
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilized boronic acid for Suzuki–Miyaura coupling: application to the generation of pharmacologically relevant molecules†
M. Martinez-Amezaga,
C. M. L. Delpiccolo and
E. G. Mata *
*
Instituto de Química Rosario (CONICET-UNR), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, 2000 Rosario, Argentina. E-mail: mata@iquir-conicet.gov.ar; Fax: +54 341 4370477
First published on 12th July 2017
Abstract
An synthetic strategy for the generation of a variety of biaryl and related derivatives, based on Suzuki–Miyaura coupling using immobilized boronic acid, is described. The importance of the methodology was demonstrated by its further application to biologically interesting compounds such as 4-biaryl-β-lactams, descripted as cholesterol absorption inhibitors and anti-MRSA active agents, neoflavonoids, imidazoles, isoxazolines, among others.
Introduction
Small organic molecules are of particular interest for medicinal chemistry and drug discovery not only because they constitute most of the medicines marketed today, but also because they are useful as probes to dissect biological systems providing information which eventually leads to the discovery of new drug targets.1 Through recent decades, solid-phase organic synthesis (SPOS) has become an important tool for the generation of structurally diverse small organic molecules including application to a series of very creative strategies such as diversity-oriented synthesis (DOS).2 Apart from the well-known advantages, like the fast isolation of resin-bound intermediates/products by filtering the resin beads, the use of high-boiling solvents and the easy manipulation,3 solid-phase organic synthesis has raised interest in metal-catalyzed cross-coupling reactions since undesirable soluble homodimers can be removed by filtration providing chemoselectivity, while anchoring one of the substrates makes its homodimerization a discouraging process due to the site isolation.4 An extra benefit can be considered within the principles of green chemistry: SPOS significantly reduces solvent waste by avoiding many chromatographic purifications (Scheme 1).5 | ||
| Scheme 1 Cross-coupling reaction on solid-phase synthesis. | ||
The Suzuki–Miyaura coupling is, arguably, one of the most versatile and successful synthetic tool for formation of carbon–carbon bonds. The Suzuki–Miyaura reaction is basically the reaction of arylboronic acids with aryl halides or pseudo halides under palladium catalyst to obtain biaryl fragments, which are present in many biologically relevant molecules. The mild reaction conditions and the broad functional groups tolerance are among the most remarkable features of this transformation, together with the fact that the non-toxic organoboron reagents can be easily synthesized by different procedures and exhibit high stability to air and moisture.6 One of the main drawbacks of Suzuki–Miyaura cross-coupling reaction is the formation of undesirable boronic acid homocoupling by-products.7 This problem can be avoided by a rigorous exclusion of oxygen giving, however, less support to the claim that Suzuki–Miyaura coupling is an “easy to handle” reaction. Site segregation in the solid-supported version of this reaction, makes self-coupling between immobilized boronic acid moieties a less favorable process. Many examples of this transformation on solid phase have been reported in the literature,8 however, very few are based on immobilized boronic acids.9
Results and discussion
For some years, our research group has been focused on the mentioned advantage of the combination of solid-phase synthesis and metal-catalyzed chemistry to build libraries of pharmacologically relevant molecules.10 In the search for alternative source of immobilized biaryl-containing β-lactam derivatives we decided to study the underdeveloped but promising solid-phase Suzuki–Miyaura reaction with a boronic acid as the immobilized substrate9 and apply the metholodogy to the generation of biaryl derivatives of privileged structures such as β-lactams, neoflavonoids, imidazoles, isoxazolines, etc.In order to find the optimal conditions, we first synthesized the immobilized 4-carboxyphenylboronic acid 1a from 4-formylphenylboronic acid by oxidation with sodium permanganate and anchoring to Wang resin by standard procedure. We then analyzed the reaction of 1a with 4-iodoacetophenone (2a) (Table 1 and Fig. 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Equiv. of iodide 2a | Pd catalyst (mol%) | Base (equiv.) | Solvent | Conditionsa | Yieldb (%) |
| a Except noted otherwise, all reactions were carried in a sealed tube.b Yields were determined by 1H NMR using an internal standard (mesitylene). Conversion by 1H NMR in brackets.c No starting material detected by 1H NMR.d Mixture of products, mostly starting material.e Open system, reflux conditions. | ||||||
| 1 | 1 | Pd(OAc)2 (5) | K2CO3 (1.2) | MeCN | 75 °C, 4 h | 30 (60) |
| 2 | 1 | PdCl2dppf (5) | K2CO3 (1.2) | DMF | 75 °C, 4 h | 22 (40) |
| 3 | 4 | Pd(PPh3)4 (5) | Cs2CO3 (1.2) | Diglyme | 150 °C, 3 h | 37c |
| 4 | 1 | Pd(OAc)2 (5) | K2CO3 (1.2) | MeCN | 75 °C, 7 h | 32 (63) |
| 5 | 4 | Pd(OAc)2 (10) | K2CO3 (2.4) | MeCN | MW, 10′, 100 °C | d |
| 6 | 4 | Pd(OAc)2 (10) | K2CO3 (2.4) | MeCN | 100 °C, 5 h | 40 (76) |
| 7 | 4 | Pd(OAc)2 (10) | K2CO3 0.5 M (2.4) | MeCN | 100 °C, 5 h | 96 |
| 8 | 4 | Pd(OAc)2 (10) | K2CO3 0.5 M (2.4) | Diglyme | 150 °C, 3 h | 50 |
| 9 | 4 | Pd(OAc)2 (10) | K2CO3 0.5 M (2.4) | MeCN | Reflux, 5 he | 88 |

 | ||
| Fig. 1 Structures and numbering of building blocks. | ||
When the immobilized boronic acid (1a) was treated with 1 equiv. of 4-iodoacetophenone (2a), 1.2 equiv. of K2CO3, under “ligand-free” conditions [Pd(OAc)2 (5 mol%)], in anhydrous acetonitrile for 4 h at 75 °C,9b an incomplete conversion was observed, and the expected product 3aa was obtained in 30% yield, after cleavage with TFA and methylation with diazomethane (entry 1). In the case of using PdCl2dppf11 (5 mol%) as catalyst, a similar result was obtained, albeit conversion and yield were lower (entry 2). Other conditions reported in literature were also attempted, thus, Wang resin-bound phenylboronic acid (1a) was treated with the iodide 2a, Pd(PPh3)4 (5 mol%) and Cs2CO3 (1,2 equiv.) in diglyme at 150 °C for 3 h,12 giving the expected biaryl 3aa in 37% yield but no starting material was detected by 1H NMR (entry 3). Afterwards, we used the combination of a ligandless catalyst such as Pd2(dba)3 and a hindered phosphine ligands [P(o-tolyl)3]13 in order to increase Pd catalytic activity, but a small amount of the required biaryl derivative was obtained together with an intractable mixture of products. Taking as reference the conditions of entry 1, we proceeded to increase the reaction time, but conversion and yield were hardly affected (entry 4). Although is clear that heating effects in microwave irradiation are just thermal,14 it is still an interesting alternative as a rapid way to reach high reaction temperatures15 in order to improve yields. However, in our case, low conversion together with a mixture of unidentified compounds was obtained under microwave irradiation (entry 5). Going back to conventional heating, we increased the equivalents of the iodide, reagents and temperature, having a slightly better yield (40%, entry 6). Finally, in our initial attempts we have noticed the difficulty of solubilising the carbonate and, since the addition of a base is considered to be crucial for the success of the reaction,16 we decided to study the addition of an small amount of water to increase dissolution, hoping that would not affect in any significant way the polymer support swelling. Thus, we dissolved the K2CO3 (2.4 equiv.) in a small amount of water and added to the suspension of resin 1a, 4-iodoacetophenone (2a) (4 equiv.) and 10 mol% of Pd(OAc)2 in acetonitrile. To our delight, after 5 h at 100 °C in a sealed tube, followed by cleaving from the resin and methylation with diazomethane, the expected product was achieved in 96% yield (entry 7). At the same time, a variation using more harsh conditions such as heating in diglyme at 150 °C for 5 h, was analyzed giving lower yield, probably due to starting material/product decomposition (entry 8). Good results were obtained using an open system (entry 9), although yield of entry 7 was still the optimum.
Subsequently, the reaction of a variety of aryl halides and pseudohalides under the optimized coupling conditions was examined (Table 2 and Fig. 1). Generally speaking, yields are very high for electron-withdrawing aryl halides and non-substituted aryl halides (entries 1–7), independently of using an iodide or a bromide derivative. In the case of electron-donating substituents on the aryl halides, Suzuki coupling proved to be less efficient, again with little influence from the halide used (entries 8–12). As expected, 2-iodothiophene gave the corresponding heterobiaryl derivative in low yield probably due to the poisoning of the catalyst (entry 13).17 In order to apply the methodology to structures of greater complexity and interest, biaryl-containing β-lactam derivatives were synthesized using the optimized conditions. β-Lactam unit is a well-known privileged structure present in numerous bioactive compounds. Aside from their antibacterial properties,18 β-lactams also show other biological activities, most significantly inhibition of cholesterol absorption, that include the commercial drug ezetimibe,19 which is one of the most prescribed medicine in US.
| Entry | Halides/pseudohalides | Product | Yieldb (%) |
|---|---|---|---|
| a Conditions: halide or pseudo-halide (4 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (2.4 equiv.) in H2O (0.2 mL), MeCN, 100 °C, 5 h, in a sealed tube.b Determined by 1H NMR vs. internal standard (mesitylene), isolated yields after column chromatography are reported in brackets.c ND = not determined. | |||
| 1 |  |
 |
96 |
| 2 |  |
 |
99 |
| 3 |  |
 |
96 |
| 4 |  |
 |
99 |
| 5 |  |
 |
99 |
| 6 |  |
 |
87 |
| 7 |  |
 |
81 (78) |
| 8 |  |
 |
77 |
| 9 | 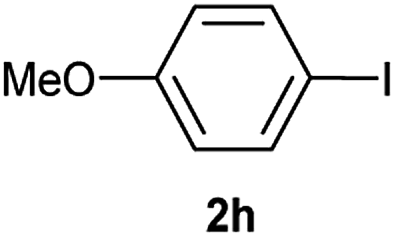 |
 |
52 |
| 10 | 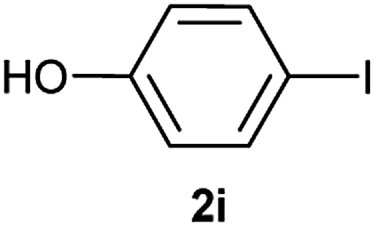 |
 |
37 |
| 11 |  |
 |
20 |
| 12 |  |
 |
8 |
| 13 |  |
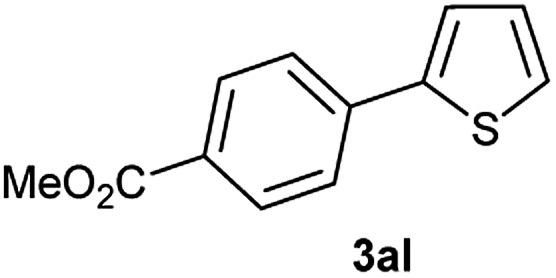 |
15 |
| 14 |  |
 |
79 (76) |
| 15 | 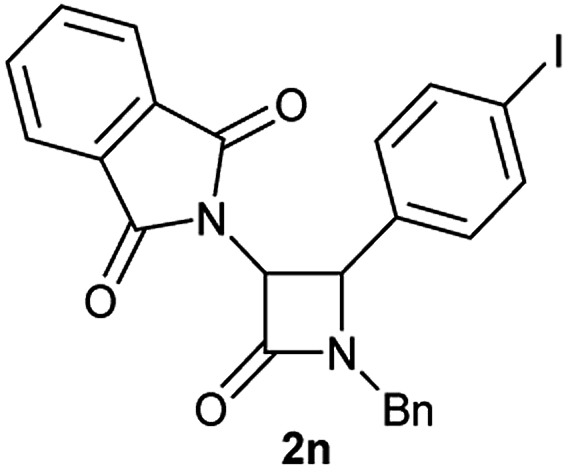 |
 |
65 (60) |
| 16 |  |
 |
NDc (90) |
| 17 |  |
 |
83 (86) |
| 18 |  |
 |
NDc (48) |
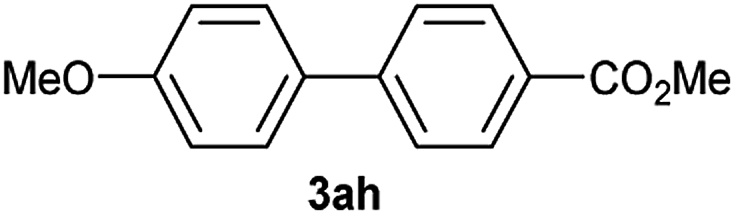
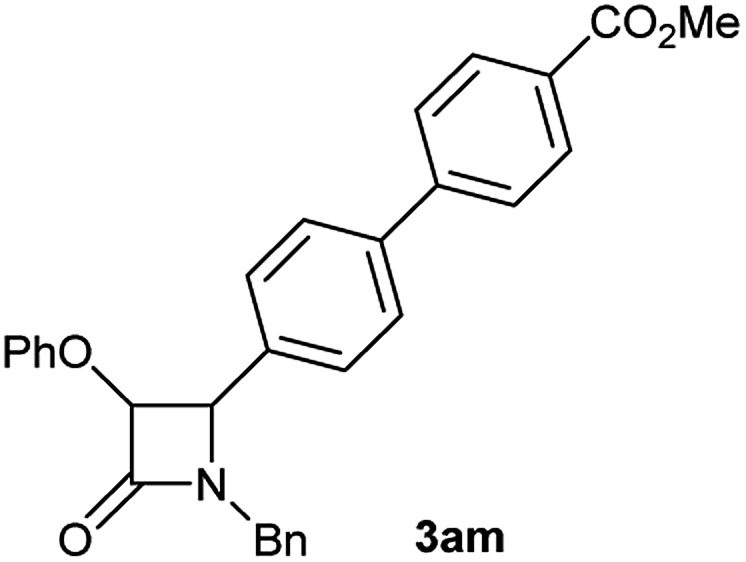
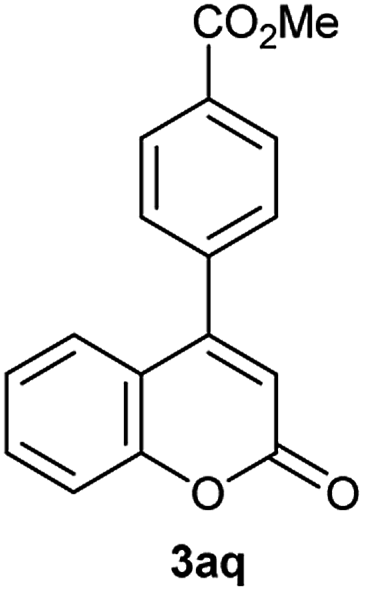
Particularly, 4-biaryl-β-lactams have been reported as cholesterol absorption inhibitors20 and antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA).21 Thus, biaryl-β-lactams (3am–ap) were obtained in very high isolated yield (entries 14 to 17, Table 2). Another synthetic objective of great interest in medicinal chemistry are the coumarin derivatives which are well known for their diverse pharmacological properties.22 Neoflavonoids (4-arylcoumarins) in particular, have shown relevant anticancer activity,23 while this moiety is found in a variety of plants belonging to the families Guttiferae, Rubiaceae, Leguminosae, Passifloraceae and Compositae.24 In this case, the triflate derivative of 4-hydroxycoumarin (2q′′)25 was subjected to the optimized conditions in the presence of the immobilized boronic acid (1a), to give 48% isolated yield of the expected 4-arylcoumarin (3aq) (entry 18).
Solid-phase chemistry is an appreciated tool for the development of many alternative synthetic route to diverse structures. Biaryl derivatives, like the ones we have synthesized, can be interesting precursors in that sense. Thus, the immobilized version of aldehyde 3af (supported 3af) was used as a versatile intermediate for the generation of other biologically promising compounds. For instance, we carried out the synthesis of secondary amines such as the N-benzyl-1-(biphenyl-4-yl)methanamine derivative (5af) by a straightforward two step reductive amination (Scheme 2). The imine formation was performed by treating supported 3af with benzylamine in the presence of trimethyl orthoformate as dehydrating agent. Reduction of the imine 4af with sodium borohydride gave the corresponding secondary N-benzyl amine 5af in a very good overall isolated yield of 60%, based on loading level of resin 1a. Supported 3af was also the starting point for the synthesis of biologically interesting heterocycles such as isoxazolines and imidazoles. Thus, this compound was first treated with hydroxylamine/triethylamine26 to generate the Wang resin-bound oxime 6af (Scheme 2). A one-pot oxidation/1,3-dipolar cycloaddition was then performed using solution of sodium hypochlorite and acrylic acid as dipolarophile.27 The biphenyl-isoxazoline 7af was obtained in a regioselective manner after removing from the resin with TFA followed by methylation (21% overall isolated yield, always based on loading level of resin 1a). Isoxazolines are a well-known privileged structure and the corresponding 3,5-disubstituted derivatives have been lately described as anti-inflamatory,28 antibacterial/antifungal agents,29,30 CFTR activators,31 and, particularly, the (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1) has been reported as a potent inhibitor of MIF tautomerase activity.32
 | ||
| Scheme 2 Further transformation of supported aldehyde 3af to biologically interesting heterocycles. | ||
Alternatively, biologically promising imidazole33 can be readily obtained by the four-component condensation of aldehydes, 1,2-diketones, amines and ammonium acetate in refluxing acetic acid as ammonia source. As an example, supported 3af was treated with excess of benzil (10) and benzylamine, in the presence of NH4OAc in acetic acid at reflux to give the supported product 8af that afforded the 1-benzyl-2-substituted-4,5-diphenyl-1H-imidazole derivative 9af after removing from the resin and methylation (20% overall isolated yield).34
In summary, we report herein an efficient application of the Suzuki–Miyaura coupling to the solid-phase synthesis of a variety of biaryl and related derivatives using the under-recognized but promising alternative based on immobilized boronic acid. Yields were very high for electron-withdrawing or unsubstituted aryl halides regardless of the halide used. We have demonstrated that biologically relevant structures, including heterocycles such as 4-biaryl-β-lactams, 4-arylcoumarins, imidazoles and isoxazolines, can be easily generated by this methodology. Biological evaluation of the synthesized compounds are currently in progress.
Experimental procedures
General
Chemical reagents were purchased from commercial sources and were used without further purification unless noted otherwise. Solvents were analytical grade or were purified by standard procedures prior to use. Resins were purchased from Sigma-Aldrich. 1H NMR spectra were recorded in a Bruker Avance spectrometer at 300 MHz in CDCl3 with tetramethylsilane (TMS) as internal standard. 13C NMR spectra were recorded on the same apparatus at 75 MHz with CDCl3 as solvent and reference (76.9 ppm). The chemical shifts (δ) are reported in ppm downfield from TMS and coupling constants (J) are expressed in hertz. Gas Chromatography-Mass Spectra (GC-MS) were recorded on a Shimadzu QP2010 Plus apparatus at an ionization voltage of 70 eV equipped with a SPB™-1 capillary column (internal diameter 0.25 mm, length 30 m). The High Resolution Mass Spectra (HRMS) were obtained with a Bruker MicroTOF-Q II instrument (Bruker Daltonics, Billerica, MA). Detection of the ions was performed in electrospray ionization, positive ion mode. Solvents were dried using a MBraun solvent system (SPS-800). Analytical thin-layer chromatography (TLC) was carried out with silica gel 60 F254 pre-coated aluminum sheets. Flash column chromatography was performed using Merck silica gel 60 (230–400 mesh). Elution was carried out with hexane–EtOAc mixtures, under positive pressure and employing gradient of solvent polarity techniques.Solid-phase reactions were carried out in polypropylene cartridges equipped with a frit (Supelco, Bellefonte, PA), unless reflux conditions were required, in that cases standard glassware was used. All solid-phase reaction mixtures were stirred at the slowest rate.
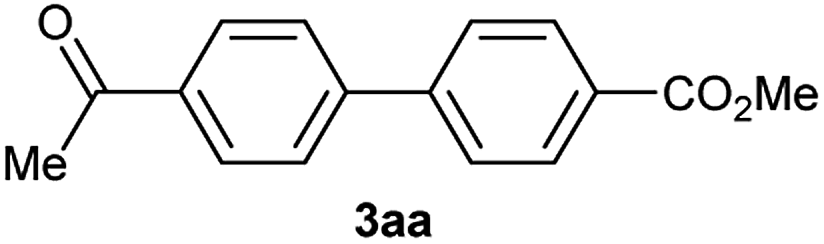
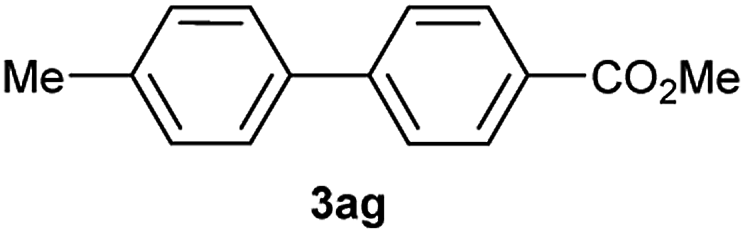
Except 2m, 2n, 2o, 2p, 3ak, 3am, 3an, 3ao and 3ap which are new compound and thoroughly characterized by 1H NMR, 13C NMR, HSQC, HMBC, HH-COSY and HRMS, the other compounds obtained have been previously described in literature: 3aa,35 3ab,36 3ac,37 3ad,36 3ae,35 3af,38 3ag,39 3ah,36 3ai,40 3aj,10f 3al36 and 3aq.41
General procedure for the preparation of β-lactams as starting materials (procedure A)42
A mixture of 4-iodobenzaldehyde (290 mg, 1.25 mmol), few grains of molecular sieves (4 Å) and the corresponding amine (1 mmol) in dichloromethane (5 mL) was stirred at 0 °C under nitrogen atmosphere for 1 h. The solution was filtered, the solvent evaporated and the residue analyzed by 1H NMR to ensure complete consumption of the amine. The crude thus obtained was dissolved in dry dichloromethane (3 mL) and cooled to 0 °C under nitrogen atmosphere. To the resulting solution were successively added triethylamine (0.5 mL, 3.5 mmol) and, dropwise, the corresponding acyl chloride (1.5 mmol). The resulting mixture was stirred overnight at room temperature and then was washed with water (3 × 5 mL), 0.1 N HCl (3 × 5 mL) and a saturated solution of NaHCO3 (5 mL). The organic layer was dried over MgSO4 and filtered; the solvent was evaporated under reduced pressure to give the corresponding crude β-lactam, which was further purified by column chromatography (hexane/EtOAc).Procedure for the preparation of phthalylglycyl chloride (procedure B)43
Oxalyl chloride (386 μL, 4.5 mmol) was added to a solution of anhydrous N-phthaloylglycine (615 mg, 3 mmol) in toluene (8 mL) and the reaction mixture was stirred at 60 °C for 3 h. The excess of solvent and oxalyl chloride were removed by evaporation under reduced pressure and that crude was immediately used for the formation of the corresponding β-lactam.Characterization of 2m. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.87 (d, J = 14.7 Hz, 2H), 4.68 (d, J = 4.5 Hz, 2H), 4.87 (d, J = 14.7 Hz, 2H), 5.39 (d, J = 4.5 Hz, 2H), 6.72 (d, J = 7.8 Hz, 2H), 6.89 (t, J = 7.1 Hz, 1H), 7.01 (d, J = 8.3 Hz, 2H), 7.10–7.16 (m, 4H), 7.29–7.33 (m, 3H), 7.61 (d, J = 8.3 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 44.3, 60.9, 82.0, 94.7, 115.5, 122.2, 128.1, 128.7, 128.9, 129.3, 130.5, 132.7, 134.5, 137.5, 156.8, 165.4. HRMS (ESI) m/z 478.0279 [(M + Na+); calcd for C22H18INNaO2: 478.0274].
Characterization of 2n. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 4.15 (d, J = 14.8 Hz, 1H), 4.76 (d, J = 5.3 Hz, 1H), 5.03 (d, J = 14.9 Hz, 1H), 5.23 (d, J = 5.2 Hz, 1H), 6.95 (d, J = 8.2 Hz, 2H), 7.20–7.25 (m, 2H), 7.30–7.34 (m, 3H), 7.51 (d, J = 8.3 Hz, 2H), 7.62–7.70 (m, 4H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 45.6, 59.7, 60.2, 94.3, 123.6, 128.1, 128.6, 129.0, 129.3, 131.1, 132.8, 134.4, 137.6, 163.6, 166.7. HRMS (ESI) m/z 531.0177 [(M + Na+); calcd for C24H17IN2NaO3: 531.0176].
Characterization of 2o. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 3.76 (s, 3H), 5.31 (d, J = 4.9 Hz, 1H), 5.55 (d, J = 4.9 Hz, 1H), 6.78–6.83 (m, 4H), 6.92–7.00 (m, 2H), 7.11 (d, J = 8.3 Hz, 2H), 7.11 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 7.1 Hz, 2H), 7.62 (d, J = 8.3 Hz, 2H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 55.5, 61.7, 81.2, 94.6, 114.5, 115.7, 118.8, 122.4, 129.4, 129.6, 130.1, 132.7, 137.6, 156.7, 156.9, 162.3. HRMS (ESI) m/z 494.0226 [(M + Na+); calcd for C22H18INNaO3: 494.0224].
Characterization of 2p. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 2.29 (s, 3H), 5.32 (d, J = 4.9 Hz, 1H), 5.55 (d, J = 4.9 Hz, 1H), 6.79 (d, J = 8.3 Hz, 2H), 6.94 (t, J = 7.4 Hz, 1H), 7.07–7.12 (m, 4H), 7.16–7.24 (m, 4H), 7.62 (d, J = 8.3 Hz, 2H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 20.9, 61.5, 81.1, 94.6, 115.9, 117.4, 122.4, 129.4, 129.8, 129.9, 132.6, 134.2, 134.6, 137.6, 156.9, 162.6. HRMS (ESI) m/z 478.0268 [(M + Na+); calcd for C22H18INNaO2: 478.0274].
Procedure for the synthesis of Wang resin-supported aryl boronic acid 1a (procedure C)
0.4 g of Wang resin (0.9 mmol g−1, 0.36 mmol) was swelled by gentle stirring in anhydrous dichloromethane (20 mL). Then, 4-carboxyphenylboronic acid (0.179 g, 1.08 mmol), DCC (N,N′-diisopropylcarbodiimide) (0.223 g, 1.08 mmol) and DMAP (catalytic amount) were added at that suspension. The mixture was shaked at 250 rpm overnight at room temperature. After filtration, the resin was sequentially washed with CH2Cl2 (3 × 10 mL), MeOH (3 × 10 mL), THF (3 × 10 mL), CH2Cl2 (1 × 10 mL) and finally dried under high vacuum. Mass recovery was used to determine resin loading after cleavage of an aliquot with 10% TFA/CH2Cl2: 0.45 mmol g−1.General procedure for the solid-phase Suzuki coupling (procedure D)
0.1 g of the supported arylboronic acid 1a (0.45 mmol g−1, 0.045 mmol) was suspended in MeCN (2 mL) in a dram vessel. Arylhalide or pseudo-halide 2 (0.18 mmol), Pd(OAc)2 (1 mg, 0.0045 mmol) and a solution of K2CO3 in H2O (0.54 M, 0.108 mmol) were added. The reaction was stirred 5 h at 100 °C. After cooling to room temperature, the resin was filtered, washed with MeCN (2 × 5 mL), H2O (2 × 5 mL), MeOH (3 × 5 mL) and DCM (3 × 5 mL) drying under high vacuum, the compound was cleaved from the resin with 5 mL of a 10% solution of TFA in DCM for 50 minutes at room temperature. Then it was filtered and washed with MeOH (2 × 3 mL) and CH2Cl2 (2 × 3 mL). The product-containing solution was concentrated under reduced pressure and dried under high vacuum. Esterification with diazomethane afforded the crude product that was analyzed by 1H NMR and GC/MS and then purified by flash column chromatography (hexane/EtOAc).Characterization of 3ab. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.95 (s, 3H), 4.00 (s, 3H), 7.40 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.5 Hz, 2H), 8.29 (dd, J = 8.0, 1.7 Hz, 1H), 8.55 (d, J = 1.7 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 52.3, 52.9, 125.5, 127.9, 130.0, 130.5, 131.0, 132.1, 133.1, 139.5, 141.1, 148.9, 164.7, 166.5.
Characterization of 3ac. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.95 (s, 6H), 7.69 (d, J = 8.5 Hz, 4H), 8.12 (d, J = 8.5 Hz, 4H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 52.2, 127.2, 129.7, 130.2, 144.3, 166.8.
Characterization of 3ad. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.95 (s, 3H), 7.36–7.51 (m, 3H), 7.60–7.70 (m, 4H), 8.11 (d, J = 8.6 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 52.1, 127.0, 127.3, 128.1, 128.9, 130.1, 140.0, 145.6, 167.0.
Characterization of 3af. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.95 (s, 3H), 7.70 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H), 7.98 (d, J = 8.3 Hz, 2H), 8.14 (d, J = 8.5 Hz, 2H), 10.08 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 52.3, 127.4, 128.0, 130.3, 130.3, 144.0, 145.9, 166.7, 191.8.
Characterization of 3ak. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.20 (t, J = 7.0 Hz, 6H), 3.40 (q, J = 7.0 Hz, 4H), 3.91 (s, 3H), 6.75 (d, J = 8.9 Hz, 2H), 7.53 (d, J = 8.9 Hz, 2H), 7.61 (d, J = 8.9 Hz, 2H), 8.04 (d, J = 8.9 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 12.6, 44.4, 51.9, 111.8, 125.5, 126.3, 127.1, 128.1, 130.0, 145.7, 147.8, 167.3. HRMS (ESI) m/z 284.1656 [(M + H+); calcd for C18H22NO2: 284.1645].
Characterization of 3am. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 3.92 (d, J = 14.5 Hz, 1H), 3.94 (s, 3H), 4.81 (d, J = 4.4 Hz, 1H), 4.92 (d, J = 14.7 Hz, 1H), 5.44 (d, J = 4.4 Hz, 1H), 6.75 (d, J = 7.8 Hz, 2H), 6.87 (t, J = 7.3 Hz, 1H), 7.11 (t, J = 8.3 Hz, 2H), 7.18 (m, 2H), 7.33 (m, 2H), 7.37 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 7.62 (d, J = 8.3 Hz, 2H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 44.3, 52.2, 61.1, 82.3, 115.6, 122.1, 127.0, 127.1, 128.0, 128.7, 128.9, 129.1, 129.2, 130.1, 132.8, 134.7, 140.2, 156.9, 165.5, 166.9. HRMS (ESI) m/z 486.1670 [(M + Na+); calcd for C30H25NNaO4: 486.1676].
Characterization of 3an. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 3.91 (s, 3H); 4.21 (d, J = 14.8 Hz, 1H), 4.90 (d, J = 5.2 Hz, 1H), 5.10 (d, J = 14.8 Hz, 1H), 5.52 (t, J = 5.2 Hz, 1H), 7.28–7.36 (m, 7H), 7.47 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.59–7.69 (m, 4H), 8.02 (d, J = 8.3 Hz, 2H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 45.7, 52.1, 60.0, 60.3, 123.5, 126.8, 127.3, 128.0, 128.1, 128.7, 129.0, 129.1, 130.0, 131.2, 133.0, 134.2, 134.8, 139.9, 144.5, 163.7, 166.8. HRMS (ESI) m/z 539.1587 [(M + Na+); calcd for C32H24N2NaO5: 539.1577].
Characterization of 3ao. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 3.75 (s, 3H), 3.93 (s, 3H), 5.41 (d, J = 4.8 Hz, 1H), 5.65 (d, J = 4.8 Hz, 1H), 6.82 (d, J = 8.9 Hz, 2H), 6.92 (t, J = 7.3 Hz, 1H), 7.16 (t, J = 8.0 Hz, 2H), 7.33 (d, J = 8.9 Hz, 2H), 7.46 (d, J = 8.2 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.3 Hz, 2H), 8.07 (d, J = 8.3 Hz, 2H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 52.2, 55.5, 61.8, 81.4, 114.5, 115.8, 118.9, 122.3, 127.0, 127.2, 128.7, 129.1, 129.3, 130.1, 130.4, 132.8, 140.3, 144.8, 156.6, 157.0, 162.4, 166.9. HRMS (ESI) m/z 502.1634 [(M + Na+); calcd for C30H25NNaO5: 502.1625].
Characterization of 3ap. 1H NMR: (CDCl3, 300 MHz) δ (ppm): 2.28 (s, 3H); 3.93 (s, 3H); 5.43 (d, J = 4.8 Hz, 1H), 5.60 (d, J = 4.8 Hz, 1H), 6.82 (d, J = 8.0 Hz, 2H), 6.92 (t, J = 7.4 Hz, 1H), 7.09 (d, J = 8.2 Hz, 2H), 7.17 (t, J = 8.0 Hz, 2H), 7.29 (d, J = 8.3 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H), 8.07 (d, J = 8.3 Hz, 2H). 13C NMR: (CDCl3, 75 MHz) δ (ppm): 21.0, 52.2, 61.7, 81.3, 115.8, 117.5, 122.3, 126.9, 127.2, 128.7, 129.1, 129.3, 129.7, 130.1, 132.8, 134.5, 134.5, 140.2, 144.8, 157.0, 162.8, 166.9. HRMS (ESI) m/z 486.1677 [(M + Na+); calcd for C30H25NNaO4: 486.1676].
Characterization of 3aq. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.98 (s, 3H), 6.40 (s, 1H), 7.24 (m, 1H), 7.38–7.45 (m, 2H), 7.52–7.60 (m, 3H), 8.20 (d, J = 8.3 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ (ppm): 52.5, 115.6, 117.5, 118.6, 124.4, 126.7, 128.6, 130.1, 131.4, 132.2, 139.6, 154.2, 154.6, 160.4, 166.3.
Procedure for the solid-phase reductive amination (procedure E)
0.3 g of the supported compound 3af (0.438 mmol g−1, 0.135 mmol) was suspended in anhydrous THF (2 mL) in a dram vessel. Benzylamine (75 μL, 0.673 mmol) and methyl orthoformate (15 μL, 0.135 mmol) were added and the reaction was stirred 18 h at room temperature. After that time, the imine-resin was filtered, washed with THF (2 × 5 mL), absolute MeOH (2 × 5 mL), DCM (2 × 5 mL) and MeOH (2 × 5 mL) and dried under high vacuum. This resin was suspended in a mixture of THF-MeOH 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v and treated with NaBH4 (51 mg, 1.347 mmol) for 6 to 8 hours. The resin was washed with THF (2 × 5 mL), EtOH (2 × 5 mL), H2O (2 × 5 mL), EtOH (2 × 5 mL), THF (2 × 5 mL) and MeOH (2 × 5 mL) and dried under high vacuum. The compound was cleaved from the support with 5 mL of a 10% solution of TFA in DCM for 50 minutes at room temperature. Then it was filtered and washed with MeOH (2 × 3 mL) and CH2Cl2 (2 × 3 mL). The product-containing solution was concentrated under reduced pressure and dried under high vacuum. Esterification with diazomethane afforded the crude product that was analyzed by 1H NMR and GC/MS and then purified by column chromatography (hexane/EtOAc).
:1 v/v and treated with NaBH4 (51 mg, 1.347 mmol) for 6 to 8 hours. The resin was washed with THF (2 × 5 mL), EtOH (2 × 5 mL), H2O (2 × 5 mL), EtOH (2 × 5 mL), THF (2 × 5 mL) and MeOH (2 × 5 mL) and dried under high vacuum. The compound was cleaved from the support with 5 mL of a 10% solution of TFA in DCM for 50 minutes at room temperature. Then it was filtered and washed with MeOH (2 × 3 mL) and CH2Cl2 (2 × 3 mL). The product-containing solution was concentrated under reduced pressure and dried under high vacuum. Esterification with diazomethane afforded the crude product that was analyzed by 1H NMR and GC/MS and then purified by column chromatography (hexane/EtOAc).
Characterization of 5af. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.85 (s, 2H), 3.87 (s, 2H), 3.94 (s, 3H), 7.27–7.37 (m, 5H), 7.52–7.60 (m, 3H), 7.45 (d, J = 8.3 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 8.10 (d, J = 8.3 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ (ppm): 52.1, 52.7, 53.2, 126.9, 127.0, 127.3, 128.2, 128.5, 128.8, 130.1, 138.7, 140.1, 140.4, 145.4, 167.0. HRMS (ESI) m/z 332.1659 [(M + H+); calcd for C22H22NO2: 332.1645].
Procedure for the solid-phase synthesis of Δ2-isoxazoline (procedure F)
0.24 g of the supported 3af (0.736 mmol g−1, 0.178 mmol) was suspended in a mixture of anhydrous MeOH (1 mL) and DCM (1 mL) in a dram vessel. Anhydrous triethylamine (124 μL, 0.89 mmol) and hydroxylamine hydrochloride (62 mg, 0.89 mmol) were added and the reaction was stirred 36 h at room temperature. After that time, the oxime-resin was filtered, washed with DMF (2 × 5 mL), DCM (2 × 5 mL), MeOH (2 × 5 mL) and DCM (2 × 5 mL) and dried under high vacuum. This oxime-resin was suspended in THF and treated with bleach (6 mL, 0.0018 mmol) and acrylic acid (62 μL, 0.9 mmol) at room temperature for 4 hours. The resin was washed with MeOH (2 × 5 mL), H2O (2 × 5 mL), MeOH (2 × 5 mL) and DCM (2 × 5 mL) and dried under high vacuum. The compound was cleaved from the support with 5 mL of a 20% solution of TFA in DCM for 50 minutes at room temperature. Then it was filtered and washed with MeOH (2 × 3 mL) and CH2Cl2 (2 × 3 mL). The product-containing solution was concentrated under reduced pressure and dried under high vacuum. Methylation with diazomethane afforded the crude product that was analyzed by 1H NMR and GC/MS and then purified by column chromatography (hexane/EtOAc).Characterization of 7af. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.69 (ABX, J = 17.1, 11.5, 9.5, 5.2 Hz, 2H), 3.84 (s, 3H), 3.95 (s, 3H), 5.23 (dd, J = 10.7, 7.4 Hz, 1H), 7.67 (d, J = 8.3 Hz, 4H), 7.78 (d, J = 8.6 Hz, 2H), 8.12 (d, J = 8.3 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 38.8, 52.2, 52.9, 78.1, 127.0, 127.5, 127.6, 128.2, 129.5, 130.2, 142.0, 144.4, 155.7, 166.8, 170.7. HRMS (ESI) m/z 362, 0998 [(M + Na+); calcd for C19H17NNaO5: 362, 0999].
Procedure for the solid-phase synthesis of imidazole (procedure G)
0.13 g of the supported 3af (0.736 mmol g−1, 0.097 mmol) was suspended in Glacial AcOH in a dram vessel. Benzylamine (212 μL, 1.94 mmol), benzil (408 mg, 1.94 mmol) and ammonium acetate (9 μL, 0.136 mmol) were added and the reaction was stirred 4 h at 100 °C. After that time, the resin was filtered, washed MeOH (2 × 5 mL), H2O (2 × 5 mL), MeOH (2 × 5 mL) and DCM (2 × 5 mL) and dried under high vacuum. The compound was cleaved from the support with 5 mL of a 20% solution of TFA in DCM for 50 minutes at room temperature. Then it was filtered and washed with MeOH (2 × 3 mL) and CH2Cl2 (2 × 3 mL). The product-containing solution was concentrated under reduced pressure and dried under high vacuum. Methylation with diazomethane afforded the crude product that was analyzed by 1H NMR and then purified by column chromatography (hexane/EtOAc).Characterization of 9af. 1H NMR (CDCl3, 300 MHz) δ (ppm): 3.94 (s, 3H), 5.16 (s, 3H), 6.86 (m, 2H), 7.20 (m, 8H), 7.32 (m, 3H), 7.59 (m, 2H), 7.67 (m, 4H), 7.77 (d, J = 8.4 Hz, 2H), 8.11 (d, J = 8.0 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 48.4, 52.2, 125.9, 126.5, 126.8, 127.0, 127.6, 128.1, 128.7, 128.8, 129.2, 129.5, 130.2, 130.4, 130.6, 130.9, 131.0, 134.3, 137.5, 138.3, 140.3, 144.8, 147.5, 166.9. HRMS (ESI) m/z 521, 2222 [(M + H+); calcd for C36H29N2O2: 521, 2223].
Acknowledgements
Support from CONICET, ANPCyT and Universidad Nacional de Rosario from Argentina is gratefully acknowledged. MMA thanks CONICET for fellowship.Notes and references
-
(a) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48 CrossRef CAS PubMed
; (b) C. J. O'Connor, L. Laraia and D. R. Spring, Chem. Soc. Rev., 2011, 40, 4332 RSC
-
(a) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed
- E. Schütznerová, P. Verdía and V. Krchnak, ACS Comb. Sci., 2016, 18, 655 CrossRef PubMed
- D. D. Young and A. Deiters, Solid-phase organic synthesis: concepts, strategies, and applications, ed. P. H. Toy and Y. Lam, John Wiley & Sons, Inc., Hoboken, New Jersey, 2012, pp. 171–201 Search PubMed
- G. S. Yellol and C.-M. Sun, Solid-Supported Synthesis en Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue Jr, John Wiley & Sons, Ltd, 2012, p. 394 Search PubMed
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS
- For articles related to the suppression of the oxidative homocoupling, see:
(a) W. D. Miller, A. H. Fray, J. T. Quatroche and C. D. Sturgill, Org. Process Res. Dev., 2007, 11, 359 CrossRef CAS
-
(a) S. Bräse, J. H. Kirchhoff and J. Köbberling, Tetrahedron, 2003, 59, 885 CrossRef
- To the best of our knowledge, two examples have been reported dealing with the use of immobilized boronic acid in organic synthesis, in both cases, the sticky PEG-PS resin was the favorite support used:
(a) B. Ruhland, A. Bombrun and M. A. Gallop, Tetrahedron Lett., 2006, 47, 19 CrossRef
-
(a) S. A. Testero and E. G. Mata, Org. Lett., 2006, 8, 4783 CrossRef CAS PubMed
- The large P-Pd-P angle of the dppf ligand is believed to make the reductive elimination of the coupling product much faster than the β-elimination. See: T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158 CrossRef CAS
- M. Ruda, N. Kann, S. Gordon, J. Bergman, W. Nelson, P. Agback, L. Hagberg and K. F. Koehler, J. Comb. Chem., 2005, 7, 567 CrossRef CAS PubMed
- V. Cerezo, M. Amblard, J. Martinez, P. Verdié, M. Planas and L. Feliu, Tetrahedron, 2008, 64, 10538 CrossRef CAS
- A. A. Poeylaut-Palena and E. G. Mata, Org. Biomol. Chem., 2010, 8, 3947 CAS
- C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71 CrossRef CAS PubMed
- C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082 CrossRef CAS PubMed
- Y. Liu, C. Khemtong and J. Hu, Chem. Commun., 2004, 398 RSC
- S. A. Testero, J. F. Fisher and S. Mobashery, β-Lactam Antibiotics in Burger's Medicinal Chemistry, Drug Discovery and Development, Vol. 7 (Antiinfectives), ed. D. J. Abraham and D. P. Rotella, Wiley and Sons, 2010, pp. 259–404 Search PubMed
- D. A. Burnett, Curr. Med. Chem., 2004, 11, 1873 CrossRef CAS PubMed
- E. Martinez, J. J. Talley, S. Antonelli, T. Barden, R. Lundrigan-Soucy, W. Schairer, J. J. Yang and D. Zimmer, 4-Biarylyl-1-phenylazetidin-2-ones, US Pat., 2005/0209165 A1, 22 September 2005
- E. Turos, C. Coates, J.-Y. Shim, Y. Wang, J. M. Leslie, T. E. Long, G. S. K. Reddy, A. Ortiz, M. Culbreath, S. Dickey, D. V. Lim, E. Alonso and J. Gonzalez, Bioorg. Med. Chem., 2005, 13, 6289 CrossRef CAS PubMed
- D. Ashok, B. V. Lakshmi, S. Ravi, A. Ganesh and S. Adam, Chem. Heterocycl. Compd., 2015, 51, 462 CrossRef CAS
-
(a) W. Kemnitzer, S. Kashibhatla, S. Jiang, H. Zhang, J. Zhao, S. Jia, L. Xu, C. Crogan-Gundy, R. Denis, N. Rarriault, L. Vaillancourt, S. Charron, J. Dodd, G. Attardo, D. Labreque, S. Lamothe, H. Gourdeau, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2005, 15, 4745 CrossRef CAS PubMed
- M. L. Yao and M. Z. A. Deng, Chem. Heterocycl. Compd., 2000, 11, 380 CAS
- S. Wattanasin, Synth. Commun., 1988, 18, 1919 CrossRef CAS
- N. Zou and B. Jiang, J. Comb. Chem., 2000, 2, 6 CrossRef CAS PubMed
- J. F. Cheng and A. M. Mjalli, Tetrahedron Lett., 1998, 39, 939 CrossRef CAS
- A. Alam, C. Pal, M. Goyal, M. K. Kundu, R. Kumar, M. S. Iqbal, S. Dey, S. Bindu, S. Sarkar, U. Pal, N. C. Maiti, S. Adhikari and U. Bandyopadhyay, Bioorg. Med. Chem., 2011, 19, 7365 CrossRef CAS PubMed
- B. S. Priya and K. S. Rangappa, Heterocycl. Commun., 2006, 12, 35 CrossRef CAS
- S. L. Gaonkar, K. L. Rai and B. Prabhuswamy, Med. Chem. Res., 2007, 15, 407 CrossRef CAS
- R. E. Sammelson, T. Ma, L. J. Galietta, A. S. Verkman and M. J. Kurth, Bioorg. Med. Chem. Lett., 2003, 13, 2509 CrossRef CAS PubMed
-
(a) Y. Al-Abed and S. VanPatten, Future Med. Chem., 2011, 3, 45 CrossRef CAS PubMed
- For recent reviews on the biological activity of imidazoles, see:
(a) M. M. Heravi, M. Daraie and V. Zadsirjan, Mol. Diversity, 2015, 19, 577 CrossRef CAS PubMed
-
(a) G. V. M. Sharma, Y. Jyothi and P. S. Lakshmi, Synth. Commun., 2006, 36, 2991 CrossRef CAS
- F. Izquierdo, M. Corpet and S. P. Nolan, Eur. J. Org. Chem., 2015, 1920 CrossRef CAS
- C. Liu, T. Li and N. L. Rosi, J. Am. Chem. Soc., 2012, 134, 18886 CrossRef CAS PubMed
- Y. A. Chen and C. Y. Liu, RSC Adv., 2015, 5, 74180 RSC
- T. Cornilleau, P. Hermange and E. Fouquet, Chem. Commun., 2016, 52, 10040 RSC
- C. I. Traficante, E. G. Mata and C. M. L. Delpiccolo, RSC Adv., 2015, 5, 26796 RSC
- K. Kawabata, M. Takeguchi and H. Goto, Macromolecules, 2013, 46, 2078 CrossRef CAS
- N. P. Cheval, A. Dikova, A. Blanc, J. M. Weibel and P. Pale, Chem.–Eur. J., 2013, 19, 8765 CrossRef CAS PubMed
- C. Palomo, M. Oiarbide, A. Esnal, A. Landa, J. I. Miranda and A. Linden, J. Org. Chem., 1998, 63, 5838 CrossRef CAS PubMed
- D. L. Boger and J. B. Myers Jr, J. Org. Chem., 1991, 56, 5385 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data: 1H NMR and 13C NMR spectra. See DOI: 10.1039/c7ra06662g |
| This journal is © The Royal Society of Chemistry 2017 |