DOI:
10.1039/C7RA06514K
(Paper)
RSC Adv., 2017,
7, 37706-37719
Water soluble Ru(II)–arene complexes of the antidiabetic drug metformin: DNA and protein binding, molecular docking, cytotoxicity and apoptosis-inducing activity†
Received
11th June 2017
, Accepted 18th July 2017
First published on 31st July 2017
Abstract
Two water soluble Ru(II)–arene complexes of the type [Ru(η6-arene)(met)Cl]Cl 1 and 2, where the arene is either p-cymene (1) or benzene (2) and met is metformin (antidiabetic drug) have been isolated and characterized by analytical and spectral methods. The X-ray crystal structure of 1 reveals that the coordination geometry around Ru(II) is described as the familiar pseudo-octahedral “piano-stool” structure. Absorption and emission spectral studies reveal that the complexes interact with calf thymus DNA through hydrophobic and hydrogen bonding interactions of coordinated ligands with the DNA base pairs. Molecular docking studies show that complex 1 effectively docks in the major groove of DNA. The decrease in viscosities of CT DNA upon binding to 1 and 2 suggest the covalent mode of DNA binding of complexes. Further, the covalent mode of binding is validated by the retardation of the mobility of supercoiled (SC) plasmid DNA by the formation of covalent adducts observed in gel electrophoretic mobility studies. The protein binding affinity of the complexes depends upon the arene ligand and follows the order of p-cymene (1) > benzene (2), which is the same as that for DNA binding affinity. Docking studies with BSA and transferrin show that the complex–protein interaction is stabilized by hydrophobic as well as hydrogen bonding interactions. The α-amylase inhibition assay of 1 and 2 indicates that they have the potency to exhibit the antidiabetic activity in vitro. A study of cytotoxicity of 1 and 2 against human breast carcinoma (MDA-MB-231), human lung carcinoma (A549), human ovarian carcinoma (A2780) cell lines and non-tumorigenic human embryonic kidney (HEK293) cells reveals that they are specifically toxic to cancerous cells and non-toxic to normal cells. Remarkably, complexes 1 and 2 exhibit cytotoxicity with potency more than the metformin suggesting that the incorporation of antidiabetic drug with the organometallic Ru-arene frameworks is potential approach to develop effective anticancer drugs. The morphological changes observed by employing AO/EB and Hoechst 33258 staining methods reveal that the complexes 1 and 2 induce an apoptotic mode of cell death in breast cancer cells.
Introduction
Platinum-based anticancer agents such as cisplatin, carboplatin and oxaliplatin are utilized as the most active metal based drugs for chemotherapeutic treatment.1,2 However the efficacy of these drugs is limited by acquired resistance against breast and prostate cancer cells and they exhibit several side effects such as myelosuppression, immune-suppression, neuro- and nephrotoxicity.3,4 This provides an impetus for the development of non-platinum based drugs with a broader spectrum of activity and lower toxicity for cancer treatment.5 In this context, several ruthenium (Ru) complexes have shown potent anticancer activity, especially two Ru(III) complexes which are entered in the clinical trials. The complex imidazolium trans-[tetrachlorido(dimethylsulfoxide)(1H-imidazole)ruthenate(III)] (NAMI-A) is active against solid metastatic tumors6 and indazolium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] (KP1019) or its sodium salt (KP1339) are active against a number of primary human tumors.7 Following the encouraging clinical studies of Ru-based anticancer agents, current attention is focused on the evaluation of anticancer activity of organometallic Ru-arene complexes, because, by varying the arene, substituent ligand and leaving group, the “piano-stool” framework of the Ru-arene provides a handle for optimizing the design of anticancer agents with improved pharmacological properties.8–10 The Ru(II)–arene–PTA complex (RAPTA-C, PTA is 1,3,5-triaza-7-phosphaadamantane) is identified as the lead structure that has been extensively studied for its antimetastatic and antiangiogenic properties.11 Sadler and co-workers found that Ru(II)–arene complexes of the type [Ru(η6-arene)(en)Cl]+, where η6-arene is biphenyl, dihydroanthracene or tetrahydroanthracene and en is ethylenediamine, are involved in covalent mode of binding with DNA considerably enhancing the cytotoxicity of the complexes against different tumor cells.12 Palaniandavar et al. have found that the Ru(II)–arene complexes with intercalating anthracenyl–methyldiazepane ligand moiety show higher DNA and protein binding affinity and exhibit prominent cytotoxicity than those containing simple diazepane ligand.13
The study of metal complexes bearing a bioactive drug is gaining more interest now than before because of the synergetic effect of drugs on coordination with a metal.14–16 In particular, several organometallic Ru-arene complexes incorporated with some biocompatible drugs have been reported to exhibit increased anticancer activity. [Ru(η6-benzene)(metronidazole)Cl2], where metronidazole is 1-(b-hydroxyethyl)-2-methyl-5-nitroimidazole, is the first organometallic Ru(II)–arene complex with the anti-infective agent metronidazole evaluated for cytotoxic properties.17 Turel et al. have established the anti-proliferative activity of Ru(II)–p-cymene complex of the antibacterial drug nalidixic acid18 and its thionated derivative19 against three different cancer cell lines in vitro. [Ru(η6-p-cymene)(ox)Cl], where H(ox) is oxicam (nonsteroidal anti-inflammatory drug), has shown cytotoxicity against human colon carcinoma with the IC50 value of 80 μM.20 Combining organometallic Ru(II)–p-cymene fragment with the anti-inflammatory drug motifs like indomethacin and diclofenac induces more cytotoxic activity than the original drugs and also displays higher cytotoxicity and selectivity than cisplatin towards the human ovarian carcinoma (A2780) cell lines.21
Metformin (N,N′-dimethylbiguanide), which belongs to the family of oral hypoglycemic agents, is the most commonly prescribed antidiabetic drug for the treatment of type II diabetes world wide.22 Interestingly, recent clinical data reveals that the usage of metformin for diabetic treatment reduces the risk of cancer23 in such a way that it exhibits significant growth inhibitory and proapoptotic activity against several cancer cells alone or in combination with the well-known chemotherapeutic drug cisplatin.24,25 The mechanism of antitumor effect of metformin is still unclear; the main effect at the molecular level is thought to be involved in the activation of AMP (5′-adenosine monophosphate)-activated protein kinase, which plays a vital role in cellular sensing of metabolism and oxidative stress.26
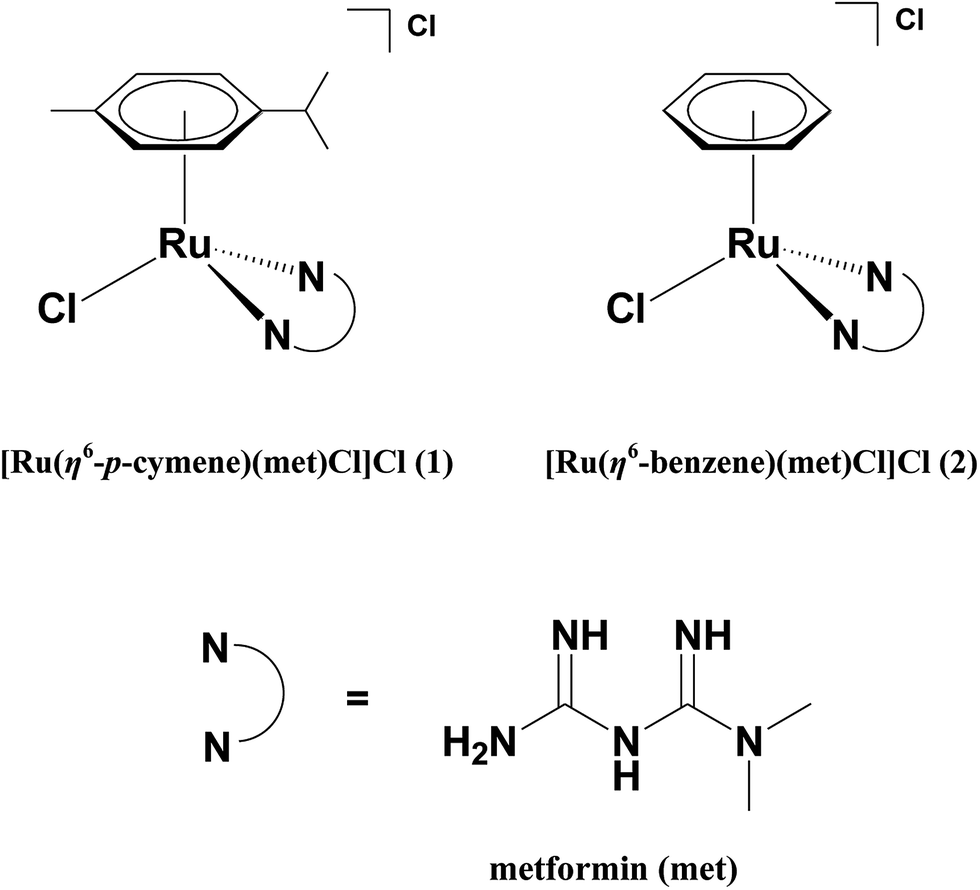
By keeping in mind the various established biological properties of metformin, the incorporation of metformin with the organometallic Ru(arene) pharmacophore is a promising approach, which can further enhance the efficacy of the metformin leading to the development of new anticancer chemotherapeutics. In this article, we have isolated water soluble Ru(II)–arene complexes of the type [Ru(η6-arene)(met)Cl]Cl 1 and 2 where arene is p-cymene (1) or benzene (2) and met is metformin (Scheme 1) and studied their interaction with DNA, which is considered as the primary pharmacological target of many anticancer drugs. We have also studied the protein binding affinity of the complexes using bovine serum albumin (BSA), since the serum albumins are recognized as major proteins to manipulate the transportation, distribution, and efficacy of the many anticancer drugs.27,28 The hydrophobicity of the arene ligands enhances the ability of complexes to exhibit cytotoxicity more potent than metformin alone against three different human cancer cell lines and induces the apoptotic mode of cell death in MDA-MB-231 human breast carcinoma cells. To the best of our knowledge, the complexes 1 and 2 are the first organometallic Ru(II)–arene complexes of antidiabetic drug metformin reported so far to exhibit apoptotic mode of cell death.
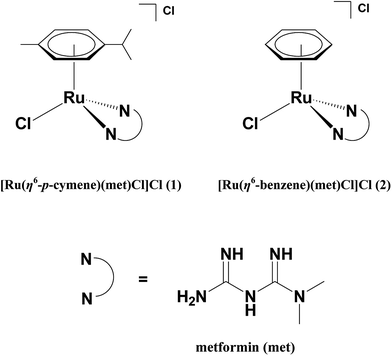 |
| | Scheme 1 Structures of the complexes and ligand used. | |
Experimental section
Reagents and materials
The reagents and chemicals were obtained from commercial sources (Sigma-Aldrich, USA; Himedia, India; Merck, India; SRL, India). RuCl3·3H2O, α-phellandrene, 1,3-cyclohexadiene, ethidium bromide (EthBr), distamycin, calf thymus (CT) DNA (highly polymerized, stored at −20 °C) (Aldrich), pUC19 supercoiled DNA, agarose and BSA (SRL) were used as received. Human breast carcinoma (MDA-MB-231), human lung carcinoma (A549), human ovarian carcinoma (A2780) and human embryonic kidney (HEK293) cells were obtained from American Type Culture Collection (ATCC), USA. Roswell Park Memorial Institute (RPMI) medium, fetal bovine serum (FBS) were purchased from GIBCO (Grand Island, NY). Glutamine and penicillin–streptomycin were obtained from Life Technologies, USA. Ultrapure MilliQ water was used in all experiments. Tris(hydroxymethyl)aminomethane·HCl (Tris–HCl) (pH 7.1) was prepared by the reported procedure.29 Commercial solvents were distilled and then used for preparation of complexes.
Preparation of ruthenium(II) precursor complexes
The starting materials [(η6-arene)RuCl2]2, where arene is p-cymene or benzene, were prepared by using the procedures already reported.30,31
Preparation of [Ru(η6-p-cymene)(met)Cl]Cl (1)
This complex was prepared by adding methanolic solution of metformin (0.26 g, 2 mmol) to a solution of ruthenium dimer precursor [(η6-p-cymene)RuCl2]2 (0.61 g, 1 mmol) in methanol and then mixture was stirred at 40 °C for 5 h. The orange-brown solution obtained was filtered and the volume of solvent was reduced. The product formed as an orange-brown microcrystalline solid on leaving the above reaction mixture to stand at 4 °C for 24 h. The product separated out was collected by suction filtration, washed with small amount of cold methanol and excess of ether and then dried in vacuum. The orange-brown colored crystals of 1 suitable for X-ray diffraction studies were obtained by dissolving the complex in aqueous methanol and allowing it to crystallize. Yield: 84%; mp: 176 °C. Anal. calcd for [Ru(η6-p-cymene)(met)Cl]Cl: C, 38.62; H, 5.79; N, 16.09. Found: C, 38.41; H, 5.36; N, 16.82%. ESI-MS: [Ru(η6-p-cymene)(met)Cl]+ displays a peak at m/z 400.0 (calcd 400.08). 1H NMR (500 MHz, D2O): δ 1.11–1.33 (m, CH(C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3)2), 2.04 (s, C6H4(C3)), 2.63–2.76 (m, C(CH3)2), 2.97 (s, N(C3)2), 3.10 (s, N2), 5.44–5.45 (d, CH of C64), 5.68–5.70 (d, CH of C64) ppm. 13C NMR (125 MHz, D2O): δ 17.70 (C6H4(
3)2), 2.04 (s, C6H4(C3)), 2.63–2.76 (m, C(CH3)2), 2.97 (s, N(C3)2), 3.10 (s, N2), 5.44–5.45 (d, CH of C64), 5.68–5.70 (d, CH of C64) ppm. 13C NMR (125 MHz, D2O): δ 17.70 (C6H4(![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3)), 21.35–21.49 (CH(H3)2), 30.32–30.37 (H(CH3)2), 38.33 (N(H3)2), 80.98, 83.40, 96.83, 98.57, 99.88 (η6-6H4) ppm. FT-IR (KBr, cm−1): 3406 (br; ν(N–H)), 1674, 1641 (s; ν(C
H3)), 21.35–21.49 (CH(H3)2), 30.32–30.37 (H(CH3)2), 38.33 (N(H3)2), 80.98, 83.40, 96.83, 98.57, 99.88 (η6-6H4) ppm. FT-IR (KBr, cm−1): 3406 (br; ν(N–H)), 1674, 1641 (s; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)). ΛM (acetonitrile, 10−3 M): 64 Ω−1 cm2 mol−1.
N)). ΛM (acetonitrile, 10−3 M): 64 Ω−1 cm2 mol−1.
Preparation of [Ru(η6-benzene)(met)Cl]Cl (2)
This complex was prepared by adopting the procedure used for the preparation of 1 but using benzene dimer precursor [(η6-benzene)RuCl2]2 (0.50 g, 1 mmol) instead of p-cymene dimer. Yield: 76%; mp: 173 °C. Anal. calcd for [Ru(η6-benzene)(met)Cl]Cl: C, 31.67; H, 4.52; N, 18.47 Found: C, 31.29; H, 4.33; N, 18.72%. ESI-MS: [Ru(η6-benzene)(met)Cl]+ displays a peak at m/z 344.0 (calcd 344.02). 1H NMR (500 MHz, D2O): δ 5.73 (s, η6-C66), 3.01 (s, N-(C3)2), 3.17 (s, N2), ppm. 13C NMR (125 MHz, D2O): δ 38.25 (N(H3)2), 80.5, 82.8 (η6-6H6) ppm. FT-IR (KBr, cm−1): 3406 (br; ν(N–H)), 1678, 1638 (s; ν(CN)). ΛM (acetonitrile, 10−3 M): 62 Ω−1 cm2 mol−1.
Experimental methods
Microanalysis (C, H, and N) was carried out with a Vario EL elemental analyzer. UV-Vis spectroscopy was recorded on a Specord 210 UV-Vis spectrophotometer using cuvettes of 1 cm path length. 1H NMR spectra were recorded on a Bruker 500 MHz NMR spectrometer. Mass spectrometry was performed on a Shimadzu LCMS-2020 spectrometer. IR spectra were recorded from 4000 to 400 cm−1 on a Agilent Carry 600 FT-IR spectrophotometer. Electrical conductivity measurements of the complexes were recorded using SYSTRONICS 304 conductivity meter at room temperature. Melting point values were recorded on a PPI-96 Inlab equipment. Emission intensity measurements were carried out using Perkin Elmer LS-45 spectrofluorometer. Viscosity measurements were carried out using Brookfield viscometer.
Solutions of calf thymus (CT) DNA in 5 mM Tris HCl/50 mM NaCl buffer gave a ratio of UV absorbance at 260 and 280 nm, A260/A280, of 1.9,32 indicating that the DNA was sufficiently free of protein. Concentrated stock solutions of DNA (20.5 mM) were prepared in buffer and sonicated for 25 cycles, where each cycle consisted of 30 s with 1 min intervals. The concentration of DNA in nucleotide phosphate (NP) was determined by UV absorbance at 260 nm after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilutions by taking the extinction coefficient, ε260 nm, as 6600 M−1 cm−1. Stock solutions of DNA were stored at 4 °C and used after no more than 4 days. Supercoiled plasmid pUC19 DNA was stored at −20 °C, and the concentration of DNA in base pairs was determined by UV absorbance at 260 nm after appropriate dilutions taking ε260 nm as 13100 M−1 cm−1.
:100 dilutions by taking the extinction coefficient, ε260 nm, as 6600 M−1 cm−1. Stock solutions of DNA were stored at 4 °C and used after no more than 4 days. Supercoiled plasmid pUC19 DNA was stored at −20 °C, and the concentration of DNA in base pairs was determined by UV absorbance at 260 nm after appropriate dilutions taking ε260 nm as 13100 M−1 cm−1.
X-ray crystallography
A BRUKER Venture X-ray diffractometer was employed for crystal screening, unit cell determination and data collection of complex 1 (ESI†).33–37 A Leica MZ 75 microscope was used to identify a suitable brown block with very well defined faces from a representative sample of crystals of the same habit. The crystal 1 of suitable size selected was mounted on a nylon loop and then placed in a cold nitrogen stream (Oxford) maintained at 110 K. The goniometer was controlled using the APEX3 software suite.33 The sample was optically centered with the aid of a video camera such that no translations were observed as the crystal was rotated through all positions. The X-ray radiation employed was generated from a Cu-IμsX-ray tube (Kα = 1.5418 Å with a potential of 50 kV and a current of 1.0 mA). 45 data frames were taken at widths of 1°. These reflections were used to determine the unit cell and the unit cell was verified by examination of the hkl overlays on several frames of data. No super-cell or erroneous reflections were observed. After careful examination of the unit cell, an extended data collection procedure (10 sets) was initiated using omega and phi scans.
Molecular docking studies
Molecular docking studies were performed to identify the possible binding site of complex with DNA, BSA and transferrin. The 3D crystal structures of B-DNA dodecamer d(CGCAAATTTCGC)2 (PDB ID: 1BNA), BSA (PDB ID: 3V03) and transferrin (PDB ID: 1D3K) were downloaded from the RCSB Protein Data Bank website (http://www.rcsb.org/pdb) and used. Docking studies were performed with Discovery Studio (Accelrys) by simulation of the complex into the binding site of the biomolecules. The docking was run using Ligand Fit dock protocol of Discovery Studio program and the receptors were typed with CHARMm force field. Ligands and water molecules were removed from the binding sites and default parameters were used for the docking calculations.
DNA binding and electrophoretic mobility studies
DNA binding and electrophoretic mobility studies were carried out by using the procedures reported already (ESI†).13
Tryptophan fluorescence quenching
The tryptophan fluorescence quenching studies were carried out as reported previously (ESI†).13
In vitro antidiabetic activity
α-Amylase inhibitory activity was studied for control and sample solution based on starch-iodine method.38 α-Amylase enzyme (1% w/v) solution was added to the sample tube containing 1 mL of different concentration of sample in 0.1 M sodium acetate/30 mM NaCl buffer (pH 7.2) and the mixture was incubated for 15 min. After pre-incubation, aliquots (1 mL) of that incubate were transferred to sample tubes containing starch substrate (1% w/v) in acetate buffer and the mixture was re-incubated for 15 min. 0.1 mL of the reaction mixture was withdrawn from each tube after mixing and discharged into 10 mL of iodine solution. Solutions were thoroughly mixed and the absorbance was measured immediately at 565 nm. Percentage inhibition was calculated according to the formula:
| Inhibition of α-amylase (%) = (Acontrol − Asample)/Acontrol × 100 |
where, Acontrol is the absorbance of the control and Asample is the absorbance of the test sample. All experiments were performed in triplicate (n = 3) and results were expressed as mean ± standard error mean (SEM).
Cell culture
Human breast carcinoma cells (MDA-MB-231), human lung carcinoma cells (A549), human ovarian carcinoma cells (A2780) and human embryonic kidney cells (HEK293) were cultured in RPMI (Gibco) media with 5% fetal calf serum (FCS, Gibco), 100 U mL−1 penicillin and 100 mg mL−1 streptomycin at 37 °C and 5% CO2.
Cytotoxicity study: resazurin assay
The cytotoxicity of the complexes 1 and 2 were studied in three different human cancer cell lines, namely, human breast carcinoma (MDA-MB-231), human lung carcinoma (A549), human ovarian carcinoma (A2780) cells and in one non-tumor human embryonic kidney cells (HEK293). The cytotoxicity of the compounds was measured by a fluorimetric cell viability assay using Resazurin (Sigma, USA). Cells were plated in triplicates in 96-well plates at a density of ∼5 × 103 cells per well in 100 μL of media 24 h prior to treatment. Cells were then treated with increasing concentrations of compounds for 24 h. After 24 h in the incubator, the medium was replaced by 100 μL complete medium containing resazurin (0.2 mg mL−1 final concentration). After 4 h of incubation at 37 °C, fluorescence of the highly red fluorescent resorufin product was quantified at 590 nm emission with 540 nm excitation wavelength in a Versa max tunable microplate reader.
Morphological studies
AO/EB staining. For both suspension and adherent cells, 96-well plates were centrifuged at 1000 rpm for 5 min using an Eppendorf 5810 centrifuge with inserts for 96-well plates. An AO/EB dye mix (8 μL) was added to each well, and cells were viewed under Nikon Eclipse90i upright microscope equipped with a Nikon DS-Ri1 12MP color camera. Tests were done in triplicate, counting a minimum of 100 total cells each.
Hoechst 33258 staining. The cell pathology was detected by staining the nuclear chromatin of trypsinized cells (4.0 × 104 per mL) with 1 μL of Hoechst 33258 (1 mg mL−1) for 10 min at 37 °C. Staining of suspension cells with Hoechst 33258 was used to detect apoptosis.39 A drop of cell suspension was placed on a glass slide, and a coverslip was overlaid to reduce light diffraction. At random, 300 cells were observed in a fluorescent microscope (Nikon Eclipse90i upright microscope) equipped with a Nikon DS-Ri1 12MP color camera and observed at 40× magnification, and the percentage of cells reflecting pathological changes was calculated. Data were collected for triplicate and used to calculate the mean and standard deviation.
Results and discussion
Synthesis and characterization of Ruthenium(II) complexes
Two water soluble half-sandwich organometallic complexes of the type [Ru(η6-arene)(met)Cl]Cl (1, 2), where arene is p-cymene (1) or benzene (2) and met is metformin, have been isolated by the reaction of the appropriate dimeric complex [(η6-arene)RuCl2]2 with a slight excess of equimolar amount of metformin ligand in methanol at 40 °C and the desired products were isolated as chloride salts in good yield. The formulae of the complexes [Ru(η6-arene)(met)Cl]Cl, as determined by elemental analysis and mass spectral studies, are supported by structural elucidation of 1 by using X-ray crystallography (cf. below). The complexes are soluble in water, Tris–HCl/NaCl (5:50 mM) buffer (pH 7.1), methanol and acetonitrile. The UV-Vis absorption spectra of complexes 1 and 2 show low energy ligand field band due to low-spin d6 configuration of Ru(II) complexes and exhibit absorbance tail around 350–410 nm, which is assigned to mixed metal-centered and metal-to-ligand charge transfer transitions.40 The weak shoulder observed for 1 (240 nm) and 2 (236 nm) is ascribed to the characteristic intra-ligand transitions. (Table S1 and Fig. S1†). The mass spectral data confirm the formation of complexes and the retention of identity of complexes even in solution (Fig. S2 and S3†). This is supported by the values of molar conductivity (ΛM) in acetonitrile (1, 64; 2, 62 Ω−1 cm2 mol−1), falling in the range for 1:1 electrolytes.41 The 1H NMR spectra of 1 and 2 in D2O show downfield shifts in positions of the arene protons as compared to the corresponding precursor complexes.42a This may be attributed to the incorporation of metformin ligand, which modifies the electron density around metal center. The N-CH3 protons of the metformin are observed as singlet in the range of 2.9–3.0 ppm (Fig. S4 and S5†). 13C NMR spectra of 1 and 2 were recorded in D2O to support the formation of the respective complexes (Fig. S6†). Infra-red spectra of complexes 1 and 2 exhibit characteristic bands due to ν(N–H) and ν(CN) stretching vibrations of metformin (Fig. S7†). The coordination of metformin ligand to metal center is confirmed by the broad band observed in the region of 3300–3400 cm−1 due to ν(N–H) stretching vibrations, which exhibit medium intensity for free metformin.42b
Description of the crystal structure of [(η6-p-cymene)Ru(met)Cl]Cl·H2O 1
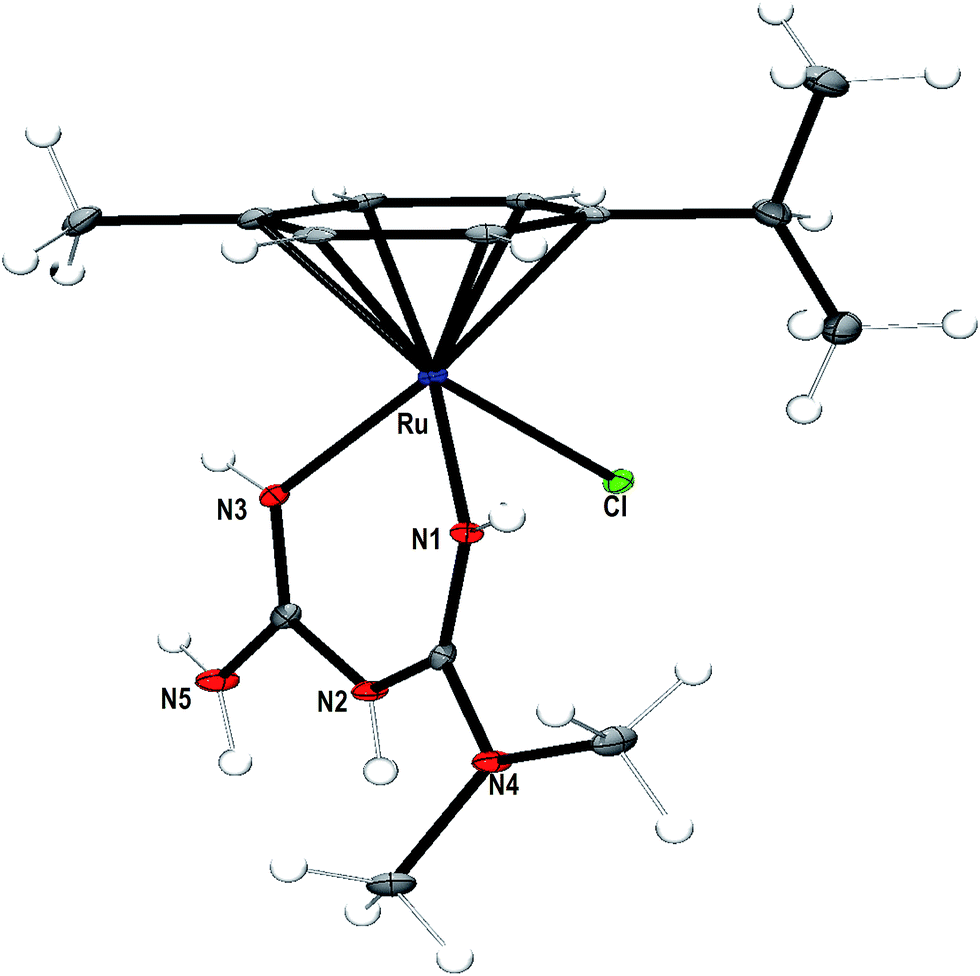
An ORTEP representation of the coordination environment of the complex monocation 1 including the atom numbering scheme is shown in Fig. 1. The crystallographic data and selected bond lengths and bond angles are listed in Tables 1 and 2 respectively. The asymmetric unit of 1 contains a cationic complex, a lattice chloride anion and water molecule. The complex cation adopts the familiar pseudo-octahedral coordination geometry43,44 by coordinating all the six carbon atoms (C5–C10) of p-cymene ring, both the nitrogen atoms (N1, N3) of metformin and chlorido ligand (Cl1). The bond angles made by N1 and N3 atoms of metformin and Cl− constitute the three legs of the ‘piano-stool’ structure [N(1)–Ru(1)–Cl(1) (84.56(8)°) and N(3)–Ru(1)–Cl(1) (84.97(8)°)]. The six Ru–C bonds have almost similar bond distances (2.155(3)–2.207(3) Å) with an average bond length of 2.178 Å forming the seat of the piano-stool.45 The N1 and N3 atoms of metformin are coordinated to the metal center with almost equal bond distance (Ru–N(1), 2.078(3); Ru–N(3), 2.073(3) Å) indicating that the tautomerisation is not involved in between –N(1)H/–N(3)H and –N(2)H groups, when the coordination complex is formed. The bite angle of metformin in 1 (N(1)–Ru(1)–N(3): 83.10(10)°) is lower than the bond angles involving chlorido ligand (N(1)–Ru(1)–Cl(1), 84.56(8)°, N(3)–Ru(1)–Cl(1) (84.97(8)°)), as expected.13 The Ru–Nimine bonds (Ru–N1, 2.078(3), Ru–N3, 2.073(3) Å) formed by metformin in 1 are shorter than Ru–Namine bonds (Ru–N1, 2.130, Ru–N2, 2.136 Å) formed by en in [(η6-p-cymene)Ru(en)Cl](PF6)12 on account of sp3 and sp2 hybridizations of the amine and imine nitrogen atoms respectively. The bite angle of metformin (83.10(10)°) in 1 is higher than that of en (78.9(10)°) in [(η6-p-cymene)Ru(en)Cl](PF6).12 This is expected because the steric hindrance formed by six membered chelate ring of metformin in 1 is less than that of five membered chelate ring formed by en ligand in [(η6-p-cymene)Ru(en)Cl](PF6). The hydrogen atom of the imine nitrogen (N3) in metformin is involved in hydrogen bonding with the oxygen atom of the neighbouring water molecule (N(3)–H⋯O: 2.163 Å).
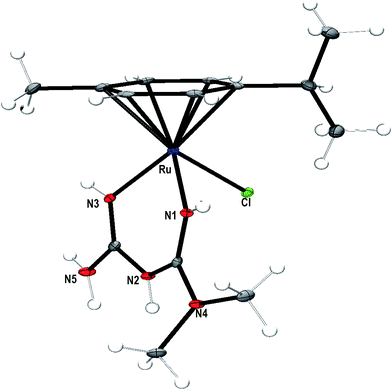 |
| | Fig. 1 ORTEP representation of the crystal structure of [Ru(η6-p-cymene)(met)Cl]+ (1) showing atom numbering scheme and displacement ellipsoid (50% probability level) (water molecule and Cl anion are omitted for clarity). | |
Table 1 Crystal data and structure refinement for complex 1
| |
1 |
| Formula |
C14H27Cl2N5ORu |
| Formula weight |
453.37 |
| Crystal system |
Monoclinic |
| Space group |
P121/n1 |
| a, Å |
16.876(4) |
| b, Å |
12.155(3) |
| c, Å |
19.495(4) |
| α, deg |
90 |
| β, deg |
111.576(2) |
| γ, deg |
90 |
| Volume [Å] |
3718.6(14) |
| Z |
8 |
| D(calc) [mg m−3] |
1.620 |
| θ, [min–max deg] |
1.369–27.495 |
| Goodness-of-fit on F2 |
1.050 |
| Final R indices [I > 2sigma(I)] |
R1 = 0.0416, wR2 = 0.0699 |
| R indices (all data) |
R1 = 0.0611, wR2 = 0.0767 |
Table 2 Selected interatomic distances [Å] and bond angles [deg] for complex 1
| |
1 |
| Bond distance [Å] |
| Ru(1)–N(1) |
2.078(3) |
| Ru(1)–N(3) |
2.073(3) |
| Ru(1)–Cl(1) |
2.431(9) |
| Ru(1)–C(5) |
2.199(3) |
| Ru(1)–C(6) |
2.160(3) |
| Ru(1)–C(7) |
2.174(3) |
| Ru(1)–C(8) |
2.207(3) |
| Ru(1)–C(9) |
2.177(3) |
| Ru(1)–C(10) |
2.155(3) |
|
| Bond angle [deg] |
| N(1)–Ru(1)–N(3) |
83.10(10) |
| N(1)–Ru(1)–Cl(1) |
84.56(8) |
| N(3)–Ru(1)–Cl(1) |
84.97(8) |
DNA binding studies: absorption spectral titration
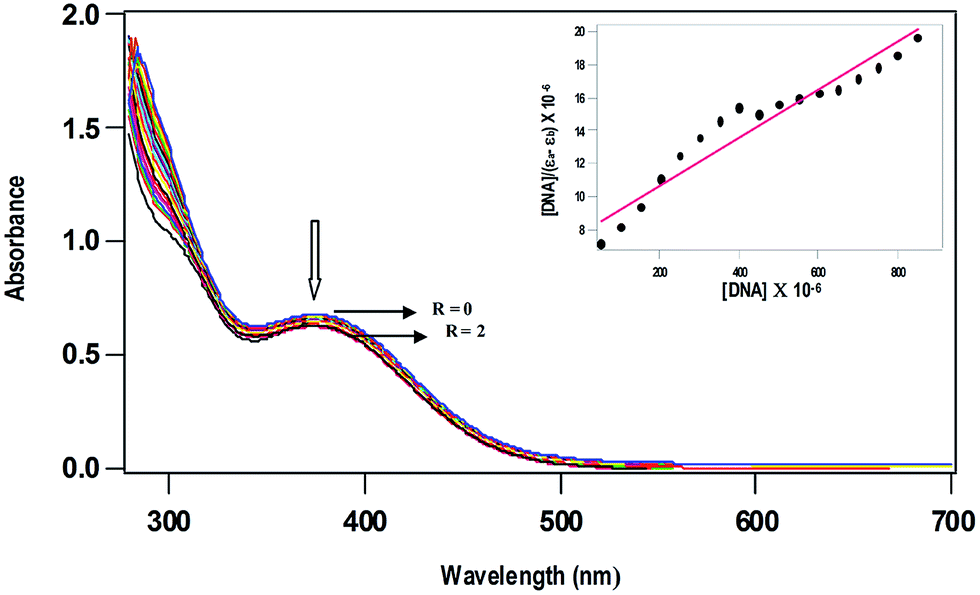
The less intense absorption spectral band observed for 1 (378 nm) and 2 (367 nm) was used to characterize the interaction of the complexes with CT DNA in 5 mM Tris–HCl/50 mM NaCl buffer at pH 7.1. Upon incremental addition of solution of CT DNA to complexes 1 and 2, a less decrease in absorption intensity (Fig. 2) with negligible shift in band position was observed suggesting weak electrostatic binding of the complexes with DNA.46–48 In order to quantitatively compare the DNA binding affinities, the intrinsic binding constant Kb was calculated using the equation,49
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) |
where [DNA] is the concentration of DNA in base-pairs, εa is the apparent extinction coefficient calculated as Aobs/[complex], εf corresponds to the extinction coefficient of the complex in its free form and εb refers to the extinction coefficient of the complex in the bound form. Each set of data, when fitted into the above equation, gives a straight line with a slope of 1/(εb − εf) and an y-intercept of 1/Kb(εb − εf) and Kb is determined from the ratio of the slope to intercept. The values of Kb vary in the order 1 (1.7 ± 0.08 × 103 M−1) > 2 (0.9 ± 0.04 × 103 M−1), illustrating that the DNA binding affinity of p-cymene complex is higher than that of benzene complex (Table 3). Both the enhanced hydrophobic interaction of methyl and isopropyl groups on arene ring in 1 and that of N-methyl groups on the metformin ligand lead to confer a higher DNA binding affinity of p-cymene complex than that of benzene complex. The Ru(II)–arene complexes like [Ru(η6-p-cymene)(L)Cl](PF6), where L is N-1-(anthracen-10-ylmethyl)-4-methylhomopiperazine (Kb, 7.8 × 104 M−1)13 and [(η6-C6H6)Ru(pmpzdpm)Cl], where pmpzdpm is 5-(2-pyrimidylpiperazine)phenyldipyrromethene (Kb, 9.5 × 104 M−1)50 show higher order of DNA binding obviously because of intercalation of appended ligand moiety with DNA base pairs. Thus, the order of DNA binding affinity of the complexes 1 and 2 is expected to be low due to non-intercalative mode of DNA binding. However, the strong hydrogen bonding propensity of the donor or acceptor groups of metformin with nucleobases or phosphate groups present in the edge of DNA, which is identified from molecular docking studies (cf. below), would also contribute to the DNA binding affinity of complexes. Thus, it is possible that the ability of the metformin ligand with strong hydrogen bonding interaction enables the Ru(II) metal center to be located close to the site of nucleobases for covalent mode of DNA binding by losing labile chloride ion. Similar observations have been made by Sadler et al. earlier for the complex [Ru(η6-p-cymene)(en)]2+, which contains a non-intercalating en ligand exhibiting a remarkably high preference for covalent binding to the N7 position of guanine. Such binding is stabilized by hydrogen bonding between the C6O of guanine and NH of the en ligand.51 Thus, the mode and extent of DNA binding of [Ru(η6-arene)(met)Cl]Cl complexes are contributed by both hydrophobic interaction of arene ligand and hydrogen bonding propensity of metformin ligand.
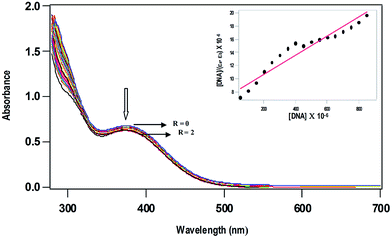 |
| | Fig. 2 Absorption spectra of [Ru(η6-p-cymene)(met)Cl]+ (0.5 × 10−3 M) in 5 mM Tris–HCl buffer at pH 7.1, in the absence (R = 0) and presence (R = 0–2) of increasing amounts of CT DNA (R = [DNA]/[complex]). | |
Table 3 Absorption spectral propertiesa and fluorescence spectral propertiesb of ruthenium(II) complexes bound to CT DNA
| Complexes |
λmax (nm) |
R |
Change in absorbance |
Kb (×103 M−1) |
Kappb (×103 M−1) |
| Measurements were made at R = 2, where R = [DNA]/[complex], concentration of solutions of Ru(II) complexes = 0.5 × 10−3 M (1 and 2). Apparent DNA binding constant from competitive DNA binding studies using 0–60 μM increasing concentration of 1 and 2. |
| [Ru(η6-p-cymene)(met)Cl]+ 1 |
378 |
2 |
Hypochromism |
1.7 ± 0.08 |
8.6 |
| [Ru(η6-benzene)(met)Cl]+ 2 |
367 |
2 |
Hypochromism |
0.9 ± 0.04 |
7.8 |
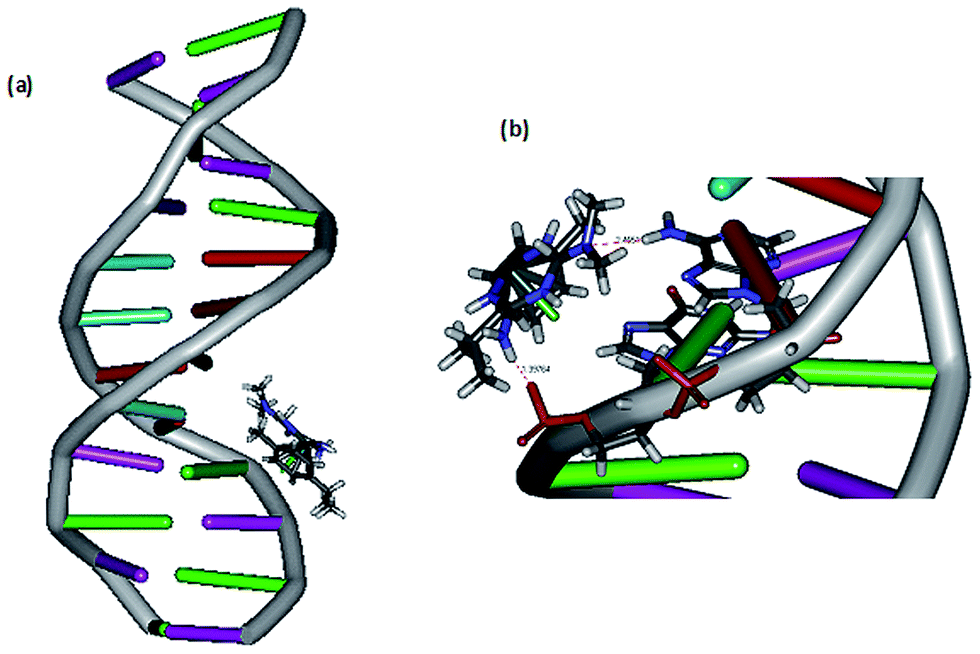
DNA docking studies. The mode of DNA binding is further investigated by performing docking analysis of 1 with B-DNA dodecamer d(CGCAAATTTCGC)2 (Fig. 3) by using Discovery Studio molecular graphics program. The calculated binding energy of the complex with B-DNA (−32.773 kcal mol−1) indicates that the complex effectively binds with DNA.10 Interestingly, the complex docks in the major groove of DNA, which supports the mode of DNA binding affinity of complex observed from absorption spectral titration (cf. above). The preference of the complex to bind in major groove rather than minor groove is due to the bulky p-cymene ligand otherwise it would cause immense strain if it is docked on minor groove. The complex is situated in major groove of DNA in such a way that the –NH2 and –N(CH3)2 groups of metformin are involved in hydrogen bonding interaction with the phosphate oxygen acceptor in guanine16 (1.397 Å) and –NH2 donor in adenine17 (2.455 Å) respectively. It is important to note that the Discovery Studio program is capable of identifying the feasible binding site of DNA by assuming that DNA and complex molecules are rigid and hence it is not suitable for detecting covalent mode of DNA binding. Thus, the covalent mode of DNA binding is further evidenced by viscosity measurements and electrophoretic mobility shift assay (cf. below).
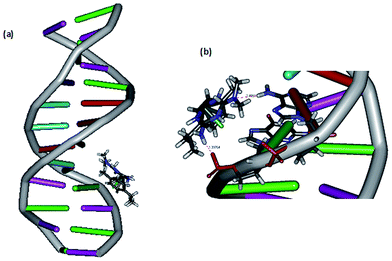 |
| | Fig. 3 (a) Docking of complex 1 at the major groove of the B-DNA dodecamer d(CGCAAATTTCGC)2; (b) hydrogen bonding interaction of complex 1 with phosphate group and adenine17 of B-DNA. | |
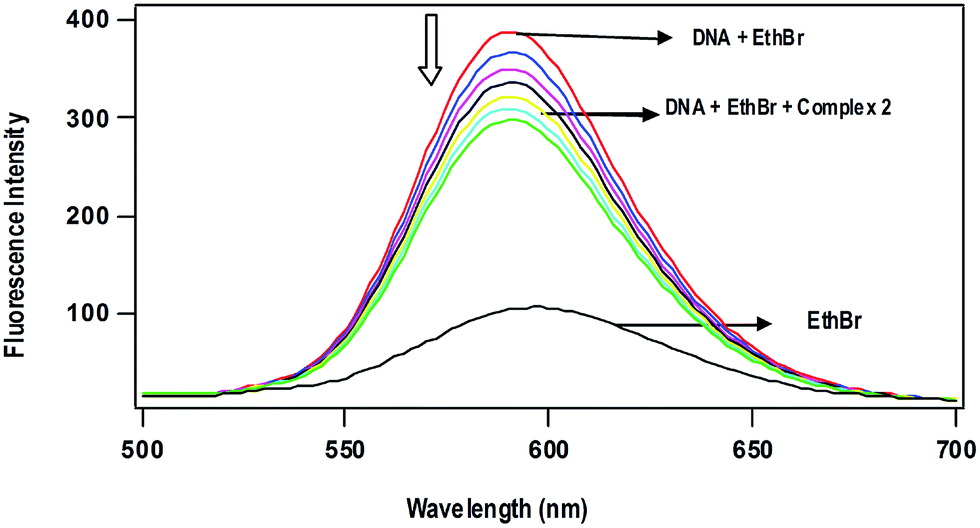
Competitive DNA binding studies. The Ru(II)–arene complexes 1 and 2 fail to show steady-state emission in all the solvents studied and lack emission even in the presence of CT DNA. When they are added (0–60 μM) to CT-DNA (125 μM) pretreated with EthBr (12.5 μM) ([DNA]/[EthBr] = 10) in 5 mM Tris–HCl/50 mM NaCl buffer at pH 7.1, the emission intensity of DNA-bound EthBr (λex, 450; λem, 595 nm) decreases (Fig. 4). From the plot of the intensity vs. complex concentration, the values of apparent DNA binding constant (Kapp) were calculated using the equation,52
| KEthBr[EthBr] = Kapp[complex] |
where KEthBr is 4.94 × 105 M−1,53 the concentration of EthBr is 12.5 μM and the concentration of the complex is that used to obtain a 50% reduction of fluorescence intensity of EthBr. Addition of a second DNA binding molecule either replaces the DNA-bound EthBr (if it binds to DNA more strongly than EthBr) and/or destabilize the excited state of DNA-bound EthBr by means of molecular collision,54 which would quench the DNA-bound EthBr emission. The observed Kapp values of the complexes decrease in the order, 1 (8.6 × 103) > 2 (7.8 × 103 M−1), which is consistent with the order of the Kb values obtained by UV-Vis absorption spectral titration (Table 3). The observed low order Kapp values reveal that the complexes are not able to involve intercalation with DNA to replace the strong intercalating molecule EthBr. However, the complexes 1 and 2 at higher concentrations would destabilize the DNA-bound EthBr more efficiently through enhanced hydrophobic interaction of arene ring and metformin with the hydrophobic DNA surface.
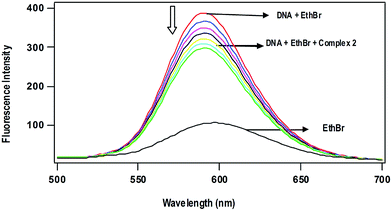 |
| | Fig. 4 Effect of addition of complex 2 on the emission intensity of the CT DNA-bound ethidium bromide (12.5 μM) at different concentrations (0–60 μM) in 5 mM Tris-HCl buffer (pH 7.1). | |
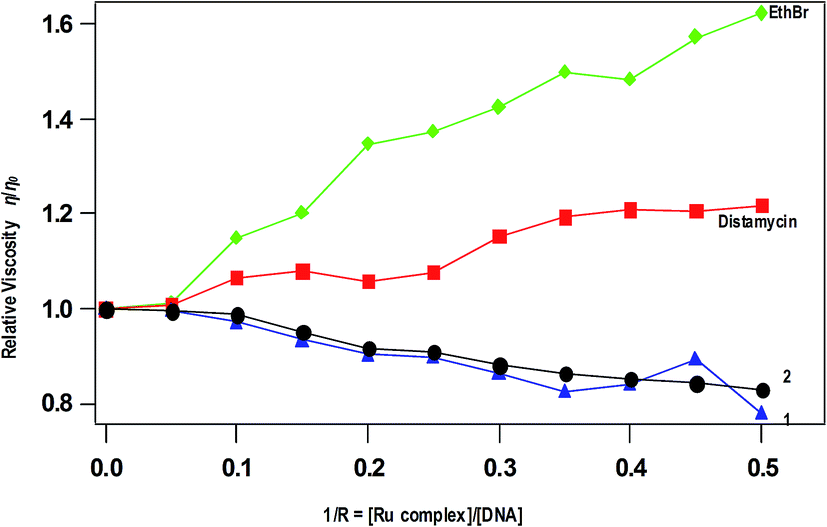
Viscosity measurements. The viscosity measurement of CT DNA upon treatment with varying concentrations of complexes reveals the DNA binding mode of the complexes. The values of relative specific viscosity (η/η0), where η and η0 are the specific viscosities of DNA in the presence and absence of the complex respectively, are plotted against 1/R (=[Ru complex]/[DNA]) = 0–0.50, along with that of EthBr and distamycin treated (Fig. 5). The intercalation of EthBr in between the DNA base pairs increase the overall DNA contour length resulting increase in viscosity whereas the viscosity of DNA is slightly increased upon the addition of distamycin, which is known to involve in minor groove DNA binding. In contrast, decrease in viscosity is observed for 1 and 2 due to kinking or bending of the DNA helices upon covalent binding of complexes with DNA base pairs. A similar decrease in viscosity has been observed for [Ru(η6-p-cymene)(L)Cl](PF6) (L = N-(2-pyridylmethyl)glycine),55 which is reported as a covalently DNA binding molecule. The decrease in viscosity is also observed for partially intercalating complex like [Ru(dmb)2(pdpt)](ClO4)2 (dmb = 4,4′-dimethyl-2,2′-bipyridine, pdpt = 3-(pyridine-2-yl)-5,6-diphenyl-as-triazine).56 However, the possibility of partial intercalation of complexes 1 and 2 in between DNA base pairs is excluded because of absence of extended aromatic ring. Thus, the covalent mode of DNA binding of the present complexes is similar to other monofunctional Ru(II)–arene-en complexes reported.51
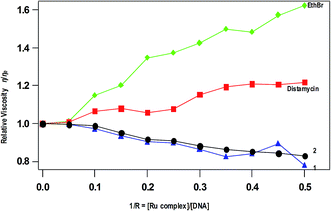 |
| | Fig. 5 Effect of addition of complexes 1, 2, EthBr and distamycin on the viscosity of CT DNA; relative specific viscosity vs. 1/R; [CT DNA] = 500 μM. | |
Electrophoretic mobility studies
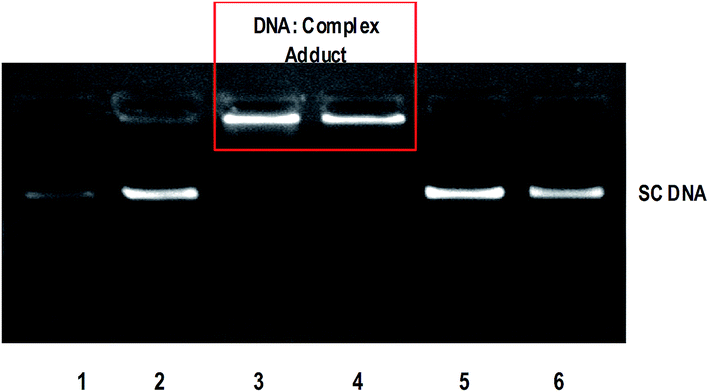
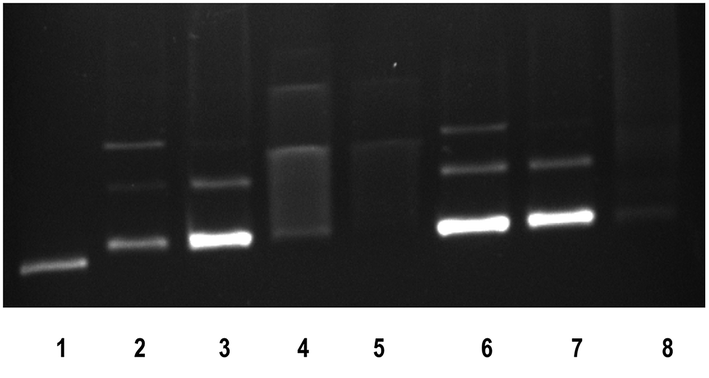
The anticancer activity of most of the complexes is related with the cellular response of metal induced DNA damage or DNA distortion, which is caused by formation of inter or intrastrand cross linking adducts with metal complexes.3,8 The gel electrophoresis technique provides an insight into the mode of interaction of DNA with metal complexes by observing the migration of plasmid DNA in different conformations. The covalent DNA binding metal complex like [Ru(η6-p-cymene)(en)Cl]+ involve local unwinding which leads to retardation of migration of DNA.57 The intercalating complexes first unwind negatively supercoiled DNA and then convert it into positively supercoiled DNA.55 The major/minor groove DNA binding and partial intercalating complexes convert the supercoiled DNA into nicked form, if one strand of DNA is cleaved13 and linear form, if both strands are cleaved.58 In order to explore the DNA interaction of 1 and 2, supercoiled (SC) pUC19 DNA (40 μM) was incubated with the complexes (100 μM) in 5 mM Tris–HCl/50 mM NaCl buffer (pH 7.1) at 37 °C for 1 h in the absence of an activator. The complexes 1 and 2 fail to retard the mobility of SC DNA with an incubation period of 1 h (Fig. S8†). However, when the incubation period is increased from 1 h to 3 h, the retention of SC DNA occurred in the well (Fig. 6) illustrating that these monocationic complexes involve covalent adduct formation with DNA. This covalent DNA-complex adduct would significantly reduce the overall negative charge of the DNA, which could stop the migration of DNA from the well. When the concentration of complexes is varied from 50 to 200 μM with an increasing incubation period of 5 h, the rate of migration of SC DNA decreases in comparison with the control DNA. Also, the maximum degradation with undetectable fragments occurs at 200 μM concentration of 1 (Lane 5, Fig. 7) where it occurs at 150 μM concentration for complex 2 (Lane 8, Fig. 7). From the analysis of the DNA gel electrophoresis data, it is proposed that the present Ru(II)–arene complexes of the type [Ru(arene)(met)Cl]+ undergo rapid aquation to form [Ru(arene)(met)(H2O)]2+ in an aqueous environment.12 Then the aqua product covalently binds to nitrogen of a DNA heterocyclic base by replacing the aqua ligand forming DNA–complex adduct. Further, with a prolonged incubation period, the adduct formed undergoes multiple fragmentation of DNA. However, detailed investigation is required to analyze the fragments formed in DNA cleavage. Thus, the Ru(II)–arene complexes containing metformin ligand are able to form covalent adduct with DNA as that of previously reported Ru(II)–arene-en complexes.57
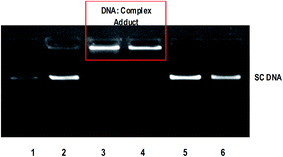 |
| | Fig. 6 Retardation of mobility of supercoiled pUC19 DNA (40 μM) by ruthenium(II) complexes 1 and 2 (100 μM) in absence of an external agent in 5 mM Tris HCl/50 mM NaCl buffer at 37 °C. Lane 1: DNA; Lane 2: DNA + metformin (100 mM); Lane 3: DNA + 1 (100 mM, 3 h incubation); Lane 4: DNA + 2 (100 mM, 3 h incubation); Lane 5: DNA + 1 (100 mM, 1 h incubation); Lane 6: DNA + 2 (100 mM, 1 h incubation). | |
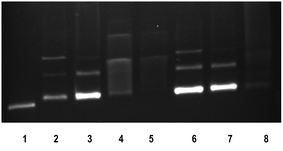 |
| | Fig. 7 Cleavage of supercoiled pUC19 DNA (40 μM) by ruthenium(II) complexes 1 and 2 in absence of an external agent in 5 mM Tris HCl/50 mM NaCl buffer at 37 °C in 5 h incubation. Lane 1: DNA; Lane 2: DNA + 1 (50 mM); Lane 3: DNA + 1 (100 mM); Lane 4: DNA + 1 (150 mM); Lane 5: DNA + 1 (200 mM); Lane 6: DNA + 2 (50 mM); Lane 7: DNA + 2 (100 mM); Lane 8: DNA + 2 (150 mM). | |
Protein binding studies
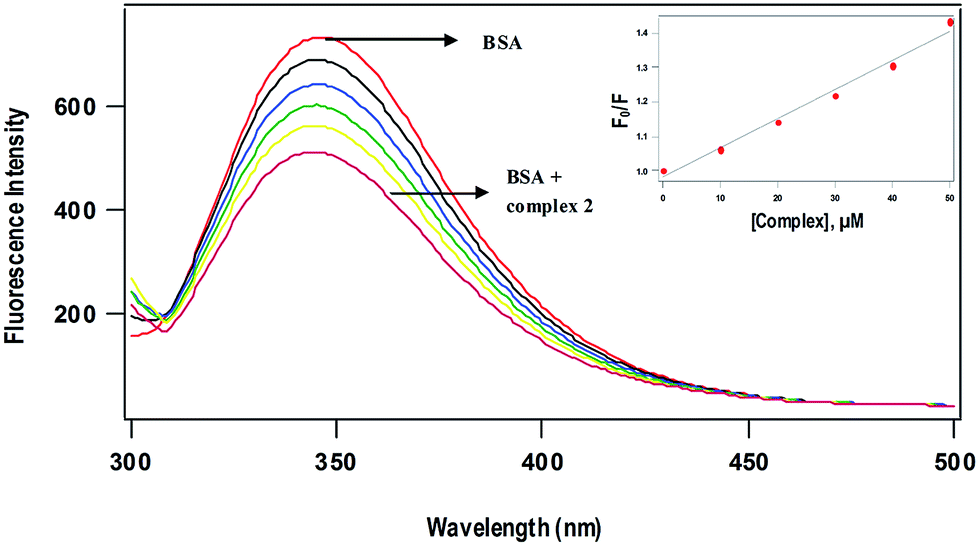
BSA has been taken as model serum albumin59 in order to determine the protein binding affinity of complexes. The major intrinsic fluorescence of BSA when excited at 295 nm is provided by tryptophan amino acid residues.60,61 Changes in the emission spectra of tryptophan residue are common in response to conformational transitions, subunit associations, substrate binding or denaturation.62 The tryptophan emission quenching experiments were carried out by adding the complexes with increasing concentration (10–50 μM) to BSA (5 μM) at 298 K and following the decrease in fluorescence intensities. Generally, fluorescence quenching can be illustrated by the well-known Stern–Volmer equation,63
where F0 and F are the fluorescence intensities of BSA in the absence and presence of the complexes respectively, KSV is the Stern–Volmer quenching constant and [Q] is the concentration of the quenching complex. The value of Ksv obtained as slope of the linear plot of F0/F vs. [Q] follows the order 1 (12.6 ± 0.3 × 103) > 2 (8.5 ± 0.5 × 103 M−1) (Fig. 8), which is in agreement with the trend in DNA binding affinities. The p-cymene complex (1) is capable of quenching the tryptophan fluorescence 1.5 times more strongly than benzene complex (2) due to enhanced hydrophobic interaction of p-cymene ligand moiety with the tryptophan site of BSA. The mechanism of tryptophan emission quenching takes place through either dynamic or static mode. In dynamic quenching, a collision between the complex and the excited state of fluorophore takes place. In contrast, the complex forms a non-fluorescent adduct with the fluorophore in the ground state in case of static quenching.13 UV-Visible absorption spectra of BSA in the absence and presence of the complexes (Fig. S9†) show that the absorption intensity of BSA decreased with no shift in band position. It reveals that there is no static interaction between BSA and the added complex, and quenching occurs through dynamic mechanism.
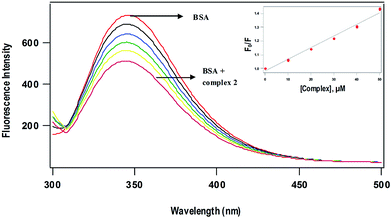 |
| | Fig. 8 Fluorescence quenching spectra of BSA (5 μM) at different concentrations of complex 2 (10–50 μM) at 298 K; excitation wavelength, 295 nm. (Inset) Plots of relative integrated emission intensity (F0/F) vs. [complex 2]. | |
Molecular docking studies with BSA and transferrin
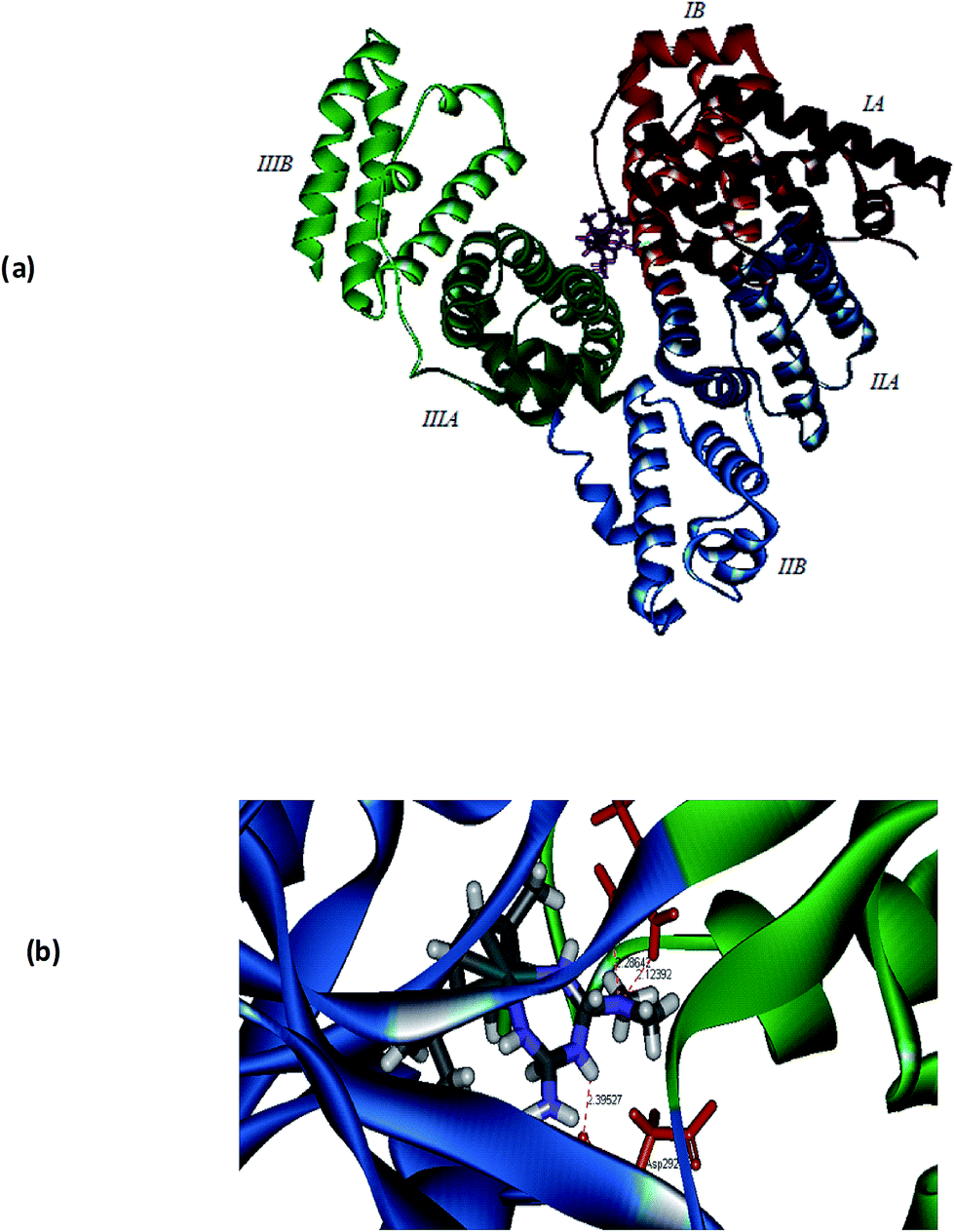
In order to get further understanding of the complex–protein interaction, molecular docking analysis was performed by docking the p-cymene complex 1 with BSA (PDB ID: 3v03) and transferrin (PDB ID: 1D3K) (Fig. 9). The globular BSA protein consists of three homologous domains (I, II, and III), which are made up of 583 amino acid residues and each primary domain is further divided into sub-domains A and B along with 17 disulphide linkages.64–66 The p-cymene complex is bound at the interface between the two sub domains IIA and IIIA, which are identified as the hydrophobic pocket of interaction of various drugs. The binding site of the complex is comprised of many amino acid residues like Asp108, Arg144, His145, Leu189, Ser192, Ala193, Arg196, Glu424, Ser428, Lys431, Tyr451, Leu454, Ile455 and Arg458. From the docking simulations, it is observed that the enhanced hydrophobic interaction of methyl and isopropyl groups on p-cymene and that of N-methyl groups on metformin lead to confer strong binding affinity with BSA. Also, the complex–protein binding is further stabilized by hydrogen bonding interaction, which is evident from the formation of hydrogen bond between –NH2 group of metformin and oxygen atom of serine residue (NH⋯Oserine: 2.676 Å).
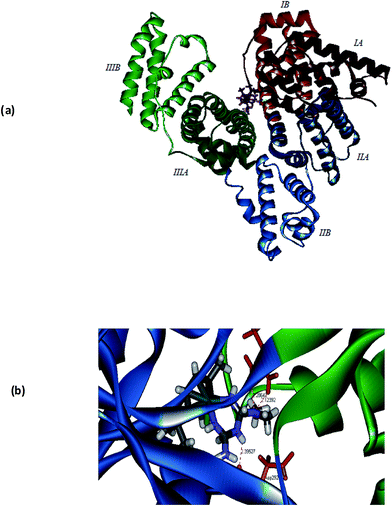 |
| | Fig. 9 Molecular docked model of 1 located within the hydrophobic pocket of BSA (a) and transferrin (b). | |
Experimental evidences show that the accumulation of antitumor ruthenium complex KP1019 might be mediated by iron uptake plasma protein transferrin. Since the neoplastic cells require high concentration of iron for its metabolism, ruthenium can bind to other sites on transferrin and ‘piggy-back’ into the cell when iron is taken into the cell.67–69 In order to ascertain the binding mode of complex 1 with transferrin, the docking simulation was performed. Transferrin is a glycoprotein containing two N and C lobes which are further divided into two sub-domains coordinating with Fe3+. The crystal structure of transferrin (1D3K) contains only the N lobes, which are divided into N1 and N2 sub-domains. The complex is bound at the interface between N1 and N2 sub-domains in a hydrophobic pocket and the binding site is lined with Trp8, Cys9, Ala10, Val11, Glu15, Tyr45, Val60, Thr61, Leu62, Leu66, Arg124, Phe186, Gly187, Tyr188, His249, Asp292, Leu294, Phe295 and Lys296 amino acid residues (Fig. 9). It is interesting to note that the hydrogen bonds are formed between –N(CH3)2 group of 1 and –NH2 group of Arg124 (2.286 Å) as well as –NH2 group of 1 and the amide oxygen of Asp292 (2.123 Å). From the calculated values of binding energy, it can be depicted that the binding affinity of 1 with BSA (−46.5 kcal mol−1) is higher than that of transferrin (−26.9 kcal mol−1).
In vitro antidiabetic activity
The present Ru(II)–arene complexes incorporated with the well-known antidiabetic drug metformin are expected to show enhanced antidiabetic activity. α-Amylase, which is an enzyme present in the pancreatic juice and saliva is involved in the process of breakdown of insoluble starch into blood absorbable sugars. The inhibition of α-amylase activity would delay the rate of absorption of glucose by blood thereby maintaining the serum blood glucose in hyperglycemic individuals.70 The α-amylase inhibition activity of 1 and 2 for 30 min incubation in sodium acetate buffer pH 7.2 has been studied in comparison with the standard drug acarbose, which is a well-known α-amylase inhibitor. The inhibitory activity of 1 is comparable to that of 2, but both the complexes show the inhibitory activity lower than the standard drug. (IC50: 1, 174.9 ± 1.6; 2, 147.5 ± 1.5; acarbose, 65.8 ± 1.5 μM). Further in vivo studies on animal models are required to understand the mechanism of antidiabetic activity, which could be related to the mechanism of cytotoxicity of complexes (cf. below).71
Cytotoxicity studies: resazurin assay
The cytotoxic activity of various Ru-arene complexes has been associated with their DNA binding affinity43,72 and/or significant reactivity against emerging protein targets.73 Since the present Ru(II)–arene complexes involve efficient interaction with DNA and protein, the cytotoxic activity against human breast carcinoma (MDA-MB-231), human lung carcinoma (A549), human ovarian carcinoma (A2780) and non-tumorigenic human embryonic kidney (HEK293) cells has been investigated for 24 h incubation by adopting resazurin assay. The ability of the complexes to kill the cancer cells vary as 1 > 2 (Table 4) reflects the ability of interaction of the complexes with DNA and protein. Interestingly, the complexes 1 and 2 show significant cytotoxicity against three different human cancer cells respectively, MDA-MB-231, A549, A2780 and the complexes exhibit potency to kill the breast cancer cells approximately 2.0 times more than to kill ovarian and lung cancer cells. Moreover, they were non-toxic to healthy human kidney cells up to the higher concentration tested (>100 μM). This clearly indicates that the cell killing activity of the complexes are more specific towards cancer cells and are non-toxic to non-cancerous cells. Also, the cytotoxicity of p-cymene complex 1 (IC50: 16.1 (1.8) μM) and benzene complex 2 (IC50: 24.4 (2.6) μM) is comparable with that of [Ru(η6-p-cymene)(en)Cl]+ (IC50, 10.0 (1.1) μM) and [Ru(η6-benzene)(en)Cl]+ (IC50, 17.0 (8.3) μM) against human ovarian carcinoma (A2780) cells respectively.74 Sadler et al. established that the cytotoxicity of [Ru(η6-arene)(en)Cl]+ complexes is due to distortion of DNA by forming monofunctional adduct, which leads to recognition of DNA binding protein and repair mechanism.75 Similar mechanism is expected for the cell killing activity of the present complexes obviously because of similar mode of DNA binding and comparable cytotoxicity observed. Previous studies have shown that the lower cationic charge of complex and hydrophobicity of ligands confer enhanced lipophilicity, which encourages the permeation of complexes across cell membrane to exhibit cytotoxicity.43,76 Hence, the efficient cytotoxicity of complexes may be ascribed, in addition to their DNA and protein binding affinity, to the monocationic charge of complex and hydrophobicity of arene and metformin ligands.
Table 4 In vitro cytotoxicity assay for complexes 1 and 2 against human breast carcinoma (MDA-MB-231), human lung carcinoma (A549), human ovarian carcinoma (A2780) and non-tumorigenic human embryonic kidney (HEK293) cells for 24 h incubation. Inhibitory concentration (IC50) values are in μM (data are mean ± SD of three replicates each)
| Complexes |
IC50a, μM |
| MDA-MB-231 human breast carcinoma cells |
A549 human lung carcinoma cells |
A2780 human ovarian carcinoma cells |
HEK293-human embryonic kidney cells |
| IC50 = concentration of drug required to inhibit growth of 50% of the cancer cells (in μM). Growth inhibition assay (ref. 74). |
| [Ru(η6-p-cymene)(met)Cl]Cl 1 |
7.9 ± 0.7 |
15.5 ± 2.8 |
16.1 ± 1.8 |
>100 |
| [Ru(η6-benzene)(met)Cl]Cl 2 |
21.0 ± 1.3 |
29.2 ± 1.7 |
24.4 ± 2.6 |
>100 |
| [Ru(η6-p-cymene)Ru(en)Cl]PF6 |
— |
— |
10.0 + 1.1b |
— |
| [Ru(η6-benzene)Ru(en)Cl]PF6 |
— |
— |
17.0 + 8.3b |
— |
| Metformin |
>350 |
>500 |
>500 |
>800 |
It is interesting to note that the present Ru(II)–p-cymene complex bearing antidiabetic drug shows cell killing activity (IC50: 1, 7.9 (0.7) μM) higher than those with the anti-inflammatory drug derived ligands (IC50: >140–170 μM) against human breast carcinoma (MDA-MB-231) cells20 and those of antibacterial quinolone drugs (IC50: >320 μM) against human ovarian cancer cells,18 irrespective of the methodology employed. Also, the complexes 1 and 2 are remarkable in displaying cytotoxicity against breast, lung and ovarian cancer cells more prominent than metformin (IC50, >350–500 μM). This clearly demonstrates the importance of coordination of antidiabetic drug to Ru-arene framework, which confers versatility of the complexes leading to improved cytotoxic activity.
Morphological assay: fluorescence and phase contrast microscopic studies
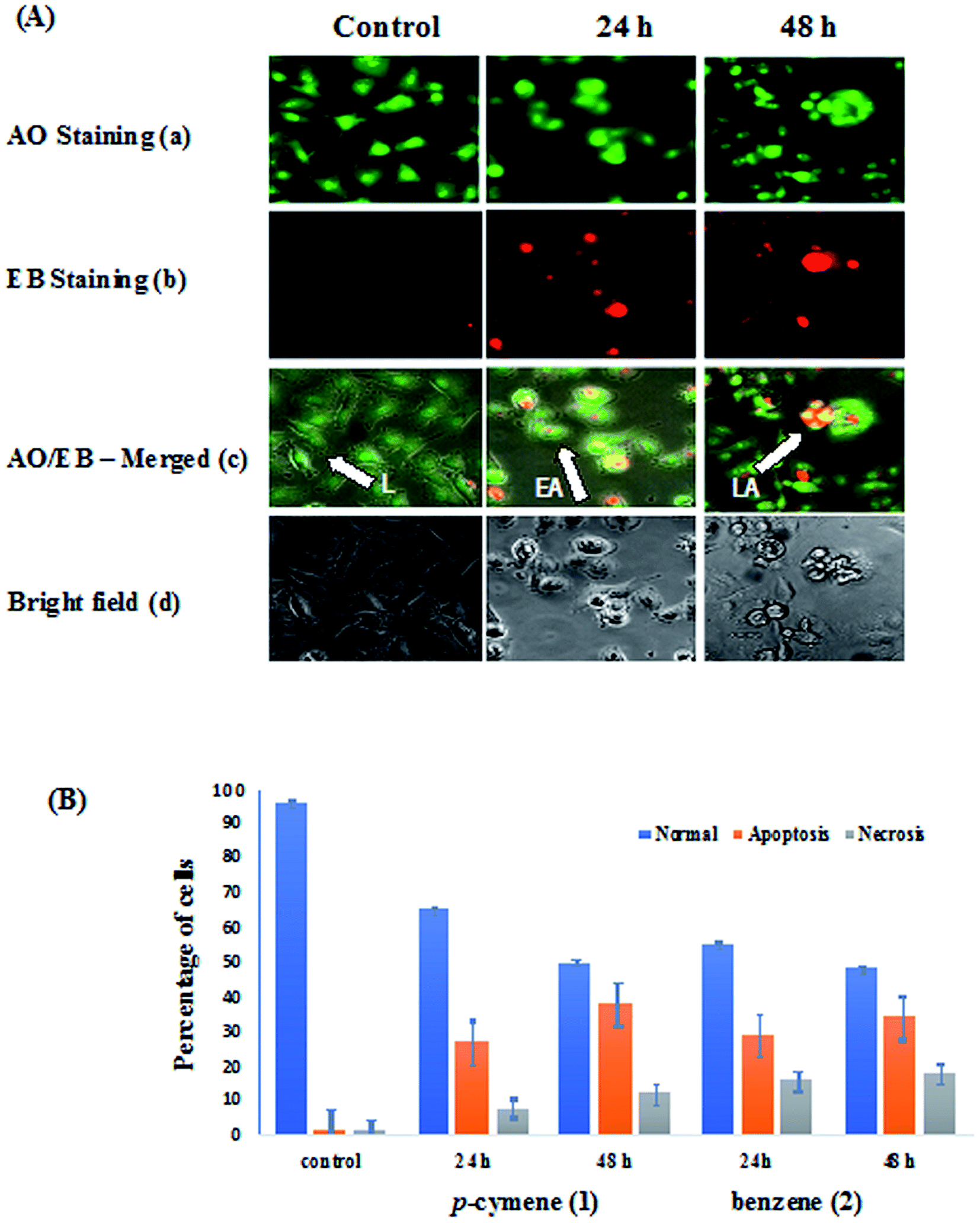
The human breast carcinoma cells (MDA-MB-231) were treated with IC50 concentration of complexes 1 and 2 for 24 h and 48 h incubation and the morphological alterations brought out by the complexes have been evaluated by adopting fluorescence microscopic analysis of AO/EB-stained cancer cells.77 Acridine orange (AO) permeates the cells and makes the nuclei appear green. Ethidium bromide (EB) is taken up by cells only when cytoplasmic membrane integrity is lost, and stains the nucleus red. The observed cytological changes are classified into four types depending upon the fluorescence emission and morphological features of chromatin condensation in the AO/EB stained nuclei as reported previously:13,43 (i) Normal and viable cells have a uniform green fluorescing nuclei with the organized structure (ii) early apoptotic cells have bright green nuclei with perinuclear chromatin condensation (iii) late apoptotic cells display orange to red fluorescing nuclei with highly condensed or fragmented chromatin(IV) cells that have died from direct necrosis have a structurally normal orange to red fluorescing nuclei. These morphological changes were more evident from the bright field images of phase contrast microscopy also. All of these morphological changes observed for 1 and 2, suggest that the cells are committed to both apoptotic and necrotic mode of cell death in a time-dependent manner (Fig. 10). In particular, the observed morphological alterations like membrane blebbings, chromatin condensation or fragmentation and cytoplasm vacuolization indicate that the induction of apoptosis is the major mode of cell death. The potency of the complexes to induce apoptotic cell death varies as 1 > 2 revealing that the higher apoptosis-inducing ability of p-cymene complex is facilitated by its higher hydrophobicity than benzene complex. The enhanced hydrophobicity and hence lipophilicity of arene and metformin ligands facilitate the transport of the complexes across the cell membrane and their eventual release at various cellular components leading to apoptosis.
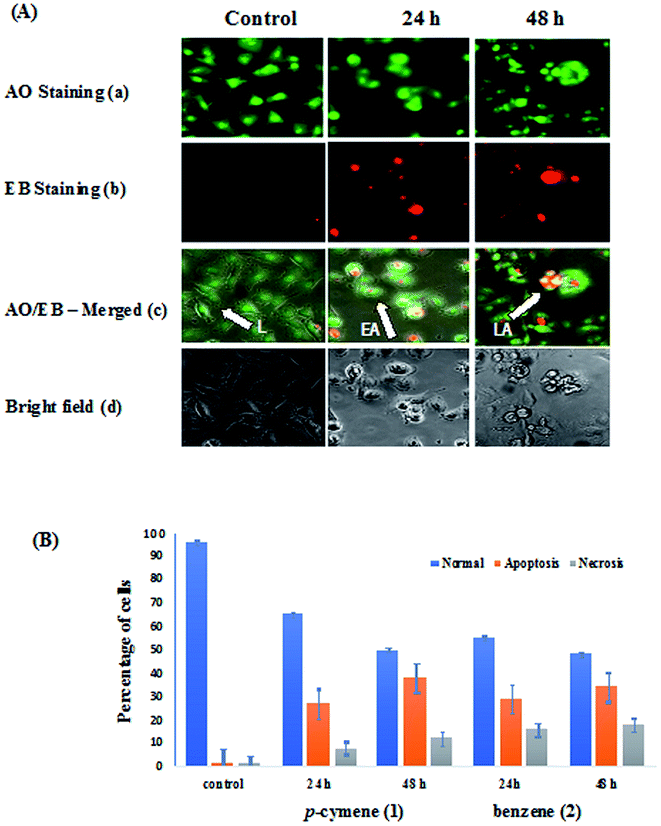 |
| | Fig. 10 (A) AO/EB-staining (a–c) and phase contrast microscopic images (d) of human breast carcinoma cells (MDA-MB-231) at 24 and 48 h incubation by the treatment of 1 (L: Live cells, EA: Early Apoptosis and LA: Late Apoptosis). (B) Graph shows manual count of percentage of normal, apoptotic and necrotic cells at 24 and 48 h treatment with compound 1 and 2 (data are mean% ± SD% of each triplicate). | |
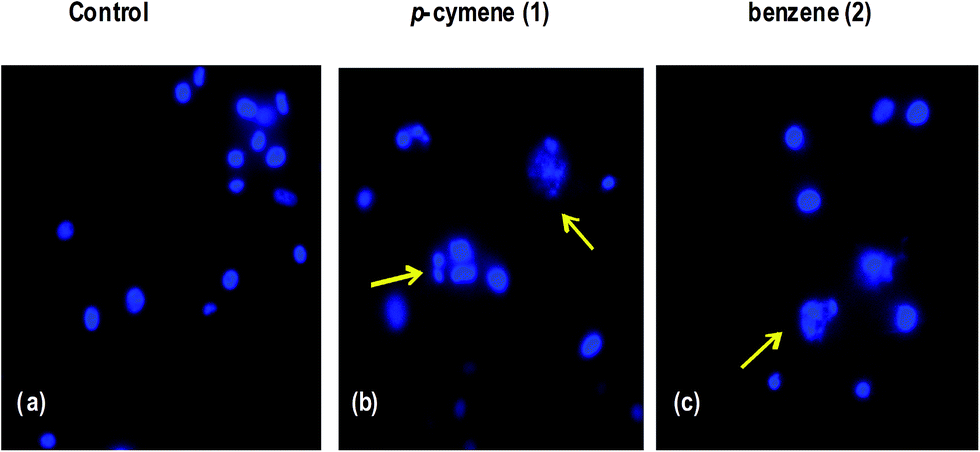
Hoechst staining studies. The molecular mechanism of cell death has been further confirmed by treating the human breast carcinoma cells (MDA-MB-231) with IC50 concentration of 1 and 2 for 24 h and then cytological changes are observed by adopting Hoechst 33258 staining. The representative morphological changes observed for 1 and 2 such as chromatin fragmentation, bi- and/or multinucleation, cytoplasmic vacuolization and late apoptosis indication of dot-like chromatin condensation are shown in Fig. 11. This clearly provides further evidence for induction of apoptosis by the complexes and the complexes could be developed as efficient anticancer drugs as the apoptosis-inducing ability is critical in determining the efficacy of an anticancer drug. However, additional biochemical experiments like cellular uptake and cell cycle analysis are to be performed to further investigate the mode of cell death observed for the complexes.
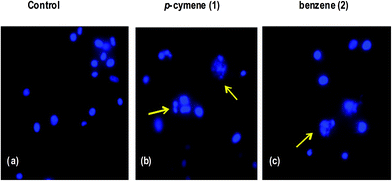 |
| | Fig. 11 Hoechst 33258 staining of human breast carcinoma cells (MDA-MB-231) were treated with IC50 concentrations of 1 and 2. (a) Untreated MDA-MB-231 cells (control), (b and c) cells were treated with 1 and 2 after 24 h incubation; apoptotic body formation is indicated by arrows. | |
Conclusions
In this study, two water soluble half-sandwich Ru(II)–arene complexes of the type [Ru(η6-arene)(metformin)Cl]Cl have been isolated and characterized. The coordination geometry around Ru(II) in the p-cymene complex is described as familiar pseudo-octahedral structure. The p-cymene complex, in addition to the donor functionalities of metformin, is involved in hydrophobic interaction with DNA through the methyl and isopropyl groups of arene ligand, which is responsible for its stronger DNA binding affinity than benzene complex. DNA docking studies reveal that p-cymene complex binds in the DNA major groove, which is stabilized by hydrogen bonding interactions. The viscosity measurements imply that the complexes are involved in the covalent mode of DNA binding, which is further supported by the retardation of migration of DNA observed from gel electrophoretic mobility studies. Remarkably, the p-cymene complex exhibits higher affinity to bind to BSA than benzene complex in the hydrophobic region and it effectively docks on BSA and transferrin. It is noteworthy that it shows lower antidiabetic activity in comparison with the standard drug in vitro. The same complex is unique in displaying more cytotoxic activity than its analogue against human breast, lung and ovarian cancer cells and is found to be non-toxic to normal kidney cells. The facile transport of the complex cation across the cell membrane on account of efficient hydrophobicity of both the arene and metformin also induces the prominent apoptotic activity of the complex. Thus, a well-known antidiabetic drug such as metformin along with coordinated p-cymene ligand may be incorporated into Ru-based organometallic anticancer agents.
Conflict of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank Science and Engineering Research Board (SERB), New Delhi for a Start-up research grant (YSS/2015/000403/CS) to M. G. and Junior Research Fellowship to D. G. M. G. sincerely thanks SRM University for providing various instrumentation facilities. We thank Dr B. Neppolian and Dr M. Mariappan, SRM University for providing the UV-Vis absorption spectral facilities. Mr K. Manikandan, SRM College of pharmacy is grateful for help to obtain the ligand. DST-FIST is greatly acknowledged for creating facilities to the Department of chemistry, SRM University, Kattankulathur, Chennai.
References
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994 Search PubMed.
- J. L. Misset, H. Bleiberg and W. Sutherland, Crit. Rev. Oncol. Hematol., 2000, 35, 75–93 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- A. J. Windebank and W. Grisold, J. Peripher. Nerv. Syst., 2008, 13, 27–46 CrossRef CAS PubMed.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2534 CrossRef CAS PubMed.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupeca, S. Zorbas-Seifrieda, C. Groessla, A. Eggera, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
-
(a) A. K. Singh, D. S. Pandey, Q. Xua and P. Braunstein, Coord. Chem. Rev., 2014, 270–271, 31–56 CrossRef CAS;
(b) Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer-Verlag, Heidelberg, 2010, vol. 32, p. 21 Search PubMed.
-
(a) F. Wang, A. Habtemariam, E. P. L. vander Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- R. K. Gupta, R. Pandey, G. Sharma, R. Prasad, B. Koch, S. Srikrishna, P. Li, Q. Xu and D. S. Pandey, Inorg. Chem., 2013, 52, 3687–3698 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnieres, H. van den Bergh, P. J. Dyson and A. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- R. E. Morris, R. E. Aird, P. D. Murdoch, H. M. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- M. Ganeshpandian, R. Loganathan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2014, 43, 1203–1219 RSC.
- E. Iniguez, A. Sánchez, M. A. Vasquez, A. Martínez, J. Olivas, A. Sattler, R. A. Sánchez-Delgado and R. A. Maldonado, J. Inorg. Biochem., 2013, 18, 779–790 CAS.
- G. Crisponi, V. M. Nurchi, D. Fanni, C. Gerosa, C. Nemolato and G. Faa, Coord. Chem. Rev., 2010, 254, 876–889 CrossRef CAS.
- G. Psomas and D. P. Kessissoglou, Dalton Trans., 2013, 42, 6252–6276 RSC.
- L. D. Dale, J. H. Tocher, T. M. Dyson, D. I. Edwards and D. A. Tocher, Anti-Cancer Drug Des., 1992, 7, 3–14 CAS.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- R. Hudej, J. Kljun, W. Kandioller, U. Repnik, B. Turk, C. G. Hartinger, B. K. Keppler, D. Miklavcic and I. Turel, Organometallics, 2012, 31, 5867–5874 CrossRef CAS.
- F. Aman, M. Hanif, W. A. Siddiqui, A. Ashraf, L. K. Filak, J. Reynisson, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2014, 33, 5546–5553 CrossRef CAS.
- E. Paunescu, S. McArthur, M. Soudani, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2016, 55, 1788 CrossRef CAS PubMed.
- J. M. Evans, L. A. Donnelly, A. M. Emslie-Smith, D. R. Alessi and A. D. Morris, BMJ, 2005, 330, 1304–1305 CrossRef PubMed.
- G. W. Landman, N. Kleefstra, K. J. van Hateren, K. H. Groenier, R. O. Gans and H. J. Bilo, Diabetes Care, 2010, 33, 322–326 CrossRef CAS PubMed.
- H. A. Hirsch, D. Iliopoulos, P. N. Tsichlis and K. Struhl, Cancer Res., 2009, 69, 7507–7511 CrossRef CAS PubMed.
- R. Erices, M. L. Bravo, P. Gonzalez, B. Oliva, D. Racordon, M. Garrido, C. Ibanez, S. Kato, J. Branes, J. Pizarro, M. I. Barriga, A. Barra, E. Bravo, C. Alonso, E. Bustamente, M. A. Cuello and G. I. Owen, Reprod. Sci., 2013, 20, 1433–1446 CrossRef PubMed.
- A. Leone, E. D. Gennaro, F. Bruzzese, A. Avallone and A. Budillon, Advances in Nutrition and Cancer, ed. V. Zappia, S. Panico, G. Luigi Russo, A. Budillon and F. D. Ragione, 2013, pp. 355–376 Search PubMed.
- F. Kratz, in Metal Complexes in Cancer Chemotherapy, ed. B. K. Keppler, VCH, Weinheim, Germany, 1993, pp. 391–429 Search PubMed.
- M. J. McKeage, Drug Saf., 1995, 13, 228–244 CrossRef CAS PubMed.
- G. Gomori, Proc. Soc. Exp. Biol. Med., 1946, 62, 33 CrossRef CAS PubMed.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233 RSC.
- R. A. Zelonka and M. C. Baird, J. Organomet. Chem., 1972, 35, C43 CrossRef CAS.
- J. Marmur, J. Mol. Biol., 1961, 3, 208 CrossRef CAS.
- APEX3, Program for Data Collection on Area Detectors, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI, USA, pp. 53711–5373 Search PubMed.
- SADABS and G. M. Sheldrick, Program for Absorption Correction of Area Detector Frames, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI, USA, pp. 53711–5373 Search PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed;
(c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed;
(d) XT, XS, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI, USA, pp. 53711–5373.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Alpha amylase, in Worthington Enzyme Manual, ed. V. Worthington, Worthington Biochemical Corp., Freehold, 1993, pp. 36–41 Search PubMed.
- G. P. A. H. Kasibhatla, D. Finucane, T. Brunner, E. B. Wetzel and D. R. Green, Protocol: Staining of suspension cells with Hoechst 33258 to detect apoptosis, Cell: A laboratory manual Culture and Biochemical Analysis of Cells, CSHL Press, Cold Spring Harbor, NY, 2000, vol. 1, p. 15.5 Search PubMed.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466 CrossRef CAS.
- J. E. Huheey, E. A. Keiter, R. L. Keiter and O. K. Medhu, Inorganic Chemistry, Principles of Structure and Reactivity, Pearson, Upper Saddle River, NJ, 2006, p. 425 Search PubMed.
-
(a) M. A. Bennett, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74 CrossRef CAS;
(b) S. Gunasekaran, R. K. Natarajan, V. Renganayaki and S. Natarajan, Indian J. Pure Appl. Phys., 2006, 44, 495–500 CAS.
- T. Khamrang, R. Kartikeyan, M. Velusamy, V. Rajendiran, R. Dhivya, B. Perumalsamy, M. A. Akbarsha and M. Palaniandavar, RSC Adv., 2016, 6, 114143–114158 RSC.
- J. Haribabu, K. Jeyalakshmi, Y. Arun, N. S. P. Bhuvanesh, P. T. Perumal and R. Karvembu, RSC Adv., 2015, 5, 46031–46049 RSC.
- S. K. Singh, M. Trivedi, M. Chandra, A. N. Sahay and D. S. Pandey, Inorg. Chem., 2004, 43, 8600 CrossRef CAS PubMed.
- V. Rajendiran, M. Murali, E. Suresh, S. Sinha, K. Somasundaram and M. Palaniandavar, Dalton Trans., 2008, 1, 148–163 RSC.
- V. Rajendiran, M. Murali, E. Suresh, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, Dalton Trans., 2008, 2157–2170 RSC.
- V. Rajendiran, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, J. Inorg. Biochem., 2010, 104, 217–220 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901–8911 CrossRef CAS.
- R. K. Gupta, A. Kumar, R. P. Paitandi, R. S. Singh, S. Mukhopadhyay, S. P. Verma, P. Das and D. S. Pandey, Dalton Trans., 2016, 45, 7163–7177 RSC.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS PubMed.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS PubMed.
- B. C. Baguley and M. LeBret, Biochemistry, 1984, 23, 937–943 CrossRef CAS PubMed.
- S. Richter, S. Singh, D. Draca, A. Kate, A. Kumbhar, A. S. Kumbhar, D. Maksimovic-Ivanic, S. Mijatovic, P. Lönnecke and E. Hey-Hawkins, Dalton
Trans., 2016, 45, 13114–13125 RSC.
- X. L. Hong, Z. H. Liang and M. H. Zeng, J. Coord. Chem., 2011, 64, 3792–4749 CrossRef CAS.
- O. Novakova, H. Chen, O. Vrana, A. Rodger, P. J. Sadler and V. Brabec, Biochemistry, 2003, 42, 11544–11554 CrossRef CAS PubMed.
- R. Loganathan, M. Ganeshpandian, N. S. P. Bhuvanesh, M. Palaniandavar, M. Amsaveni, S. K. Ghosh, A. Riyasdeen and M. A. Akbarsha, J. Inorg. Biochem., 2017, 174, 1–13 CrossRef CAS PubMed.
- C. Tan, J. Liu, H. Li, W. Zheng, S. Shi, L. Chen and L. Ji, J. Inorg. Biochem., 2008, 102, 347–358 CrossRef CAS PubMed.
- C. V. Dang, R. F. Ebert and W. R. Bell, J. Biol. Chem., 1985, 260, 9713 CAS.
- C. X. Wang, F. F. Yan, Y. X. Zhang and L. Ye, J. Photochem. Photobiol., A, 2007, 192, 23 CrossRef CAS.
- D. Senthil Raja, N. S. P. Bhuvanesh and K. Natarajan, Inorg. Chem., 2011, 50, 12852–12866 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 2nd edn, 1999 Search PubMed.
- B. K. Paul, A. Samanta and N. Guchhait, J. Phys. Chem. B, 2010, 114, 6183–6196 CrossRef CAS PubMed.
- B. Bhattacharya, S. Nakka, L. Guruprasad and A. Samanta, J. Phys. Chem. B, 2009, 113, 2143–2150 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- S. M. Page, S. R. Boss and P. D. Barker, Future Med. Chem., 2009, 1, 541–559 CrossRef CAS PubMed.
- T. Pieper and B. K. Keppler, Analusis, 1998, 26, M84 CrossRef CAS.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS PubMed.
- R. Rhabasa-Lhoret and J. L. Chiasson, Alpha-glucosidase inhibitors, in International Textbook of Diabetes Mellitus, ed. R. A. Defronzo, E. Ferrannini, H. Keen and P. Zimmet, John Wiley, UK, 3rd edn, 2004, vol. 1 Search PubMed.
- J. Kasznicki, A. Sliwinska and J. Drzewoski, Ann. Transl. Med., 2014, 6, 57 Search PubMed.
-
(a) Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, Weinheim, 2011, p. 151 Search PubMed;
(b) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed;
(c) Q. Wu, K. Zheng, S. Liao, Y. Ding, Y. Li and W. Mei, Organometallics, 2016, 35, 317–326 CrossRef CAS;
(d) Q. Wu, T. Chen, Z. Zhang, S. Liao, X. Wu, J. Wu, W. Mei, Y. Chen, W. Wu, L. Zeng and W. Zheng, Dalton Trans., 2014, 43, 9216–9225 RSC;
(e) Q. Wu, J. He, W. Mei, Z. Zhang, X. Wu and F. Sun, Metallomics, 2014, 6, 2204–2212 RSC.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrel, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- O. Novakova, J. Kasparkova, V. Bursova, C. Hofr, M. Vojtiskova, H. Chen, P. J. Sadler and V. Brabec, Chem. Biol., 2005, 12, 121–129 CrossRef CAS PubMed.
- S. W. Chang, A. R. Lewis, K. E. Prosser, J. R. Thompson, M. Gladkikh, M. B. Bally, J. J. Warren and C. J. Walsby, Inorg. Chem., 2016, 55, 4850–4863 CrossRef CAS PubMed.
- S. Khasibhatla, P. Gustavo, A. Mendes, D. Finucane, T. Brunner, E. Bossy-Wetzel and D. R. Green, Acridine orange/Ethidium Bromide (AO/EB) staining to detect apoptosis, Cold Spring Harbour Protocols, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1547296. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06514k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Rangasamy Loganathanb,
Nattamai S. P. Bhuvaneshc,
Xavier Janet Sabina
*a,
Rangasamy Loganathanb,
Nattamai S. P. Bhuvaneshc,
Xavier Janet Sabina