
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkaloids from the stems of Clausena lansium and their neuroprotective activity†
Jie Liuab,
Yi-Qian Dua,
Chuang-Jun Lia,
Li Lia,
Fang-You Chena,
Jing-Zhi Yanga,
Nai-Hong Chen a and
Dong-Ming Zhang*a
a and
Dong-Ming Zhang*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: zhangdm@imm.ac.cn; Fax: +86-10-63165227; Tel: +86-10-63165227
bBeijing Research Institute of Chinese Medicine, Beijing University of Chinese Medicine, Beijing 100029, People's Republic of China
First published on 14th July 2017
Abstract
Eight new alkaloids, including three pairs of enantiomers (+)-(2′S,3′R)-clauselansine A (1a) and (−)-(2′R,3′S)-clauselansine A (1b); (+)-(2′S,3′R)-clauselansine B (2a) and (−)-(2′R,3′S)-clauselansine B (2b); (+)-(3S,4R,5S,6S)-clauselansine C (3a) and (−)-(3R,4S,5R,6R)-clauselansine C (3b), (+)-(1′R,2′R,6′R)-claulansine B (4a), and (+)-(1′R,2′R)-claulansine D (5a), together with twelve known alkaloids (4b, 5b, 6a, 6b, 7a, 7b and 8–13) were isolated from the stems of Clausena lansium. Their structural elucidation and stereochemistry determination were achieved by spectroscopic and chemical methods including 1D and 2D NMR experiments, especially the employment of electronic circular dichroism calculation spectra, Mosher's method, and Snatzke's method expressed by the induced circular dichroism spectrum. Compounds 1b, 2a, 3b, 5a, and 5b inhibited PC12 cell damage induced by Okadaic Acid, and increased cell viability from 70.5 ± 5.4% to 83.4 ± 4.1%, 91.2 ± 10.1%, 83.5 ± 7.8%, 89.7 ± 4.8%, 83.3 ± 5.9% at 10 μM, respectively.
Introduction
Clausena is a small genus within the family Rutaceae, which is distributed throughout the south of the Yangtze River in china, such as the Yunnan, Guangxi and Guangdong regions.1 Previous phytochemical studies have revealed that the Clausena genus contains a wide variety of carbazole alkaloids, amide alkaloids, coumarins, and limonoids, many of which possess various pharmacological properties such as antibacterial, anti-inflammatory, anti-HIV, cytotoxic, hepatoprotective and neuroprotective effects.2–10 The leaves and fruits of Clausena lansium, Clausena excavata, Clausena emarginata, and Clausena anisum-molens are not only eaten as food but also used for traditional medicine.Clausena lansium (Lour.) Skeels (Rutaceae), a fruit tree, was widely distributed in southern China. In traditional Chinese medicine, the leaves and roots of C. lansium were used for cough, asthma, dermatological disease, viral hepatitis, and gastro-intestinal diseases; the seeds are used to treat acute and chronic gastro-intestinal inflammation, ulcers, etc.11 Various bioactive constituents including coumarins, carbazole alkaloids and amide alkaloids have been isolated and identified from this plant.12–14 Previously, twenty new natural products including thirteen new carbazole alkaloids,10,15 eight alkaloid glycosides,16 four new coumarins,17 a new amide and a new megastigmane glucoside18 from the leaves and skeels of C. lansium were reported by our research group, and some of these alkaloids showed selective neuroprotective effects. In order to investigate the potential neuroprotective constituents from different parts of C. lansium, n-BuOH extract of the stems of this plant were selected for investigation. This paper reported further investigation of n-BuOH extract from the stems of C. lansium which led to the isolation and characterization of four new indole alkaloids (1a, 1b, 2a, 2b), two new amide alkaloids (3a, 3b), and two carbazole alkaloids (4a, 5a) along with twelve known compounds (4b, 5b, 6a, 6b, 7a, 7b, 8–13) from the stems of C. lansium. The neuroprotective activities of 1–13 were also evaluated. We present herein the isolation and structural characterization of 1–13, as well as their bioactivities (Fig. 1).
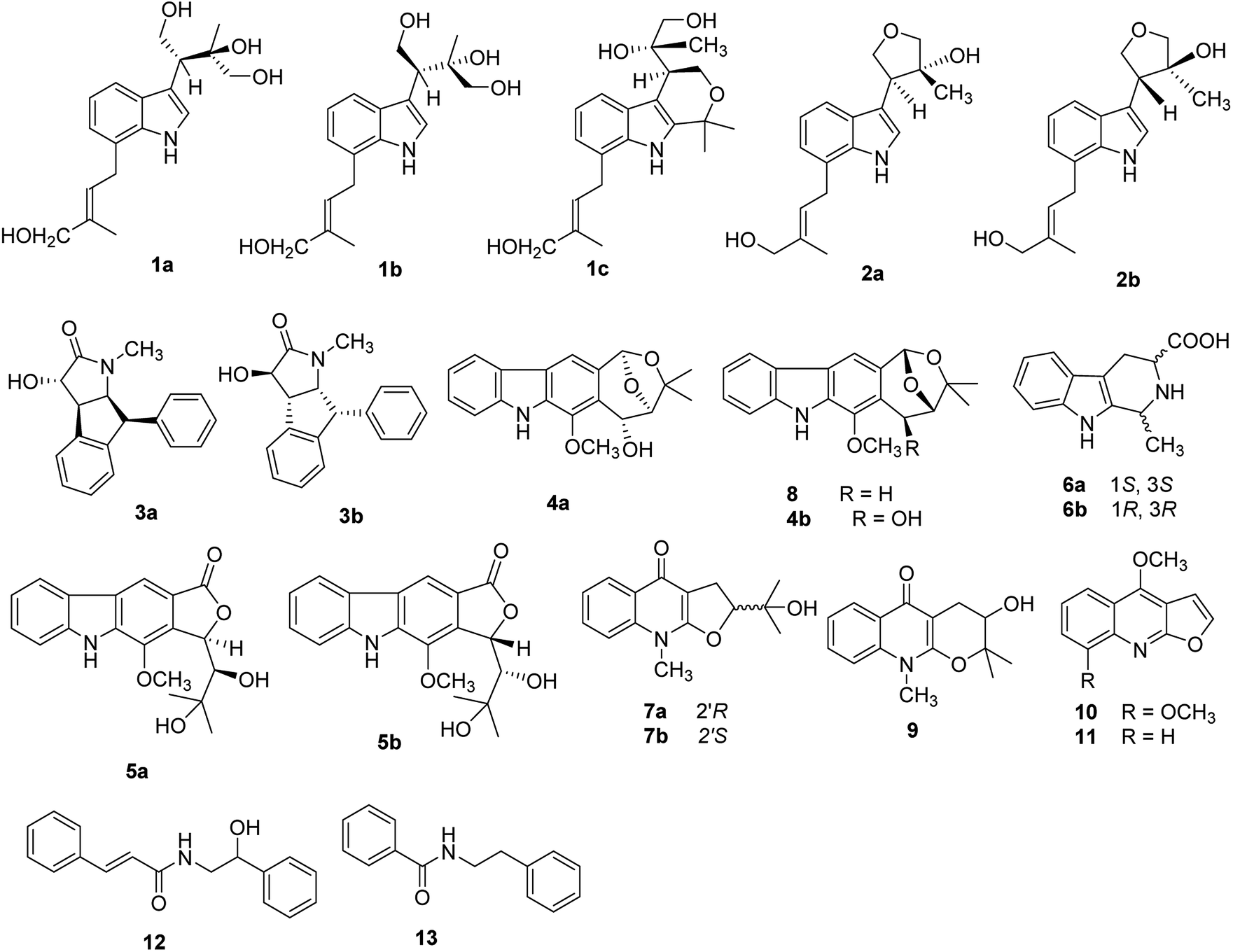 | ||
| Fig. 1 Alkaloid derivatives (1–13) obtained from the stems of C. lansium. | ||
Results and discussion
Compound 1 (1a/1b) was obtained as colourless oil. Its molecular formula was assigned as C18H25NO4 based on the 13C NMR spectroscopic data and HRESIMS (m/z 320.1854 [M + H]+, calcd for C18H26NO4, 320.1856), implying seven indices of hydrogen deficiency. The IR spectrum displayed absorptions characteristic of amino (3313 cm−1), amide (1640 cm−1) and aromatic ring (1671, 1612, and 1439 cm−1) groups, and the UV spectrum showed absorptions at λmax 202, 223, 282 nm. The 1H NMR (Table 1) spectrum showed a set of signals for 1,2,3-trisubstituted benzene ring at 7.43 (1H, d, J = 7.6 Hz, H-4), 6.90 (1H, t, J = 7.4 Hz, H-5), 6.85 (1H, d, J = 6.8 Hz, H-6), together with a doublet at δH 7.18 (1H, d, J = 2.4 Hz, H-2) and a broad signal at δH 10.80 (1H, NH), which indicated a 3,7-disubstituted indole moiety. In addition, a double bond group at δH 5.66 (1H, t, J = 7.3 Hz, H-2′′), a methylene at 3.55 (2H, d, J = 7.3 Hz, H-1′′) and two methyls at δH 1.70 (3H, s, H-5′′), 1.00 (3H, s, H-5′) were also exhibited in the 1H NMR spectrum. The 13C NMR and DEPT spectra exhibited a double bond, and four methylenes, two methines, two methyls and the remaining eight in indole moiety. The above information indicated 1 was a diprenylated indole. A comparison of the 1H and 13C NMR of 1 with those of hexalobines19 suggested that their structures are closely related, except for 1,3,4-trihydroxy-3-methylbut-2-yl and 4-hydroxy-3-methyl-2-butenyl at C-3 and C-7 in 1. In the HMBC spectrum, the cross-peaks between H-1′/C-3 (δC 114.2), C-3′ (δC 75.0), H-2′/C-2 (δC 123.0), C-3a (δC 128.3), C-5′ (δC 22.0), H-4′/C-3′ (δC 75.0), C-5′ (δC 22.0) demonstrated 1,3,4-trihydroxy-3-methylbut-2-yl group attached to C-3 of the indole moiety (Fig. 2); the cross-peaks between H-1′′/C-6 (δC 119.7), C-7a (δC 134.4), C-3′′ (δC 136.3), H-2′′/C-7 (δC 123.7), C-4′′ (δC 66.3), C-5′′ (δC 13.7) demonstrated 4-hydroxy-3-methyl-2-butenyl group attached to C-7 of the indole moiety (Fig. 2). The NOE difference experiment displayed that a strong enhancement of H-2′′ was observed when H-4′′ was irradiated while H-2′′ was no enhanced on irradiation of H-5′′, indicating an E configuration of the double bond. Thus, the structure of 1 was elucidated. The specific rotation of 1 approached zero, and no Cotton effect was found in the electronic circular dichroism (ECD) spectrum of 1, indicating a racemic mixture. Subsequent chiral resolution of 1 afforded the anticipated enantiomers 1a and 1b, which showed mirror image-like ECD curves and specific rotations {1a: [α]20D +18.7 (c 0.1 MeOH); 1b: [α]20D −17.0 (c 0.1 MeOH)}. In order to confirm the absolute configuration of the 1′,3′,4′-triol of 1b, 1b was treated with 2,2-dimethoxypropane and pyridinium p-toluene sulfonate and converted into its acetonide 1c (Fig. 3). According to 1D and 2D NMR, 1c was determined similar to pyrido[3,4-b]pyrano[3,4-b]indoles.20 In order to confirm the absolute configuration of 1c, the ECD calculations were also performed for the four configurations 2′R,3′S-, 2′R,3′R-, 2′S,3′R-, and 2′S,3′S-1c using the time-dependent density functional theory (TDDFT) method at the B3LYP/6-31G (d) level.21,22 The calculated ECD spectrum for 2′R,3′S- and 2′R,3′R-1c enantiomer agreed with the experimental ECD data (Fig. 4) of 1c. Thus, the absolute configuration at C-2′ of 1c was 2′R. In addition, the absolute configuration of the 3′,4′-diol moiety of 1c was determined using induced CD spectra by Snatzke's method.23,24 A positive Cotton effect at 323 nm (Fig. 5) in the induced CD spectrum indicated the 3′S configuration for 1c by means of the empirical helicity rule. According to the above information, the absolute configuration of 1b was 2′R,3′S, and then the absolute configuration of 1a was 2′S,3′R. Therefore, compounds 1a and 1b were given the trivial names (+)-(2′S,3′R)-clauselansine A and (−)-(2′R,3′S)-clauselansine A, respectively.| Position | 1 | 1c | 2 | |||
|---|---|---|---|---|---|---|
| δHa | δCb | δHa | δCb | δHa | δCb | |
| a In DMSO-d6 (600 MHz).b In DMSO-d6 (150 MHz). Coupling constants (J) in Hz are given in parentheses. The assignments were based on HSQC and HMBC experiments. | ||||||
| 2 | 7.18, d (2.4) | 123.0 d | 140.5 | 7.23, d (2.4) | 123.9 d | |
| 3 | 114.2 s | 106.1 | 108.4 s | |||
| 3a | 128.3 s | 127.3 | 128.3 s | |||
| 4 | 7.43, d (7.4) | 116.8 d | 7.58, d (7.5) | 117.1 | 7.42, d (7.4) | 116.5 d |
| 5 | 6.90, t (7.4) | 118.3 d | 6.86, t (7.5) | 118.7 | 6.90, t (7.4) | 118.5 d |
| 6 | 6.85, d (7.4) | 119.7 d | 6.80, d (7.5) | 119.6 | 6.85, d (7.4) | 119.9 d |
| 7 | 123.7 s | 123.4 | 123.5 s | |||
| 7a | 134.4 s | 134.4 | 134.7 s | |||
| 1′ | 4.02, m; 3.76, m | 62.6 t | 4.28, d (11.6); 3.68, dd (11.6, 3.6) | 61.0 t | 4.02, m | 71.8 t |
| 2′ | 3.21, m | 43.6 d | 2.87, d (3.6) | 38.9 d | 3.42, t (9.7) | 46.3 d |
| 3′ | 75.0 s | 74.8 s | 76.8 s | |||
| 4′ | 3.19, m | 68.4 t | 3.41, m; 3.26, m | 68.4 t | 3.79, s | 80.5 t |
| 5′ | 1.00, s | 22.0 q | 1.02, s | 22.0 q | 1.18, s | 22.9 q |
| 1′′ | 3.55, d (7.3) | 28.6 t | 3.57, d (7.4) | 28.7 t | 3.56, d (7.4) | 28.5 t |
| 2′′ | 5.66, t (7.3) | 121.2 d | 5.67, t (7.4) | 121.2 d | 5.65, t (7.4) | 121.1 d |
| 3′′ | 136.3 s | 136.3 s | 136.3 s | |||
| 4′′ | 3.84, d (5.6) | 66.3 t | 3.85, s | 66.2 t | 3.83, s | 66.2 t |
| 5′′ | 1.70, s | 13.7 q | 1.70, s | 13.7 q | 1.69, s | 13.6 q |
| NH | 10.80, br s | 10.60, s | 10.90, s | |||
| 1′′′ | 1.56, s | 28.3 | ||||
| 2′′′ | 71.7 | |||||
| 3′′′ | 1.45, s | 26.0 | ||||
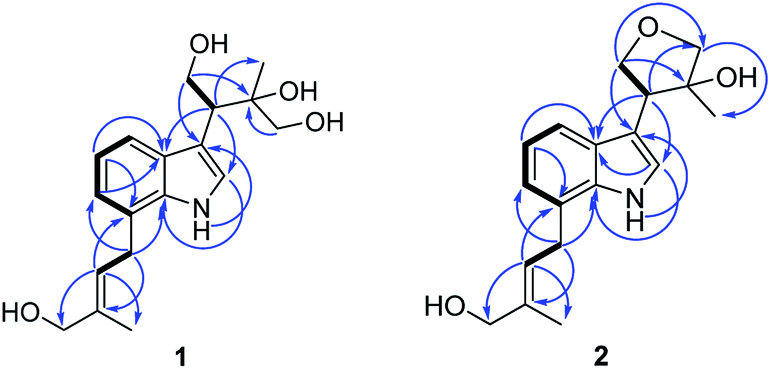 | ||
| Fig. 2 Key 1H, 1H-COSY and HMBC correlations of compound 1 and 2. | ||
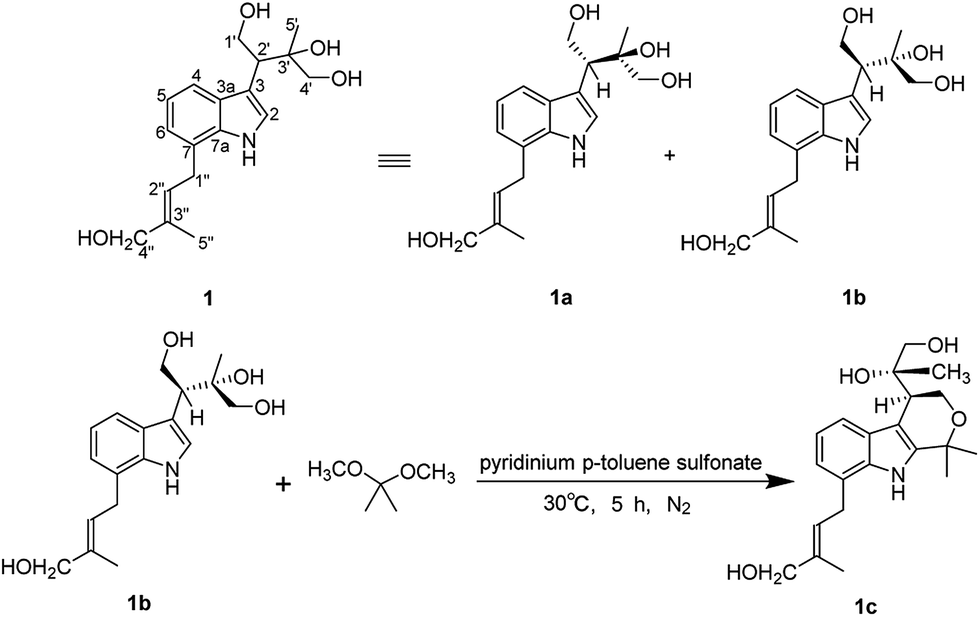 | ||
| Fig. 3 The isolation of 1 to 1a and 1b and the action of compound 1b to its acetonide derivative 1c. | ||
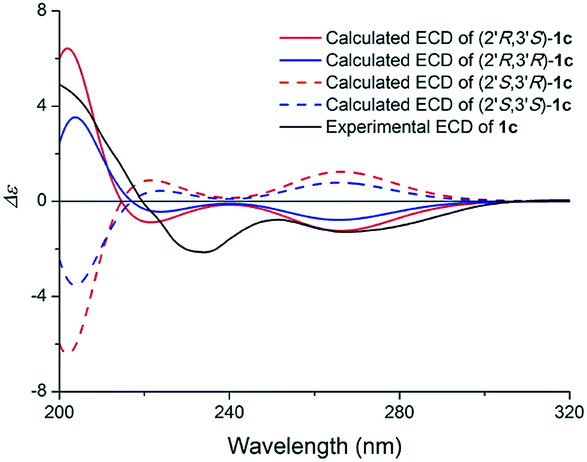 | ||
| Fig. 4 Calculated ECD spectra of (2′R,3′S)-1c, (2′R,3′R)-1c, (2′S,3′R)-1c, (2′S,3′S)-1c and the experimental ECD spectrum of (1c) in MeOH. | ||
 | ||
| Fig. 5 CD spectrum of compound 1c in a DMSO of dimolybdenum tetraacetate (the inherent CD of the diol was subtracted). | ||
Compound 2 (2a/2b) was obtained as a white powder. Its molecular formula C18H23NO3 was deduced from the HRESIMS (m/z 302.1758 [M + H]+, calcd for C18H24NO3, 302.1751) and 13C NMR spectroscopic data, corresponding with eight indices of hydrogen deficiency. The 1H and 13C NMR of 2 displayed signals characteristic of diprenylated indole, which were similar to these reported for compound 1. The only difference between 2 and 1 was that OH-1′ and OH-4′ of 1,3,4-trihydroxy-3-methylbut-2-yl group in 1 is formed five-membered oxygen ring in 2. This was supported further by the HMBC correlations between H-1′ (δH 4.02) and C-4′ (δC 80.5) and H-4′ (δH 3.79) and C-1′ (δC 71.8). The NOE difference experiment displayed that a strong enhancement of H-2′′ was observed when H-4′′ was irradiated, while H-2′′ was no enhanced on irradiation of H-5′′, indicating an E configuration of the double bond. Thus, the structure of 2 was elucidated.
The specific rotation of 2 approached zero, and no Cotton effect was found in the electronic circular dichroism (ECD) spectrum of 2, indicating a racemic mixture. Subsequent chiral resolution of 2 afforded the anticipated enantiomers 2a and 2b, which showed mirror image-like ECD curves and specific rotations {2a: [α]20D +28.0 (c 0.1 MeOH); 2b: [α]20D −32.6 (c 0.1 MeOH)}. In order to confirm the absolute configuration of the enantiomers 2a and 2b, a systematic conformational analysis and optimization were performed for 2a and 2b using the same method applied to 1a and 1b. A comparison of the theoretically calculated and experimental ECD curves (Fig. 6) demonstrated that the configuration of 2a was 2′S,3′R and the configuration of 2b was 2′R,3′S. According to the structures of 1a and 2a, we speculate that 2a was possibly generated by dehydration of 1a, which means the configuration of 2a was the same as 1a. And 2b was also possibly generated by dehydration of 1b, which means the configuration of 2b was the same as 1b. Thus, 2a has a 2′S,3′R-configuration and 2b has a 2′R,3′S-configuration. Therefore, compounds 2a and 2b were given the trivial names (+)-(2′S,3′R)-clauselansine B and (+)-(2′R,3′S)-clauselansine B, respectively.
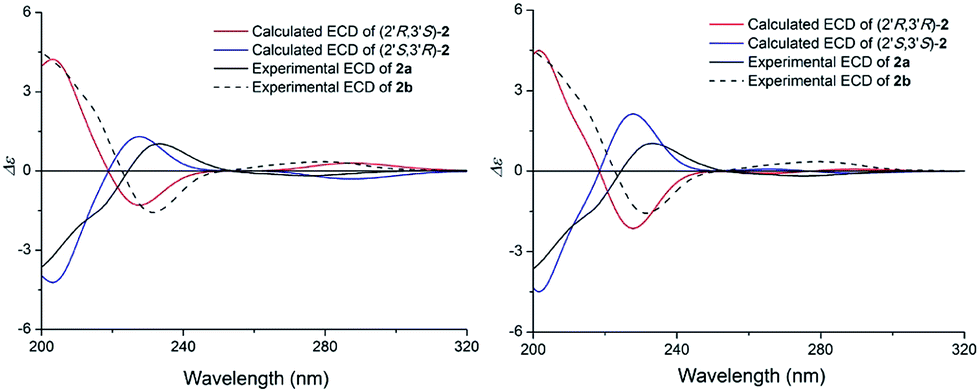 | ||
| Fig. 6 Calculated ECD spectra of (2′R,3′S)-2, (2′S,3′R)-2, (2′R,3′R)-2, (2′S,3′S)-2 and the experimental ECD spectrum of (+)- and (−)-clauselansine B (2a/2b) in MeOH. | ||
Compound 3 (3a/3b) was obtained as a white solid. Its molecular formula was assigned as C18H17NO2 based on the 13C NMR spectroscopic data and HRESIMS (m/z 280.1333 [M + Na]+, calcd for C18H17NO2Na, 280.1332), implying eleven indices of hydrogen deficiency. The IR spectrum displayed absorptions characteristic of hydroxyl (3320 cm−1), carbonyl (1678 cm−1) and aromatic groups (1601, 1483, and 1454 cm−1). The 1H NMR spectrum showed nine aromatic protons [ring A: δH 6.92 (1H, d, J = 7.6 Hz, H-3′), 7.23 (1H, overlapped, H-4′), 7.31 (1H, m, H-5′) and 6.23 (1H, d, J = 7.6 Hz, H-6′); ring B: δH 6.98 (2H, d, J = 7.1 Hz, H-2′′, 6′′), 7.27 (2H, m, H-3′′, 5′′), 7.23 (1H, overlapped, H-4′′)], together with four methine groups at δH 4.11 (1H, d, J = 8.3 Hz, H-3), 3.77 (1H, dd, J = 8.3, 5.3 Hz, H-4), 4.66 (1H, t, J = 8.3 Hz, H-5), 4.77 (1H, d, J = 8.3 Hz, H-6), and a hydroxyl group at δH 5.97 (1H, s). The 13C NMR spectrum exhibited twelve aromatic carbons, one carbonyl δC 174.1, one oxymethine δC 75.0, three methines (δC 52.3, 53.3, 65.2), and one methyl (δC 29.6). The 1H, 1H-COSY correlations (Fig. 7) suggested one OCH–CH–CH–CH fragment. The 1H and 13C NMR data (Table 2) also indicated 1,2-disubstituted and monosubstituted aromatic units (ring A and B) and one methyl group. The HMBC correlations (Fig. 6) of H-4 with C-2′ (δC 144.7) and C-6′ (δC 124.3); H-5 with C-2′ (δC 144.7) and C-1′′ (δC 140.7); H-6 with C-3′ (δC 125.8) and C-2′′/C-6′′ (δC 129.6) showed that ring A was connected at C-4 and C-6, and ring B was connected at C-6. The above information coupled with biogenetic considerations and literature references indicated 3 was similar to the dehydro-derivative of neoclausenamide.25,26 Its relative configuration was established on the basis of NOESY correlations (Fig. 7). The NOESY correlations of H-4 with H-5, H-5 with H-4, H-6, N–CH3 and H-6 with H-5, H-6′′, H-3 with H-2′′ showed that H-4, H-5 and H-6 were β orientation, while H-3 was α orientation. Thus, compound 3 (clauselansine C) was fully identified.
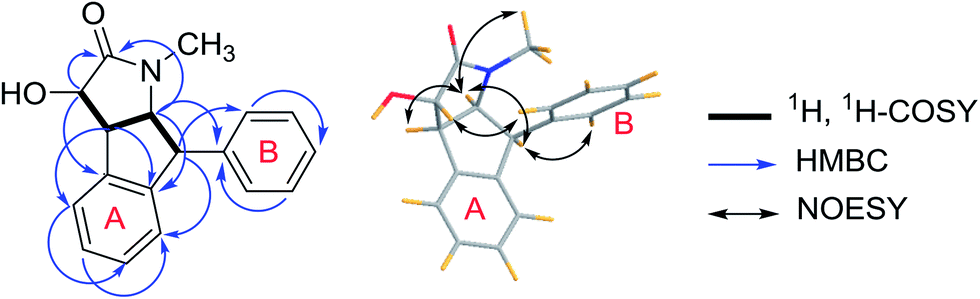 | ||
| Fig. 7 Key 1H, 1H-COSY, HMBC and NOESY correlations of compound 3. | ||
| Position | 3 | Position | 3 | ||
|---|---|---|---|---|---|
| δHa | δCb | δHa | δCb | ||
| a In DMSO-d6 (600 MHz).b In DMSO-d6 (150 MHz).c Signal overlapped. Coupling constants (J) in Hz are given in parentheses. The assignments were based on HSQC and HMBC experiments. | |||||
| 2 | 174.1 s | 5′ | 7.31, m | 127.7 | |
| 3 | 4.11, d (8.3) | 75.0 d | 6′ | 7.45, d (7.6) | 124.3 |
| 4 | 3.77, dd (8.3, 5.3) | 52.3 d | 1′′ | 140.7 | |
| 5 | 4.66, t (8.3) | 65.2 d | 2′′, 6′′ | 6.98, d (7.1) | 129.6 |
| 6 | 4.77, d (8.3) | 53.3 d | 3′′, 5′′ | 7.27, m | 128.2 |
| 1′ | 142.8 s | 4′′ | 7.23c | 126.9 | |
| 2′ | 144.7 s | N–CH3 | 2.10, s | 29.6 | |
| 3′ | 6.92, d (7.6) | 125.8 d | OH | 5.97, s | |
| 4′ | 7.23c | 128.0 d | |||
The specific rotation of 3 approached zero, and no Cotton effect was found in the electronic circular dichroism (ECD) spectrum of 3, indicating a racemic mixture. Subsequent chiral resolution of 3 afforded the anticipated enantiomers 3a and 3b, which showed mirror image-like ECD curves and specific rotations {3a: [α]20D +17.9 (c 0.1 MeOH); 3b: [α]20D −22.0 (c 0.1 MeOH)}. Compound 3a was treated with (R)- and (S)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl) in anhydrous CH2Cl2 to afforded the 3a-(S)-MTPA ester (3aa) and 3a-(R)-MTPA ester (3ab), respectively. The ΔδSRH values were calculated as shown in Fig. 8. Application of Mosher's rule27 revealed that 3a had the 3S,4R,5S,6S configuration. Meanwhile, the absolute configuration of 3b was assigned with the 3R,4S,5R,6R-configuration. In order to confirm the absolute configuration of the enantiomers 3a and 3b, the ECD calculations were also performed for the two configurations 3S,4R,5S,6S- and 3R,4S,5R,6R-3 using the time-dependent density functional theory (TDDFT) method at the B3LYP/6-31G (d) level.21,22 The calculated ECD spectrum for 3S,4R,5S,6S-3 agreed with the experimental ECD data (Fig. 9) of 3a. The calculated ECD spectrum for 3R,4S,5R,6R-3 was in good accordance with the experimental spectrum of 3b (Fig. 9). Thus, 3a has a 3S,4R,5S,6S configuration and 3b has a 3R,4S,5R,6R configuration. Therefore, compounds 3a and 3b were given the trivial names (+)-(3S,4R,5S,6S)-clauselansine C and (−)-(3R,4S,5R,6R)-clauselansine C, respectively.
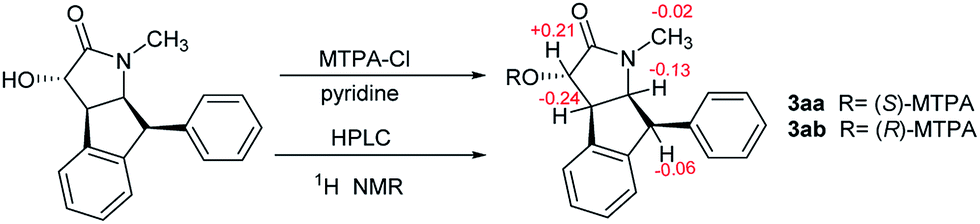 | ||
| Fig. 8 The Mosher's method of 3a. | ||
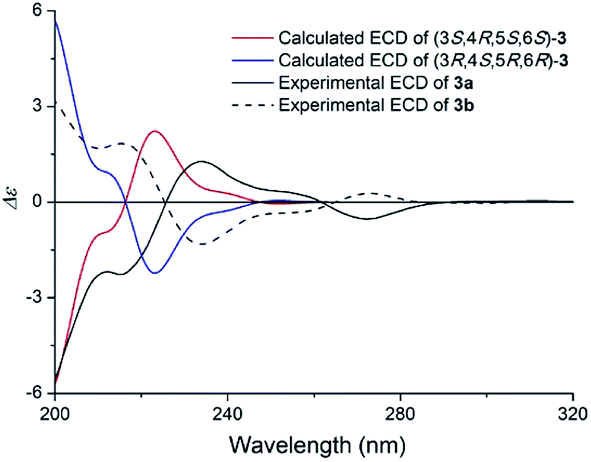 | ||
| Fig. 9 Calculated ECD spectra of (3S,4R,5S,6S)-3 and (3R,4S,5R,6R)-3 and the experimental ECD spectra of (+)- and (−)-clauselansine C (3a/3b) in MeOH. | ||
Compound 4 (4a/4b) was obtained as a white powder. Its molecular formula was determined as C19H19NO4 on the basis of its 13C NMR and HRESIMS (m/z 326.1398 [M + H]+, calcd for C19H20NO4 326.1387), corresponding with eleven indices of hydrogen deficiency. Its NMR data (Table 3) was almost identical to those of claulansine B.10 Chiral separation of 4 afforded a pair of enantiomer 4a {[α]20D +142.7 (c 0.1 MeOH)} and 4b {[α]20D −99.5 (c 0.1 MeOH)}, which had opposite ECD curves. By comparison of the calculated ECD spectra of the 1′R,2′R,6′R and 1′S,2′S,6′S configurations of 4 with the experimental data of 4a and 4b (Fig. 10), the absolute configurations of 4a and 4b were assigned as 1′R,2′R,6′R and 1′S,2′S,6′S. Thus, 4a was defined as (+)-(1′R,2′R,6′R)-claulansine B, and 4b was identified as the known compound (−)-(1′S,2′S,6′S)-claulansine B.
| Position | 4a | 5a | ||
|---|---|---|---|---|
| δHa | δCb | δHa | δCb | |
| a In DMSO-d6 (600 MHz).b In DMSO-d6 (150 MHz). Coupling constants (J) in Hz are given in parentheses. The assignments were based on HSQC and HMBC experiments. | ||||
| 1 | 144.3 | 138.6 | ||
| 1a | 132.6 s | 136.3 | ||
| 2 | 123.2 s | 135.1 | ||
| 3 | 129.1 s | 118.8 | ||
| 4 | 7.62, s | 111.2 s | 8.37, s | 112.7 |
| 4a | 122.7 d | 122.6 | ||
| 5 | 8.04, d (7.5) | 120.1 d | 8.27, d (7.5) | 121.0 |
| 5a | 123.6 | 126.3 | ||
| 6 | 7.14, t (7.5) | 118.8 | 7.22, t (7.5) | 119.6 |
| 7 | 7.38, t (7.5) | 125.8 | 7.46, t (7.5) | 126.7 |
| 8 | 7.50, t (7.5) | 111.4 | 7.55, d (7.5) | 111.7 |
| 8a | 140.1 | 140.8 | ||
| 1′ | 4.77, d (7.5) | 61.2 | 6.16, s | 77.6 |
| 2′ | 4.30, s | 86.0 | 3.89, d (7.5) | 75.8 |
| 3′ | 76.9 | 71.6 | ||
| 4′ | 1.27, s | 29.7 | 1.22, s | 28.5 |
| 5′ | 1.04, s | 23.2 | 1.26, s | 24.7 |
| 6′ | 6.09, s | 100.3 | 170.8 | |
| 1-OCH3 | 3.91, s | 61.0 | 4.06, s | 60.3 |
| NH | 11.37, br s | 11.80 br s | ||
| 1′-OH | 5.34, d (7.5) | |||
| 2′-OH | 5.71, d (6.0) | |||
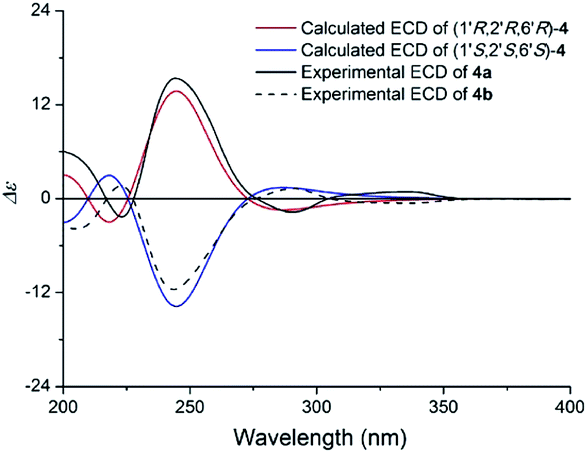 | ||
| Fig. 10 Calculated ECD spectra of (1′R,2′R,6′R)-4 and (1′S,2′S,6′S)-4 and the experimental ECD spectra of (+)- and (−)-claulansine B (4a/4b) in MeOH. | ||
Compound 5 (5a/5b) was obtained as a white powder. The HRESIMS displayed m/z 364.1163 [M + Na]+ (calcd for C19H19NO5Na 364.1155), which was consistent with a molecular formula of C19H19NO5 with eleven indices of hydrogen deficiency. Its NMR data (Table 3) was almost identical to those of claulansine D.10 Chiral isolation of 5 afforded the enantiomer 5a {[α]20D +62.0 (c 0.11 MeOH)} and 5b {[α]20D −99.2 (c 0.1 MeOH)}, which had opposite ECD curves (Fig. 11). The absolute configurations of 5a and 5b were determined using the same methods as described in 4a and 4b. Thus, 5a was defined as (+)-(1′R,2′R)-claulansine D, and 5b was identified as the known compound (−)-(1′S,2′S)-claulansine D.
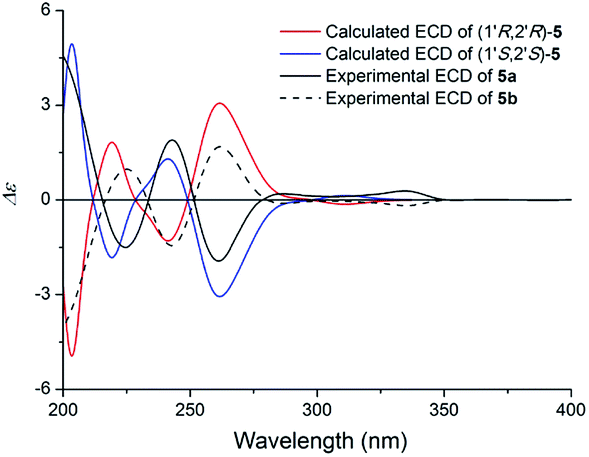 | ||
| Fig. 11 Calculated ECD spectra of (1′R,2′R)-5 and (1′S,2′S)-5 and the experimental ECD spectra of (+)- and (−)-claulansine D (5a/5b) in MeOH. | ||
The structures of nine known compounds were also identified by comparing their spectroscopic data to those found in the literature. The known compounds isolated were claulansine B (4b),10 claulansine D (5b),10 (−)-(1S,3S)-1-methyl-1,2,3,4-tetrahydro-β-carboline-3-carboxylic acid (6a),28 (+)-(1R,3S)-1-methyl-1,2,3,4-tetrahydro-β-carboline-3-carboxylic acid (6b),28 (R)-isoplatydesmine (7a),29 (S)-isoplatydesmine (7b),30 claulansine A (8),10 ribalinine (9),31 γ-fagarine (10),32 dictamnine (11),33 N-(2-hydroxy-2-phenylethyl)-cinnamamide (12),34 N-phenylethyl-benzamide (13).34
Compounds 1–13 were evaluated for their neuroprotective effect on neuron-like PC12 cells induced by Aβ25-35, and Okadaic Acid (OKA) in vitro using the MTT method. The neuron growth factor (NGF) was used as a positive control. At 10 μM, 1b, 2a, 3b, 5a, 5b increased the cell survival rate of the Okadaic acid-treated group, other compounds were inactive, while all the compounds failed to protect cells from Aβ25-35.
Clausenamide is one of novel compounds isolated from Clausena lansium (Lour) skeels. Clausenamide is unusual in that it contains 4 chiral centers yielding 8 pairs of enantiomers. In pharmacological studies numerous models and indicators showed that (−)-clausenamide is the active enantiomer, while (+)-clausenamide is inactive and elicits greater toxicity than (−)-clausenamide.35 Compounds 3 and clausenamide are very similar in structure and therefore have the same biological activity. Similarly, 3b is the active enantiomer, while 3a is inactive. The carbazole and quinolone alkaloids having neuroprotective effects were exhibited in previous researches,10,36 and carbazole alkaloids may derived from indole alkaloids. As we all know, thalidomide as a chiral racemic compound, its R-configuration has inhibitory activity of pregnancy, while S-configuration has teratogenic. One of the isomers of the enantiomer is highly active and the other isomer is inactive, or both isomers are active, or both isomers are inactive. Thus, it's reasonable that compound 1b, 2a, 5a, and 5b are active while 1a, 2b are inactive. All in all, alkaloids isolated from C. lansium are worthy of study to find more potential effects in the further (Table 4).
| a ###p < 0.001 vs. control, ***p < 0.001 vs. model, **p < 0.01 vs. model, *p < 0.1 vs. model. | ||
|---|---|---|
| Control | 10 μM | 100.0 ± 2.64 |
| Model | 10 μM | 70.5 ± 5.4### |
| 1a | 10 μM | 65.4 ± 6.2 |
| 1b | 10 μM | 83.4 ± 4.1** |
| 2a | 10 μM | 91.2 ± 10.1*** |
| 2b | 10 μM | 69.5 ± 5.3 |
| 3a | 10 μM | 71.7 ± 5.9 |
| 3b | 10 μM | 83.5 ± 7.8** |
| 4a | 10 μM | 74.4 ± 4.6 |
| 4b | 10 μM | 76.6 ± 7.5 |
| 5a | 10 μM | 89.7 ± 4.8*** |
| 5b | 10 μM | 83.3 ± 5.9*** |
Conclusions
In summary, we have reported the isolation, identification and biological study of twenty compounds (1–13) including three new enantiomers (1a, 1b, 2a, 2b, 3a, 3b), two new ones (4a, 5a), and several of them inhibited PC12 cell damage induced by Okadaic Acid. Furthermore, compounds 6–7 were isolated from the genus Clausena for the first time. The occurrence of alkaloids derivatives from C. lansium is in agreement with the previous findings, 1–7 indicating that the isolation of these enantiomer compounds might be a useful chemotaxonomic for screening activity. The results of preliminary neuroprotective effect assays suggested that several isolated alkaloids derivatives showed moderate activity. Moreover, previous findings exhibited that the carbazole, amide and quinolone alkaloids metabolites have neuroprotective effects and others biological activities such as anti-inflammatory, hepatoprotective, cytotoxic9,15–17 which indicating that the alkaloid compounds and their biological activities of C. lansium are worth studying in order to find compounds with potential activity.Experimental
Optical rotations were measured on a JASCO P2000 automatic digital polarimeter. UV spectra were recorded on a JASCO V-650 spectrophotometer, CD spectra were measured on a JASCO J-815 spectropolarimeter. IR spectra were recorded on a Nicolet 5700 spectrometer using an FT-IR microscope transmission method. NMR spectra were acquired with Bruker AVIIIHD 600, VNS-600, and Mercury-400 spectrometers in DMSO-d6 and MeOH-d6. HRESIMS spectra were collected on an Agilent 1100 series LC/MSD ion trap mass spectrometer. MPLC system was composed of two C-605 pumps (Büchi), a C-635 UV detector (Büchi), a C-660 fraction collector (Büchi), and an ODS column (450 mm × 60 mm, 50 μm, 400 g; YMC). Semi-preparative HPLC was conducted using a Shimadzu LC-6AD instrument with an SPD-20A detector and a Daicel Chiralpak AD-H column (250 × 10 mm, 5 μm). Preparative HPLC was also performed on a Shimadzu LC-6AD instrument with a YMC-Pack ODS-A column (250 × 20 mm, 5 μm). Column chromatography (CC) was performed with silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, People's Republic of China), SF-PRP 512A (100–200 mesh, Beijing Sunflower and Technology Development Co., Beijing, People's Republic of China), ODS (50 μm, YMC, Japan), and Sephadex LH-20 (GE, Sweden). TLC was carried out on glass precoated silica gel GF254 plates. Spots were visualized under UV light or by spraying with 10% sulfuric acid in EtOH followed by heating. PC12 cells (adrenal gland; pheochromocytoma) were purchased from the American Type Culture Collection. Dimethyl sulphoxide (DMSO), Aβ25–35, Okadaic acid, 3-(3,4-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were obtained from Sigma (St Louis, MO, USA). Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS) and equine serum were purchased from Gibco BRL (New York, NY, USA). All other chemicals were of analytical grade and were commercially available.Plant material
The stems of C. lansium were collected in Liuzhou, Guangxi, China, in December 2008 and identified by Engineer Guangri Long, Forestry of Liuzhou. A voucher specimen has been deposited at the Herbarium of Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College (ID-S-2320).Fraction B (180.6 g) was fractionated via silica gel column chromatography, eluting with CHCl3–MeOH–H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.05, 9:1:0.1, 8:2:0.2, 7:3:0.3, 6:4:0.4) to afford twelve fractions B1–B12 on the basis of TLC analysis. Fraction B4 (5.9 g) was further separated by reversed-phase silica MPLC with MeOH–H2O (20–50%, 50 mL min−1, 6 h) to afford B4-1–B4-47 fractions. Fractions B4-38–B4-42 was successively separated using preparative HPLC (detection at 210 nm, 18% CH3CN, 8 mL min−1) to yield 1 (20.3 mg, tR 37.50 min). Compound 1 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 6:1, 3 mL min−1) to give 1a (9.1 mg, tR 57.63 min) and 1b (8.7 mg, tR 67.99 min). Fraction B10 (5.1 g) was further separated by reversed-phase silica MPLC with MeOH–H2O (20–50%, 50 mL min−1, 7 h) to afford B10-1-B10-65 fractions. Fractions B10-15–B10-20 were successively separated using preparative HPLC (detection at 210 nm, 8% CH3CN, 8 mL min−1) to yield 6a (8.2 mg, tR 58.35 min) and 6b (2.4 mg, tR 68.93 min).
:1:0.05, 9:1:0.1, 8:2:0.2, 7:3:0.3, 6:4:0.4) to afford twelve fractions B1–B12 on the basis of TLC analysis. Fraction B4 (5.9 g) was further separated by reversed-phase silica MPLC with MeOH–H2O (20–50%, 50 mL min−1, 6 h) to afford B4-1–B4-47 fractions. Fractions B4-38–B4-42 was successively separated using preparative HPLC (detection at 210 nm, 18% CH3CN, 8 mL min−1) to yield 1 (20.3 mg, tR 37.50 min). Compound 1 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 6:1, 3 mL min−1) to give 1a (9.1 mg, tR 57.63 min) and 1b (8.7 mg, tR 67.99 min). Fraction B10 (5.1 g) was further separated by reversed-phase silica MPLC with MeOH–H2O (20–50%, 50 mL min−1, 7 h) to afford B10-1-B10-65 fractions. Fractions B10-15–B10-20 were successively separated using preparative HPLC (detection at 210 nm, 8% CH3CN, 8 mL min−1) to yield 6a (8.2 mg, tR 58.35 min) and 6b (2.4 mg, tR 68.93 min).
Fraction C (197.3 g) was fractionated via silica gel column chromatography, eluting with CHCl3, EtOAc, n-BuOH, CH3COCH3, MeOH to afford five fractions C1–C5. Fraction C1 was further separated by PRP-512A with MeOH–H2O (35–70%) to afford C1-1–C1-6 fractions. Fraction C1-3 was further separated by reversed-phase silica MPLC with MeOH–H2O (35–55%, 50 mL min−1, 6 h) to afford C1-3-1–C1-3-12 fractions. Fraction C1-3-11 was successively separated by Sephadex LH-20 and then using preparative HPLC (detection at 210 nm, 30% CH3CN, 8 mL min−1) to yield 5 (6.2 mg, tR 42.87 min), 7 (11.2 mg, tR 62.14 min) and 9 (24.4 mg, tR 51.17 min). Compound 5 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 3:1, 3 mL min−1) to give 5a (2.8 mg, tR 36.37 min) and 5b (2.1 mg, tR 45.78 min). Compound 7 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 7:1, 3 mL min−1) to give 7a (4.8 mg, tR 19.85 min) and 7b (5.3 mg, tR 25.86 min). Fraction C1-3-12 was successively separated by Sephadex LH-20 and then using preparative HPLC (detection at 210 nm, 29% CH3CN, 8 mL min−1) to yield 2 (5.2 mg, tR 48.14 min). Compound 2 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 8:1, 3 mL min−1) to give 2a (2.8 mg, tR 25.81 min) and 2b (2.1 mg, tR 34.17 min). Fraction C1-5 was further separated by reversed-phase silica MPLC with MeOH–H2O (35–55%, 50 mL min−1, 6 h) to afford C1-5-1–C1-5-8 fractions. Fraction C1-5-6 was successively separated via silica gel column chromatography and then using preparative HPLC (detection at 210 nm, 32% CH3CN, 8 mL min−1) to yield 3 (10.7 mg, tR 53.75 min), 4 (9.2 mg, tR 39.05 min), 10 (3.1 mg, tR 52.10 min), 12 (2.4 mg, tR 49.16 min) and 13 (3.2 mg, tR 65.64 min). Compound 3 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 9:1, 3 mL min−1) to give 3a (4.1 mg, tR 19.87 min) and 3b (3.9 mg, tR 25.12 min). Compound 4 was further separated by semipreparative chiral HPLC (n-hexane-2-propanol, 7:1, 3 mL min−1) to give 4a (1.5 mg, tR 21.98 min) and 4b (4.3 mg, tR 40.07 min). Fraction C1-6 was further separated by reversed-phase silica MPLC with MeOH–H2O (35–55%, 50 mL min−1, 6 h) to afford C1-6-1–C1-6-10 fractions. Fraction C1-6-2 was successively separated via silica gel column chromatography and then using preparative HPLC (detection at 210 nm, 30% CH3CN, 8 mL min−1) to yield 8 (5.2 mg, tR 33.31 min) and 11 (7.1 mg, tR 23.17 min).
(+)-(2′S,3′R)-Clauselansine A (1a). Colourless oil; [α]20D +18.7 (c 0.1 MeOH); UV (MeOH) λmax (log
ε) 202.8 (4.29), 222.6 (4.38), 281.8 (3.48) nm; ECD (MeOH) λmax (Δε) 268 (+0.75), 226 (+4.01) nm; IR (microscope) νmax 3313, 2925, 1671, 1612, 1439, 1379, 1109, 1040, 749 cm−1; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz), see Table 1; HRESIMS m/z 320.1854 [M + H]+ (calcd for C18H26NO4, 320.1856).
(−)-(2′R,3′S)-Clauselansine A (1b). Colourless oil; [α]20D −17.0 (c 0.1 MeOH); ECD (MeOH) λmax (Δε) 268 (−0.23), 226 (−0.79) nm; UV, IR, NMR, and HRESIMS were the same as those of 1a.
(+)-(2′S,3′R)-Clauselansine B (2a). White powder; [α]20D +28.0 (c 0.1 MeOH); UV (MeOH) λmax (log
ε) 203.0 (4.37), 220.0 (4.37), 281.6 (3.53) nm; ECD (MeOH) λmax (Δε) 276 (−0.18), 233 (+1.03) nm; IR (microscope) νmax 3358, 2917, 2851, 1615, 1377, 1039, 1002, 739 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 1; HRESIMS m/z 302.1758 [M + H]+ (calcd for C18H24NO3, 302.1751).
(−)-(2′R,3′S)-Clauselansine B (2b). White powder; [α]20D −32.6 (c 0.1 MeOH); ECD (MeOH) λmax (Δε) 280 (+0.35), 232 (−1.56) nm; UV, IR, NMR, and HRESIMS were the same as those of 2a.
(+)-(3S,4R,5S,6S)-Clauselansine C (3a). White powder; [α]20D +17.9 (c 0.1 MeOH); UV (MeOH) λmax (log
ε) 203.8 (4.28) nm; ECD (MeOH) λmax (Δε) 272 (−0.53), 234 (+1.27), 215 (−2.27) nm; IR (microscope) νmax 3320, 2923, 1678, 1483, 1454, 1403, 1202, 1132, 1078, 753, 703 cm−1; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz), see Table 2; HRESIMS m/z 280.1333 [M + Na]+ (calcd for C18H17NaNO2, 280.1332).
(−)-(3R,4S,5R,6R)-Clauselansine C (3b). White powder; [α]20D −22.0 (c 0.1 MeOH); ECD (MeOH) λmax (Δε) 272 (+0.27), 234 (−1.31), 215 (+1.84) nm; UV, IR, NMR, and HRESIMS were the same as those of 3a.
(+)-(1′R,2′R,6′R)-Claulansine B (4a). White powder; [α]20D +142.7 (c 0.1 MeOH); UV (MeOH) λmax (log
ε) 201.6 (4.40), 241.8 (4.24), 294.8 (3.68) nm; ECD (MeOH) λmax (Δε) 335 (+0.90), 290 (−1.71), 244 (+15.40), 223 (−2.31) nm; IR (microscope) νmax 3319, 2971, 1613, 1573, 1503, 1456, 1383, 1248, 1081, 1024, 745 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 3; HRESIMS m/z 326.1398 [M + H]+ (calcd for C19H20NO4, 326.1387).
(+)-(1′R,2′R)-Claulansine D (5a). White powder; [α]20D +62.0 (c 0.11 MeOH); UV (MeOH) λmax (log
ε) 195.8 (3.71), 231.0 (3.87), 278.2 (4.11) nm; ECD (MeOH) λmax (Δε) 335 (+0.28), 261 (−1.94), 243 (+1.89), 225 (−1.50) nm; IR (microscope) νmax 3379, 2919, 2850, 1737, 1613, 1462, 1358, 1242, 1099, 729 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 3; HRESIMS m/z 364.1163 [M + Na]+ (calcd for C19H19NNaO5, 364.1155).
Preparation of acetonide derivative (1c)
To determine the relative configuration of 1b, compound 1b (3 mg) was treated with 2,2-dimethoxypropane (1 mL) and pyridinium p-toluene sulfonate (1 mg), then stirred at 30 °C for 5 h under N2 circumstance. The reaction solution was evaporated in vacuo and purified by reversed-phase preparative HPLC using CH3OH/H2O (60:40 v/v) to yield the acetonide 1c (2.47 mg).
ε) 206 (6.24), 225 (6.40), 277 (5.75) nm; ECD (MeOH) λmax (Δε) 267 (−1.28), 251 (−0.78), 234 (−2.14) nm; Mo2(OAc)4-induced CD (DMSO) 323 (Δε +0.26) nm; IR (microscope) νmax 3307, 2923, 2854, 1645, 1542, 1452, 1400, 1240, 1099, 660 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 1; HRESIMS m/z 382.1993 [M + Na]+ (calcd for C21H29NNaO4, 382.1989).Determination of absolute configurations of the 3′,4′-diol unit in 1c
According to the published procedure, a 1:1.2 mixture of diol/Mo2(OAc)4 for 1c was subjected to CD measurements at a concentration of 0.1 mg mL−1 in anhydrous DMSO. The first CD spectrum was recorded immediately after mixing, and its time evolution was monitored until stationary (about 10 min after mixing). The inherent CD was subtracted. The observed sign of the diagnostic band at around 310 nm in the induced CD spectrum was correlated to the absolute configuration of the 3′,4′-diol unit.
Preparation of (R)- and (S)-MTPA eaters of 3a
A solution of 3a (1.31 mg) in dehydrated CH2Cl2 (2 mL) was treated with (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride [(R)-MTPA-Cl (10 mg)] in the presence of anhydrous pyridine, and the mixture was stirred at room temperature for 13 h. After cooling, the reaction mixture was poured into ice-water and extracted with EtOAC. The EtOAC extract was successively washed with 5% aqueous HCl, saturated aqueous NaHCO3, and brine, then dried over Na2SO4 and filtered. The solvent was removed from the filtrate under reduced pressure to afford a residue. The residue was purified by semi-preparative HPLC (C18 column, 3.0 mL min−1, UV 210 nm, 80% CH3CN–H2O) to yield (S)-MTPA ester derivative of 3a (compound 3aa 1.03 mg). (R)-MTPA ester derivative of 3a (compound 3ab 0.92 mg) was obtained from 3a (1.05 mg).Neuroprotection bioassays
Pheochromocytoma (PC12) cells were incubated in DMEM supplied with 5% fetal bovine serum and 5% equine serum as basic medium. PC12 cells in logarithmic phase were cultured at a density of 5000 cells per well in a 96-well microtiter plate. After 24 h incubation, the medium of the model group was changed to DMEM or basic medium with 15 μM Aβ25-35 for 48 h or basic medium with 50 nM OKA for 24 h. Test compounds dissolved in dimethyl sulfoxide (DMSO) were added to each well for >1000-fold dilution in the model medium at the same time. Each sample was tested in triplicate. After the incubation at 37 °C in 5% CO2 for 24 h, 10 μL of MTT (5 mg mL−1) was added to each well and incubated for another 4 h; then liquid in the wells was removed. DMSO (100 μL) was added to each well. The absorbance was recorded on a microplate reader (Bio-Rad model 550) at a wavelength of 570 nm.37 Analysis of variance (ANOVA) followed by Newman–Keuls post hoc test were performed to assess the differences between the relevant control and each experimental group. p-Values of <0.05, <0.01, and <0.001 were regarded as statistically significant. Data were expressed as mean ± SEM as indicated.Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the Department of Instrumental Analysis, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, for the measurement of the UV, IR, CD, NMR, and HRESIMS spectra. This research program is financially supported by the National Natural Science Foundation of China (No. 21272278) and the National Megaproject for Innovative Drugs (No. 2012ZX09301002-002).Notes and references
- R. L. Pan and Z. Y. Zhu, World Notes Plant Medicine, 1990, 5(6), 243–247 Search PubMed.
- C. Yenjai, S. Sripontan, P. Sriprajun, P. Kittakoop, A. Jintasirikul, M. Tanticharoen and Y. Thebtaranonth, Planta Med., 2000, 66, 277–279 CrossRef CAS PubMed.
- D. Y. Shen, Y. Y. Chan, T. L. Hwang, S. H. Juang, S. C. Huang, P. C. Kuo, T. D. Thang, E. J. Lee, A. G. Damu and T. S. Wu, J. Nat. Prod., 2014, 77, 1215–1223 CrossRef CAS PubMed.
- W. Maneerat, W. Phakhodee, S. Cheenpracha, T. Ritthiwigrom, S. Deachathai and S. Laphookhieo, Phytochemistry, 2013, 88, 74–78 CrossRef CAS PubMed.
- H. M. Xia, C. J. Li, J. Z. Yang, J. Ma, X. G. Chen, D. Zhang, L. Li and D. M. Zhang, J. Nat. Prod., 2014, 77, 784–791 CrossRef CAS PubMed.
- B. Kongkathip, N. Kongkathip, A. Sunthitikawinsakul, C. Napaswat and C. Yoosook, Phytother. Res., 2005, 19, 728–731 CrossRef CAS PubMed.
- W. Maneerat, W. Phakhodee, T. Ritthiwigrom, S. Cheenpracha, T. Promgool, K. Yossathera, S. Deachthai and S. Laphookhieo, Fitoterapia, 2012, 83, 1110–1114 CrossRef CAS PubMed.
- H. M. Xia, C. J. Li, J. Z. Yang, J. Ma, Y. Li, L. Li and D. M. Zhang, Phytochemistry, 2016, 130, 238–243 CrossRef CAS PubMed.
- W. W. Song, G. Z. Zeng, W. W. Peng, K. X. Chen and N. H. Tan, Helv. Chim. Acta, 2014, 97, 298–305 CrossRef CAS.
- H. Liu, C. J. Li, J. Z. Yang, N. Ning, Y. K. Si, L. Li, N. H. Chen, Q. Zhao and D. M. Zhang, J. Nat. Prod., 2012, 75, 677–682 CrossRef CAS PubMed.
- A. C. Adebajo, E. O. Iwalewa, E. M. Obuotor, G. F. Ibikunle, N. O. Omisore, C. O. Adewunmi, O. O. Obaparusi, M. Klaes, G. E. Adetogun, T. J. Schmidt and E. J. Verspohl, J. Ethnopharmacol., 2009, 122, 10–19 CrossRef CAS PubMed.
- W. Maneerat, T. Ritthiwigrom, S. Cheenpracha and S. Laphookhieo, Phytochem. Lett., 2012, 5, 26–28 CrossRef CAS.
- G. T. Liu, W. X. Li, Y. Y. Chen and H. L. Wei, Drug Dev. Res., 1996, 39, 174–178 CrossRef CAS.
- K. N. Prasad, H. H. Xie, J. Hao, B. Yang, S. X. Qiu, X. Y. Wei, F. Chen and Y. M. Yue, Food Chem., 2010, 118, 62–66 CrossRef CAS.
- H. Liu, F. Li, C. J. Li, J. Z. Yang, L. Li, N. H. Chen and D. M. Zhang, Phytochemistry, 2014, 107, 141–147 CrossRef CAS PubMed.
- J. Liu, C. J. Li, L. Ni, J. Z. Yang, L. Li, C. X. Zang, X. Q. Bao, D. Zhang and D. M. Zhang, RSC Adv., 2015, 5, 80553–80560 RSC.
- Y. Q. Du, H. Liu, C. J. Li, J. Ma, D. Zhang, L. Li, H. Sun, X. Q. Bao and D. M. Zhang, Fitoterapia, 2015, 103, 122–128 CrossRef CAS PubMed.
- Q. Zhao, J. Z. Yang, C. J. Li, N. H. Chen and D. M. Zhang, J. Asian Nat. Prod. Res., 2011, 13, 361–366 CrossRef CAS PubMed.
- H. Achenbach, C. Renner and R. Waibel, Liebigs Ann., 1995, 1327–1337 CrossRef CAS.
- K. Freter and V. J. Fuchs, J. Heterocycl. Chem., 1982, 19, 377–379 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B. 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- N. Berova, L. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- J. Frelek, M. Geiger and W. Voelter, Curr. Org. Chem., 1999, 3, 117–146 CrossRef CAS.
- L. D. Bari, G. Pescitelli, C. Pratelli, D. Pini and P. J. Salvadori, J. Org. Chem., 2001, 66, 4819–4825 CrossRef PubMed.
- N. C. Ma, K. M. Wu and L. Huang, Eur. J. Med. Chem., 2008, 43, 1781–1784 CrossRef CAS PubMed.
- N. C. Ma, K. M. Wu and L. Huang, J. Heterocycl. Chem., 2008, 45, 785–787 CrossRef CAS.
- J. M. Seco, E. Quinoa and R. Riguera, Chem. Rev., 2004, 104, 17–117 CrossRef CAS.
- G. Q. Li, Z. W. Deng, J. Li, H. Z. Fu and W. H. Lin, J. Chin. Pharm. Sci., 2004, 13(2), 81–86 CAS.
- M. F. Grundon and S. A. Surgenor, J. Chem. Soc., Chem. Commun., 1978, 14, 624–626 RSC.
- J. J. Chen, C. Y. Duh and H. Y. Huang, Planta Med., 2003, 69(6), 542–546 CrossRef CAS PubMed.
- A. Ahond, C. Poupat and J. Pusset, Phytochemistry, 1979, 18, 1415–1416 CrossRef CAS.
- Y. D. Min, H. C. Kwon, M. C. Yang, K. H. Lee, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2007, 30(1), 58–63 CrossRef CAS.
- J. Pusset, J. L. Lopez, M. Pais, M. A. Neirabeyeh and J. M. Veillon, Planta Med., 1991, 57, 153–155 CrossRef CAS PubMed.
- M. X. You, S. Purevsuren and C. Q. Hu, Nat. Prod. Res. Dev., 2008, 20, 647–649 CAS.
- F. Zhang, Y. N. Yang, X. Y. Song, S. Y. Shao, Z. M. Feng, J. S. Jiang, L. Li, N. H. Chen and P. C. Zhang, J. Nat. Prod., 2015, 78, 2390–2397 CrossRef CAS PubMed.
- S. F. Chu and J. T. Zhang, Acta Pharm. Sin. B, 2014, 4(6), 417–423 CrossRef PubMed.
- M. Pieroni, S. Girmay, D. Q. Sun, R. Sahu, B. L. Tekwani and G. T. Tan, ChemMedChem, 2012, 7(11), 1895–1900 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The spectra including 1D-, 2D-NMR, HRESIMS, calculated CD spectra of compounds 1a/b/c, 2a/b, 3a/b, 4a, 5a. See DOI: 10.1039/c7ra06753d |
| This journal is © The Royal Society of Chemistry 2017 |