor was very difficult to control.17,22 That was so because the H2Se gas was easy to be formed during dissolving selenium powder. To date, the preparation of spherical ZnSe nanocrystals with size up to 5 nm is very difficult. The large-sized ZnSe nanocrystals are easy to result in the formation of branched nanocrystals due to the anisotropic growth.14 It is not conducive to further the formation of a stable geometrical structure. Therefore, it is very necessary to study the properties of Mn:ZnSe nanocrystals with large size, good stability, and spherical morphology.
The Mn:ZnSe QDs synthesized by conventional organo-phosphine methods have good stability and highly emission efficiency because the organo-phosphine is a reducing agent.25 Recently, Pradhan et al. reported that the luminescence was easily quenched by dissolved oxygen in solution, and the surface Se atoms of the nanoparticles are easily oxidized, resulting in low luminescence efficiency and poor photochemical stability.26 DDT has also been using to synthesize chalcogen nanocrystals based on phosphine-free synthesis.27–29 These shortcomings inspire us to find a greener and phosphine-free synthesis method to prepare large-sized, spherical, and high-quality Mn-doped nanocrystals.
In this work, the high efficient Mn-doped ZnSeS (Mn:ZnSeS) nanocrystals were prepared using organic selenide salt as the precursor, which was obtained by dissolving in the ODE in the presence of alkylthiol (e.g., 1-dodecanethiol). Se powder was firstly reduced by 1-dodecanethiol to generate an alkylammonium selenide at (200–230 °C). This precursor not only induced the growth of Mn:ZnSeS alloyed nanocrystals beyond 8 nm, but also manipulated incorporation of Mn ions in the center of the host cores, leading to highly luminous efficiency and excellent photostability.
2. Experimental section
2.1. Chemicals
Manganese acetate (MnAc2), zinc acetate (ZnAc2), manganese chloride (MnCl2·4H2O, ≥99%), 1-dodecanethiol (DDT, 98%), selenium powder (200 mesh, 99.999%), oleylamine (≥70%) were purchased from Aldrich. Oleic acid (OA), 1-octadecene (ODE, 90%) was purchased from Alpha Aesar. Manganese stearate (MnSt2) was purchased from City Chemical. All chemicals were used without further purification.
2.2. Preparation of stock solutions
The manganese precursor solution was prepared by dissolving MnSt2 (0.03 g) or the same amount of MnAc2/MnCl2 in 2.5 mL of ODE and heated to 100 °C after being degassed with Ar gas. The zinc precursor solution was prepared by dissolving ZnAc2 (0.5 mmol) and OA (2 mmol) in 2 g of ODE, and then heated to 160 °C until the solution turned clear.
2.3. Typical synthesis of Mn:ZnSeS d-dots
Selenium powder (0.0237 g), DDT (0.4 mL), and ODE (3 g) were loaded into a 25 mL three-neck flask and degassed at 80 °C for 10 min under Ar flow. The reaction mixture was then heated to 210 °C and kept this temperature until the Se powder was completely dissolved, then increased the temperature to 290 °C. The manganese precursor solution (0.2 mL) was rapidly injected into the reaction flask at 290 °C. After that, the 0.2 mL of OLA was quickly added 1 min later, an additional 0.3 mL of OLA was added quickly and the reaction mixture was allowed to cool to 278 °C. Subsequently, the reaction temperature was set at 260 °C with the injection of zinc precursor (the first layer, 0.1 mL) and reacted for 5–8 min for growth of ZnSeS shell. The reaction temperature was kept at 260 °C for further growth of the ZnSeS shell (the second to eighth layer, 0.1 mL). A designated amount of oleylamine (about 0.1 mL) was injected into the reaction mixture to active the zinc precursor. The growth process was monitored by UV-vis and PL spectroscopy by taking aliquots. Finally, the reaction was cooled to room temperature and purified using ethyl acetate for further characterization. Mn:ZnSeS QDs synthesized by this method typically had PL QY up to 50%.
2.4. Characterizations
Room temperature absorption spectra and PL spectra were recorded with the UV-2450 and F-6000 fluorescence spectrophotometers, respectively. The PL QY of d-dots was determined by comparing the integrated emission of the d-dots in solution with that of a fluorescent dye (rhodamine 6G in ethanol or rhodamine 101 in 0.01% HCl ethanol solution) with an identical optical density. Transmission electron microscopy (TEM) images were taken on a Tecnai F30 transmission electron microscope with 200 kV acceleration voltage. High resolution transmission electron microscopy (HRTEM) images were recorded using a JEM-2100F electron microscopy. Phase determination of the samples were performed on a X-ray diffractometer (Bruker AXS D8) using Cu-Kα radiation (λ = 1.5418 Å). The PL decay was performed with a single photo-counting spectrometer (TCSPC, Horiba Jobin Yvon Fluoromax-4P). FTIR spectrum was performed on an IRTracer-100. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos Axis Ultra X-ray photoelectron spectrometer with an Al Kα source.
3. Results and discussion
3.1. Structural characterization of Se–organic complex
In our experiments, we found that DDT could greatly promote the dissolution of selenium powder. This new synthetic system seems to have a new formation mechanism, and DDT was used as a major reducing agent. To confirm this hypothesis and know the bonding nature, FTIR spectra of DDT, the mixture of DDT and ODE, and the corresponding dissolved products were measured (Fig. 1). As shown in Fig. 1, the FTIR spectra showed that the characteristic peaks of the vinyl group of ODE were at the 906.7, 991.6 cm−1, and the peaks located at 1639.4 cm−1 should be attributed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch, and the peak at 3079.2 cm−1 was from C–H stretch. It should be noted that the new characteristic peak appeared at 958.9 cm−1, which should be from the product of Se powder dissolving in DDT and ODE. This confirmed that a new Se-DDT precursor was formed because of the addition of DDT. In order to investigate the nature of the precursor in our experiments, we tested the UV-vis absorption and PL spectra in addition to FTIR spectra. As indicated in Fig. S1 (ESI),† the absorption spectra of the three precursors are very different. In the range of 300–450 nm, the absorption of Se-DDT–ODE precursor is between those of Se-ODE and Se-DDT. In the synthetic protocol, we just used 0.4 mL of DDT, but 3 g of ODE (about 4.5 mL). That is to say, DDT only acts as an auxiliary solvent. Due to the large amount of ODE in the synthesis, ODE should play a great role in the dissolution of Se powder. The PL spectra also indicated that the three organics do not show PL property (ESI, Fig. S2†).
C stretch, and the peak at 3079.2 cm−1 was from C–H stretch. It should be noted that the new characteristic peak appeared at 958.9 cm−1, which should be from the product of Se powder dissolving in DDT and ODE. This confirmed that a new Se-DDT precursor was formed because of the addition of DDT. In order to investigate the nature of the precursor in our experiments, we tested the UV-vis absorption and PL spectra in addition to FTIR spectra. As indicated in Fig. S1 (ESI),† the absorption spectra of the three precursors are very different. In the range of 300–450 nm, the absorption of Se-DDT–ODE precursor is between those of Se-ODE and Se-DDT. In the synthetic protocol, we just used 0.4 mL of DDT, but 3 g of ODE (about 4.5 mL). That is to say, DDT only acts as an auxiliary solvent. Due to the large amount of ODE in the synthesis, ODE should play a great role in the dissolution of Se powder. The PL spectra also indicated that the three organics do not show PL property (ESI, Fig. S2†).
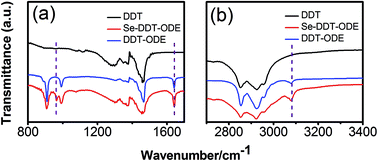 |
| | Fig. 1 FTIR spectra of the mixture obtained by dissolving Se powder in DDT and ODE (red), (a) 800–1700 nm, (b) 2700–3400 nm. For reference, FTIR spectra of DDT (black) and the mixture of DDT and ODE (blue) are also provided. Note: the dashed purple lines are only used as a guide. | |
3.2. Selection of synthetic strategies
The doping strategies mainly include the nucleation-doping and growth-doping.12 The previous reports indicated that resulting doped nanocrystals with nucleation-doping possessed higher PL QY than those with growth-doping.12 For the nucleation-doping strategy, the energy transfer and emission process can occur at the interface between the crystal nuclei containing the impurity ions and the host material, with the dopants act as the emissive center and the shell as the absorption zone. Based on this consideration, the emphasis of this work is to control the size of the crystal nuclei containing impurity ions as well as the interface of the crystal nuclei and the host shell. Therefore, controlling the size of the crystal nucleus is a prerequisite for the preparation of high-quality luminescent QDs. If the small-sized crystal nucleus will perfectly place the impurity ions in the center of nanocrystals, it will make the impurity ions from the surface as far as possible. Thus, the influence of surface defect states will be minimal, which is conducive to improve the emission efficiency. Therefore, the nucleation-doping was adopted in our study.
3.3. Effects of manganese precursor on the optical properties
The reaction activity of Mn precursor was the key to prepare small-sized crystal nucleus. The reactivity should be high enough to ensure the formation of the crystal nuclei, but also low enough to avoid the formation of the large-sized nucleus at high temperature.12 The balance of reactivity was achieved by using different manganese precursors. In our experiments, DDT could be used as a ligand, and excess DDT actually became a sulfur source, resulting in the formation of the MnSeS-alloyed crystal nucleus and ZnSeS-alloyed host shell (see the following XRD spectrum of the alloy structure). As shown in the Fig. 2a–c, with ZnSeS shell growth, high dopant emission was observed for all three manganese precursors. However, compared with the MnSt2 and MnAc2 precursors, the resulted Mn:ZnSeS QDs with MnCl2 exhibited obvious band edge emission of ZnSeS at 420 nm. This was probably due to the higher reactivity of the MnCl2 precursor. Thus, the larger MnSeS cores were formed at high temperature, resulting in the separate nucleation of ZnSeS nanocrystals. Fig. 2d shows the evolution of PL intensity of Mn:ZnSeS QDs with overcoating of the ZnSeS shell using the different manganese precursors. The reaction activity of the MnSt2 and MnAc2 precursors was almost identical, so the luminous efficiency of the resulting d-dots was relatively close. However, it should be noted that the d-dots synthesized by using MnCl2 possessed distinctly lower PL QY compared to those by using the MnSt2 and MnAc2 precursors due to significantly high reactivity of MnCl2. The highest PL QYs of Mn:ZnSeS QDs prepared using MnSt2, MnAc2, and MnCl2 are 79.8%, 69.4%, and 21.6%, respectively.
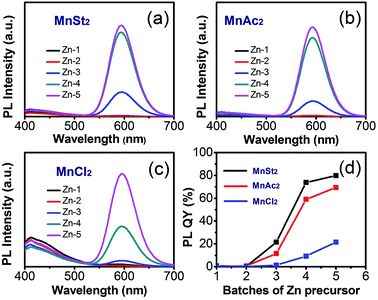 |
| | Fig. 2 PL spectra of Mn:ZnSeS nanocrystals with MnSt2 (a), MnAc2 (b), and MnCl2 (c) as the manganese precursors. Changes of PL intensity of Mn:ZnSeS nanocrystals with different manganese precursors (d). | |
3.4. Effects of growth time of MnSeS cores on the optical properties
The stable and small-sized MnSeS crystal nucleus is a prerequisite for the preparation of high-quality Mn:ZnSeS QDs based on the nucleation-doping strategy.12 The size of MnSeS cores was found to be difficult to be definitely confirmed because the size was too small (about 2 nm) and could not be observed by TEM. Therefore, the optimized size of MnSeS cores was indirectly determined through the changes of normalized PL intensity of the resulting d-dots. Form the absorption spectra of MnSe core with different growth time (ESI, Fig. S3†), the outline is very similar for the growth time of 10 s, 30 s, and 1 min due to the protective effect of DDT. As indicated in Fig. 3a, if the other experimental conditions were unchanged, the PL intensity of prepared d-dots increased with formation of the MnSeS nucleus, and reached maximum value at about 10 s. Then with further increasing the MnSeS size, the PL QY decreased gradually. This was mainly because the large-sized MnSeS nuclei made dopants closer to the surface of the d-dot, thus the luminescence was quenched by surface defects in part. The relationship between the PL intensity and the growth time is shown in Fig. 3b. If not specified, the MnSeS growth time mentioned in this article was 10 s.
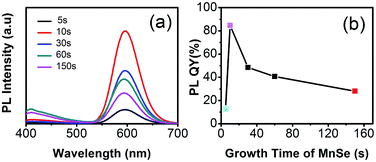 |
| | Fig. 3 PL spectra of Mn:ZnSeS d-dots using MnSt2 as the manganese precursor (a) and the corresponding evolution of PL intensity (b). | |
3.5. Effects of amounts of DDT on the optical properties
Due to the strong coordination ability of DDT to the surface free metal ions of MnSeS crystal nuclei, DDT can effectively limit the growth of nanocrystals and obtain the small size of MnSeS crystal nuclei.30 Thus, the amount of DDT has a great influence on the optical properties of the resulting d-dots.
From the UV-vis absorption spectra, the absorption bands of d-dots with different dosages of DDT are close (Fig. 4a), which indicates that the resulting d-dots have the close size. However, the different amount of DDT will lead to the difference of reactivity of Mn-DDT- and Zn-DDT-precursors, which may make the Mn ions in the different radial position of d-dots. The difference of the emission from the Mn ions was resulted by the surface defect state. As shown in the Fig. 4b and c, when the amount of DDT was 0.4 mL, the d-dots have the highest PL QY. This also meant that the Mn dopants were in the most central position, and the emission was least affected by the surface defect state. However, the specific radial position of Mn ions was still unclear and needed further study.
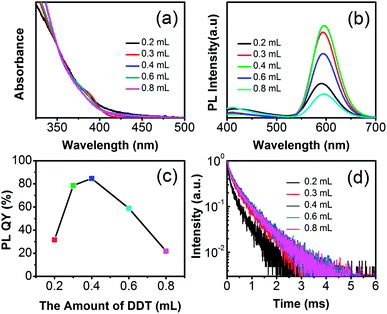 |
| | Fig. 4 UV-vis absorption (a) and PL (b) spectra, the change curve of normalized PL intensity (c), and PL decay (d) of the Mn:ZnSeS d-dots with different amount of DDT. | |
PL-decay has important value in the study of PL luminescence mechanism. The decay kinetics of Mn-doped QDs treated with different DDT amounts were measured (Fig. 4d). For all the five samples, the amount of DDT was 0.2 mL, 0.3 mL, 0.4 mL, 0.6 mL, and 0.8 mL, respectively. The decay curve was fitted by the biexponential function of I(t) = y0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2), where τ1 and τ2 are the time constants, and A1 and A2 are the normalized amplitudes of the components. The average lifetime was calculated by the formula of τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). The time constants of the decays are shown in Table 1. As seen from Table 1, the slower time constants of the two components were to be on the scale of hundreds of μs, which is attributed to the d–d transition of the electrons in the host conduction band.11 The faster one should be from the non-radiative decay. The average lifetimes of the five decays τav are 339 μs, 504 μs, 565 μs, 559 μs, and 547 μs, respectively, which are consistent with the reported results. As indicated in Table 1 and Fig. 6d, the DDT dependence of the PL lifetime agreed with the PL QY of d-dots. Thus, increasing the PL QY should come from the more central location of Mn dopants. The results also showed that the PL lifetime of d-dots with 0.4 mL DDT was longer than those of the other d-dots.
exp(−t/τ1) + A2exp(−t/τ2), where τ1 and τ2 are the time constants, and A1 and A2 are the normalized amplitudes of the components. The average lifetime was calculated by the formula of τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). The time constants of the decays are shown in Table 1. As seen from Table 1, the slower time constants of the two components were to be on the scale of hundreds of μs, which is attributed to the d–d transition of the electrons in the host conduction band.11 The faster one should be from the non-radiative decay. The average lifetimes of the five decays τav are 339 μs, 504 μs, 565 μs, 559 μs, and 547 μs, respectively, which are consistent with the reported results. As indicated in Table 1 and Fig. 6d, the DDT dependence of the PL lifetime agreed with the PL QY of d-dots. Thus, increasing the PL QY should come from the more central location of Mn dopants. The results also showed that the PL lifetime of d-dots with 0.4 mL DDT was longer than those of the other d-dots.
Table 1 The time constants of the Mn:ZnSeS/ZnS d-dots
| Amount of DDT |
A1 |
τ1 (μs) |
A2 |
τ2 (μs) |
τav (μs) |
| 0.2 mL |
0.34504 |
418 |
0.6575 |
64 |
339 |
| 0.3 mL |
0.44295 |
587 |
0.52729 |
103 |
504 |
| 0.4 mL |
0.50125 |
648 |
0.50157 |
122 |
565 |
| 0.6 mL |
0.48363 |
637 |
0.50099 |
111 |
559 |
| 0.8 mL |
0.4806 |
625 |
0.51425 |
103 |
547 |
3.6. Effects of multiple injections of Zn precursor on optical properties, morphology and crystal structure of nanocrystals
As we all know, the optical properties and the stability of QDs are extremely sensitive to their surface chemistry and external environment. In generally, this is an effective way to cover the shell in QDs for increasing the luminescent properties and stability, especially, a thick shell is needed indeed.31 Here, the multiple Zn precursors were injected, resulting in the formation of the ZnSeS shell with the excess DDT and Se power. As shown in the Fig. 5a and b, with the addition of Zn precursor, the PL QY increases gradually. However, after growing of “thicker” shells (about 5 layers), the PL QY declined gradually. The increase in fluorescence intensity should be due to a reduction in surface defects of QDs. As Mn:ZnSeS QDs further grew, the shells become thicker and more new structural defects were likely formed in the host lattice. These new defects may dominate, leading to decreasing of photoluminescence efficiency. The size of Mn:ZnSeS can reach above 8.5 nm after tenth injection of Zn precursor. The prepared QDs were of super high stability, and the PL QY could be maintained more than 80% a year later.
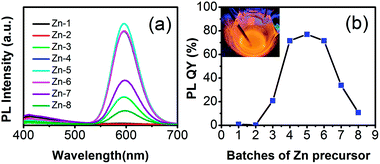 |
| | Fig. 5 PL spectra (a) and evolution of PL intensity (b) of the Mn:ZnSeS d-dots with addition of the Zn precursor. The digital picture (inset in the right panel) was taken with the radiation of a hand-held UV lamp (365 nm) at high reaction temperature. | |
To further characterize the structure of Mn:ZnSeS core/shell nanocrystals, the crystallographic structure was investigated by X-ray diffraction (XRD) (Fig. 6). The standard powder diffraction pattern of zinc blende ZnSe (red) and ZnS (blue) bulk crystals was shown in Fig. 6. Three broad diffraction peaks correspond to the (111), (220) and (311) crystal surface, respectively. As shown in Fig. 6, the peak positions were located in the middle of the bulk ZnSe and ZnS positions, which indicated the formation of host ZnSeS alloy structures.32 The resulted particle size was about 7.4 nm according to the scherrer formula. TEM images were also employed to study the morphology of Mn:ZnSeS core/shell QDs (Fig. 7). It was clear that all the NCs were basically spherical shaped and uniformly distribution. However, the structure of core/shell is still difficult to observe. That is so because lattice constants of and ZnSe and ZnS is similar MnSe(Fig. S4, ESI†). The particle radius increased gradually with deposition of the ZnSeS shell, and the size reached about 7.4 nm when the shell reached the eighth layers from the initial about 2 nm MnSeS core and the final particle size was consistent with that obtained by XRD results.
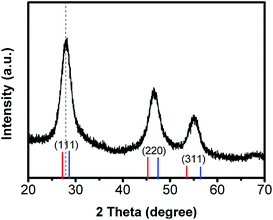 |
| | Fig. 6 XRD spectrum of Mn:ZnSeS d-dots. The labeled peak positions correspond to zinc blende ZnSe (red) and ZnS (blue), respectively. | |
 |
| | Fig. 7 TEM images of Mn:ZnSeS d-dots with ZnS-1 (a, 2.5 nm), ZnS-4 (b, 6.0 nm), and ZnS-8 (c, 7.4 nm). | |
To further obtain structural information of the Mn:ZnSeS QDs, XPS analysis was performed. Fig. 8 reveals the presence of Mn and Se in the MnSe cores and Mn, Zn, S and Se in the Mn:ZnSeS QDs, respectively. Typical peaks of Mn 2d at 641.8 and 642.0 eV were attributed to Mn(II) of MnSe cores and Mn:ZnSeS QDs (Fig. 8a),33 which indicated that Mn was successfully doped in the Mn:ZnSeS QDs. This result is in agreement with previous reports. It should be noted that S cannot be monitored in the MnSe cores (Fig. 8b), which is due to the low reactivity and the short growth time. The peaks at 1021.4 and 1044.5 eV were attributed the Zn of Mn:ZnSeS QDs (Fig. 8c).34 In Fig. 8d, Se was monitored in the MnSe cores and Mn:ZnSeS QDs, which indicated the Se was always involved in the reaction during the whole synthesis. In brief, based on XPS results, the structure Mn:ZnSeS QDs was clearly characterized, i.e., MnSe@ZnSeS core/shell structure.
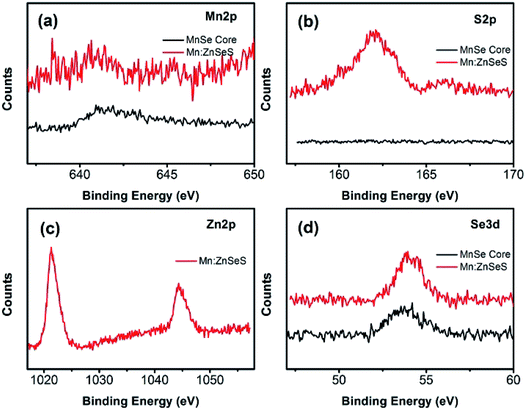 |
| | Fig. 8 XPS spectra of the as-obtained MnSe cores and Mn:ZnSeS QDs, Mn 2p (a), S 2p (b), Zn 2p (c), and Se 3d (d). | |
4. Conclusions
In summary, the solubility of Se powder was greatly enhanced by adding 1-dodecanethiol at high temperature (200–220 °C). The highly emissive Mn-doped ZnSeS QDs was obtained using this novel, greener and highly reactive Se precursor. After multiple injections of Zn precursor, the approximately spherical and large-sized Mn-doped ZnSeS QDs could be prepared, which were of super high stability. Typically, the size of QDs could reach 7.4 nm after the deposition of the eighth Zn precursor. This phosphine-free synthesis is simple, greener, and convenient, thus it should be extended to the synthesis of other selenide nanocrystals and has important application value in many practical fields such as light-emitting diode and photovoltaic devices.
Conflicts of interest
The authors declare that there are no conflicts to declare.
Acknowledgements
We acknowledge the financial supports from the National Natural Science Foundation of China (No. 21661010, 21461006, 51472053, and 61627807), project of Guangxi Innovative Research Team and Project of International Cooperation in Science and Technology of Guizhou Province ([2014]7008). We also thank Mr Sheng Cao for the measurement of PL decay and Mr Rongan Shen for the measurement of TEM images.
References
- Z. Bai, W. Ji, D. Han, L. Chen, B. Chen, H. Shen, B. Zou and H. Zhong, Chem. Mater., 2016, 28, 1085–1091 CrossRef CAS.
- R. Zeng, T. Zhang, J. Liu, S. Hu, Q. Wan, X. Liu, Z. Peng and B. Zou, Nanotechnology, 2009, 20, 095102 CrossRef PubMed.
- Z. Bai and H. Zhong, Sci. Bull., 2015, 60, 1622–1624 CrossRef CAS.
- C. Pu, H. Qin, Y. Gao, J. Zhou, P. Wang and X. Peng, J. Am. Chem. Soc., 2017, 139, 3302–3311 CrossRef CAS PubMed.
- Q. Zhou, Z. Bai, W.-g. Lu, Y. Wang, B. Zou and H. Zhong, Adv. Mater., 2016, 28, 9163–9168 CrossRef CAS PubMed.
- X. Meng, J. Du, H. Zhang and X. Zhong, RSC Adv., 2015, 5, 86023–86030 RSC.
- Y. Ma, Y. Li and X. Zhong, RSC Adv., 2014, 4, 45473–45480 CAS.
- X. Peng, Nano Res., 2009, 2, 425–447 CrossRef CAS.
- X. Peng, Acc. Chem. Res., 2010, 43, 1387–1395 CrossRef CAS PubMed.
- A. K. Guria, S. K. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2017, 2, 1014–1021 CrossRef CAS.
- S. Cao, C. Li, L. Wang, M. Shang, G. Wei, J. Zheng and W. Yang, Sci. Rep., 2014, 4 Search PubMed.
- N. Pradhan and X. Peng, J. Am. Chem. Soc., 2007, 129, 3339–3347 CrossRef CAS PubMed.
- N. Pradhan, S. Das Adhikari, A. Nag and D. D. Sarma, Angew. Chem., Int. Ed. Engl., 2017, 56, 7038–7054 CrossRef CAS PubMed.
- R. Zeng, M. Rutherford, R. Xie, B. Zou and X. Peng, Chem. Mater., 2010, 22, 2107–2113 CrossRef CAS.
- R. Zeng, R. Shen, Y. Zhao, Z. Sun, X. Li, J. Zheng, S. Cao and B. Zou, CrystEngComm, 2014, 16, 3414–3423 RSC.
- R. Zeng, R. Shen, Y. Zhao, X. Li, Z. Sun and Y. Shen, Nanotechnology, 2014, 25, 135602 CrossRef PubMed.
- R. Zeng, T. Zhang, G. Dai and B. Zou, J. Phys. Chem. C, 2011, 115, 3005–3010 CAS.
- C. Pu, J. Zhou, R. Lai, Y. Niu, W. Nan and X. Peng, Nano Res., 2013, 6, 652–670 CrossRef CAS.
- J. Jasieniak, C. Bullen, J. van Embden and P. Mulvaney, J. Phys. Chem. B, 2005, 109, 20665–20668 CrossRef CAS PubMed.
- Y. Wei, J. Yang, A. W. H. Lin and J. Y. Ying, Chem. Mater., 2010, 22, 5672–5677 CrossRef CAS.
- S. Sapra, A. L. Rogach and J. Feldmann, J. Mater. Chem., 2006, 16, 3391–3395 RSC.
- C. Bullen, J. van Embden, J. Jasieniak, J. E. Cosgriff, R. J. Mulder, E. Rizzardo, M. Gu and C. L. Raston, Chem. Mater., 2010, 22, 4135–4143 CrossRef CAS.
- O. Chen, X. Chen, Y. Yang, J. Lynch, H. Wu, J. Zhuang and Y. C. Cao, Angew. Chem., Int. Ed., 2008, 47, 8638–8641 CrossRef CAS PubMed.
- Z. T. Deng, L. Cao, F. Q. Tang and B. S. Zou, J. Phys. Chem. B, 2005, 109, 16671–16675 CrossRef CAS PubMed.
- N. Pradhan, D. M. Battaglia, Y. Liu and X. Peng, Nano Lett., 2007, 7, 312–317 CrossRef CAS PubMed.
- S. Jana, B. B. Srivastava, S. Acharya, P. K. Santra, N. R. Jana, D. D. Sarma and N. Pradhan, Chem. Commun., 2010, 46, 2853–2855 RSC.
- Y. Liu, D. Yao, L. Shen, H. Zhang, X. Zhang and B. Yang, J. Am. Chem. Soc., 2012, 134, 7207–7210 CrossRef CAS PubMed.
- D. Yao, W. Xin, Z. Liu, Z. Wang, J. Feng, C. Dong, Y. Liu, B. Yang and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 9840–9848 CAS.
- D. Yao, H. Liu, Y. Liu, C. Dong, K. Zhang, Y. Sheng, J. Cui, H. Zhang and B. Yang, Nanoscale, 2015, 7, 18570–18578 RSC.
- W. Zhang, Y. Li, H. Zhang, X. Zhou and X. Zhong, Inorg. Chem., 2011, 50, 10432–10438 CrossRef CAS PubMed.
- C. Pu and X. Peng, J. Am. Chem. Soc., 2016, 138, 8134–8142 CrossRef CAS PubMed.
- R. Zeng, Z. Sun, C. Zhou, C. Fang, G.-C. Han and Z. Chen, J. Alloys Compd., 2016, 671, 66–73 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- R. Zeng, Z. Sun, S. Cao, R. Shen, Z. Liu, Y. Xiong, J. Long, J. Zheng, Y. Zhao, Y. Shen and D. Wang, RSC Adv., 2015, 5, 1083–1090 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06873e |
| ‡ These authors contributed equally to the paper. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Bingsuo Zoub and
Zhencheng Chen*a
*a,
Bingsuo Zoub and
Zhencheng Chen*a







