
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave spectroscopic detection of flame-sampled combustion intermediates
N. Hansen *a,
J. Wullenkordb,
D. A. Obenchainc,
I. Grafb,
K. Kohse-Höinghausb and
J.-U. Grabowc
*a,
J. Wullenkordb,
D. A. Obenchainc,
I. Grafb,
K. Kohse-Höinghausb and
J.-U. Grabowc
aCombustion Research Facility, Sandia National Laboratories, Livermore, CA 94551, USA. E-mail: nhansen@sandia.gov
bDepartment of Chemistry, Bielefeld University, D-33615 Bielefeld, Germany
cInstitut für Physikalische Chemie & Elektrochemie, Gottfried-Wilhelm-Leibniz-University Hannover, D-30167 Hannover, Germany
First published on 1st August 2017
Abstract
Microwave spectroscopy probes the rotational transitions of polar molecules in the gas phase and is a proven technique for the detection and identification of short-lived molecules produced from a variety of molecular sources. In this explorative study, we demonstrate that two prerequisites can be met for microwave spectroscopy to become a quantitative tool for the analysis of high-temperature gas mixtures as found in combustion environments. First, we show that the rotational temperature of the targeted species can be sufficiently cooled to allow for a sensitive detection of low-lying rotational states after sampling from hot (∼2200 K) flames. Second, we show that signal intensity profiles can be assembled which, after correcting for the different flame temperatures at various sampling positions, agree well with mole fraction profiles obtained via flame-sampling molecular-beam mass spectrometry. Based on the described results, it is conceivable that rotational spectroscopy can contribute towards the unraveling of complex, high-temperature reaction networks.
Introduction
The extraction of precise information of species identities and concentrations in the high-temperature environment of chemically reacting flows, as found in combustion conditions, is a fundamental physical chemistry issue that is commonly addressed with gas chromatography, laser spectroscopy, mass spectrometry, and combinations of these techniques. For current examples and reviews see ref. 1–6. The obtained experimental insights are vital for developing a detailed understanding of the underlying chemical reactions and their corresponding rates. Among the laboratory-scale combustion experiments,7–10 the premixed burner-stabilized low-pressure flame is one of the preferred test stations to identify and quantify many intermediates and products of radical–radical and radical–molecule reactions under controlled reacting flow conditions.11 For this type of reactor, flame-sampling molecular-beam mass spectrometry (MBMS) is often used as an analytical tool employing mainly electron ionization (EI), laser-based multi-photon ionization, or synchrotron-based single-photon ionization (PI) as ionization techniques.10–12 Especially PI measurements have now been recognized as a very precise and multiplexed tool, because they provide the mass-to-charge ratio of the sampled species' ion and the species' ionization energy simultaneously.13However, challenges and limitations remain and they include, for example, the following ones. Dissociative ionization, which can occur when ionizing flame intermediates with high-energy electrons or photons, might preclude a complete qualitative and quantitative interpretation of the sampled mass spectra.10,11 Also, small Franck–Condon overlaps might hamper an adequate formation and sensitive detection of the corresponding ion.14,15 Furthermore, similar ionization energies and photoionization efficiency curves may prevent isomer-specific assignments.10,11,14–16 It should be mentioned that this issue, which becomes especially challenging when the targeted systems are complex and the possible species numerous, is currently being addressed by developing isomer-resolving photoelectron–photoion coincidence techniques.17–19 Not less importantly, access to synchrotron facilities for PI measurements might be very competitive and limited.
With the aim to overcome these limitations and challenges, this work explores the applicability of microwave (MW) spectroscopy as an analytical tool for combustion chemistry diagnostics. MW spectroscopy measures the energies of transitions between rotational states of rotating polar molecules in the gas phase and has been continuously developed for decades to become a high-resolution spectroscopic technique in gas-phase physical chemistry.20–25 As of today, MW spectroscopy presents the most accurate method for the determination of molecular structures of polar gas-phase molecules. The highest resolution in MW spectroscopy is achieved in an experimental setup that applies the Fourier transform (FT) technique and combines a supersonic expansion of species in a molecular jet with a Fabry–Pérot cavity in a coaxial arrangement of the MW and molecular jet.24,25 Because of the cooling effect in the supersonic expansion, rotational temperatures of the order of 10 K are typically achieved, which result in the depopulation of rotationally excited states and consequently in an increase of the relative concentrations in the population of lower rotational states. As such, the strong cooling process allows for the sensitive detection of transitions between low-lying rotational states and a simplification of the broadband rotational spectrum.
For microwave spectroscopy to be suitable for combustion diagnostics, the challenge is that rotational temperatures of only 300–400 K are typically achieved in the molecular beams of flame-sampling experiments.15,26 Because of the resulting small population differences, these temperatures are likely too high to enable sensitive detection of flame constituents via rotational spectroscopy. In this explorative work however, we demonstrate that gases that are sampled from the hot environment (∼2200 K) of a model flame can actually be efficiently cooled rotationally so that a sensitive detection of polar flame constituents becomes possible via high-resolution jet FTMW spectroscopy.
Experimental details
The experiments were performed at the Leibniz-University Hannover (Germany) using the existing high-resolution Fourier-transform microwave (FTMW) spectrometer utilizing the coaxially oriented beam-resonator arrangement (COBRA).27 This spectrometer is well suited for the experiments described here because it provides high sensitivity and resolution (ν/Δν ∼ 106) in the range from 2 to 26.5 GHz in which the pure rotational transitions of many combustion intermediates are located.The setup of the microwave spectrometer, which has been described in detail in the literature,27 has been modified in order to allow for flame-sampling experiments. The modified experimental setup is shown schematically in Fig. 1 and the experimental details are as follows: gases from two different low-pressure flames were sampled via a quartz sampling probe and transferred into a fast mixing nozzle28 where they were mixed with Ar and supersonically expanded into high vacuum along the axis of a Fabry–Pérot resonator. Microwave pulses of 1.0 μs length were used to polarize the sampled gases for optimized emission signals. Typical values for the MW power would be on the order of a fraction of a mW for species exhibiting a dipole moment of several tenth of a Debye. The resulting free-induction decay (FID) was subsequently digitized in the time domain and Fourier transformed to the frequency domain. In the spectra shown below, the molecular signal is represented as its amplitude spectrum.
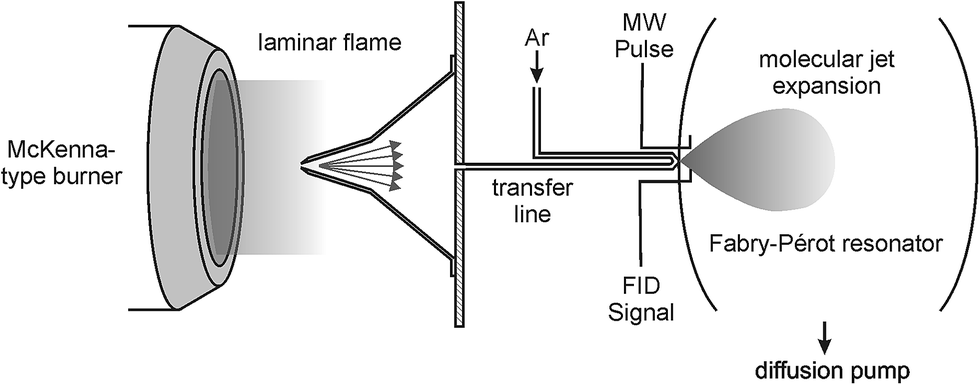 | ||
| Fig. 1 Schematic representation of the experimental setup. Gases from the low-pressure flames were sampled through a quartz probe and coupled into a fast mixing nozzle forming a molecular jet. | ||
The flames used in this exploratory study were a fuel-rich (ϕ = 1.7) C2H4/O2 flame and a fuel-rich (ϕ = 2.0) dimethyl ether (DME)/O2 flame. Both flames contained 25% Ar in the unburnt cold-gas mixture and were stabilized at a reduced pressure of p = 40 mbar (30 Torr) on a water-cooled homebuilt burner with a sintered bronze matrix (diameter of d = 65 mm). The total cold-gas flow rates were 4.0 and 4.1 standard liters per minute (slm; 273.15 K, 1 bar), respectively.
We have unambiguously identified the intermediates formaldehyde (CH2O), ketene (CH2CO), and acetaldehyde (CH3CHO) via well-established rotational frequencies. Specifically, we used the 21,1–21,2 transition at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 488.479 MHz for the detection and identification of formaldehyde,29 the 10,1–00,0 transition at 20209.201 MHz for ketene,30 and the 10,1–00,0 transition at 19262.140 MHz for acetaldehyde.31 Although not further discussed here, the fuel DME was traced using the 20,2–11,1 transition in the torsional ground states at 9118.816 MHz.32
488.479 MHz for the detection and identification of formaldehyde,29 the 10,1–00,0 transition at 20209.201 MHz for ketene,30 and the 10,1–00,0 transition at 19262.140 MHz for acetaldehyde.31 Although not further discussed here, the fuel DME was traced using the 20,2–11,1 transition in the torsional ground states at 9118.816 MHz.32
To probe the signal intensity as function of sampling position, i.e. different flame temperatures, the horizontally mounted burner assembly was moved towards or away from the quartz probe using a high-precision stepper motor. In order to verify the applicability of microwave spectroscopy to deliver quantitative information, the acquired data was then compared with mole fraction profiles obtained from accompanying flame-sampling molecular-beam mass spectrometry experiments using the instrument at Bielefeld University and analysis procedures described in ref. 33.
The flame temperatures at 20 mm away from the burner surface were determined to be (2264 ± 50) K and (2528 ± 50) K for the DME and ethylene flame, respectively, using planar laser induced fluorescence measurement of the OH rational temperature.34 The entire temperature profiles were subsequently obtained from the sampling function and the first-stage pressure in the mass spectrometry experiments after calibrating the post-flame temperature at 20 mm with the results of the laser measurements.35
Results and discussion
In this work, we demonstrate that gases that are sampled from the hot environment (∼2200 K) of a model flame can be efficiently cooled rotationally so that a sensitive detection of polar flame constituents becomes possible via high-resolution jet FTMW spectroscopy. Examples of rotational transitions of flame-sampled formaldehyde (CH2O), ketene (CH2CO), and acetaldehyde (CH3CHO) are shown in Fig. 2. For these spectra, formaldehyde and ketene were sampled from an ethylene flame at a height above the burner (HAB) of 1.25 and 0.95 mm, respectively, and the acetaldehyde intermediate was sampled from a flame fueled by DME at a HAB of 0.95 mm. The HAB values have been adjusted by 0.25 mm as described below in order to match the accompanying mass spectrometry experiments. As indicated by the vertical lines in Fig. 2, the flame-sampled spectra are in excellent agreement with the expected spectral positions.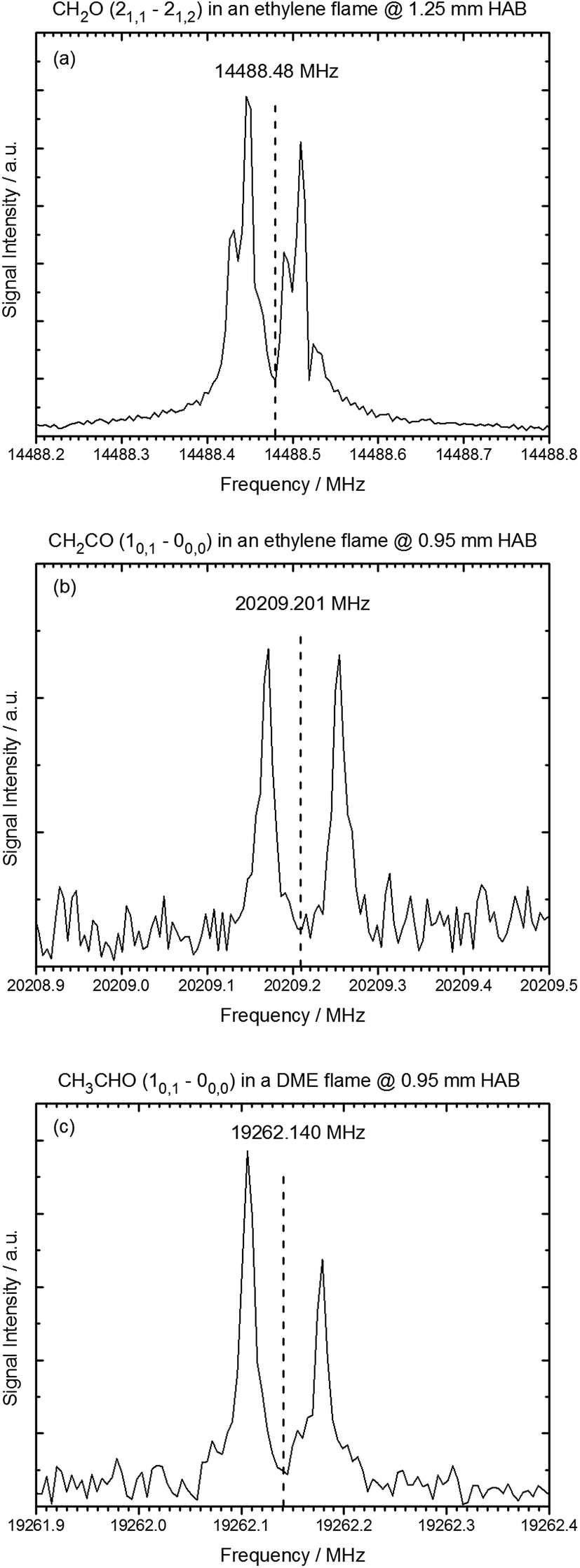 | ||
| Fig. 2 Rotational spectra of oxygenated intermediates after sampling from the ethylene and DME flame. HAB (height above burner) refers to the sampling position. (a) The 21,1–21,2 rotational transition of formaldehyde sampled from the ethylene flame at HAB = 1.25 mm. (b) The 10,1–00,0 rotational transition of ketene sampled from the ethylene flame at HAB = 0.95 mm. (c) The 10,1–00,0 rotational transition of acetaldehyde in the DME flame at 0.95 mm above the burner surface. Vertical lines indicate the reference frequencies from literature for these transitions.30–32 | ||
In the spectra, the resonance frequencies appear as a Doppler doublet because of the coaxial orientation of the jet and the propagation direction of the electromagnetic field. Hyperfine splittings were observed in the spectra of formaldehyde due to 1H nuclear–spin nuclear–spin interactions.36,37 Such hyperfine effects and other splittings in the rotational transitions, for example caused by nuclear quadrupole effects, internal rotation, or spin rotation interactions, can in general be used to unambiguously identify chemical compounds.
Especially with the advent of chirp FTMW spectroscopy,20,38 several efforts have been undertaken to establish rotational spectroscopy as an analytical tool to quantitatively probe chemical dynamics and kinetics.39–42 For MW spectroscopy to be applicable for quantitative combustion diagnostics it would be preferable if the temperature of the sampled gases does not influence the rotational temperature in the molecular jet, thus resulting in the same population differences and hence in the same nominal signal intensities. It should be kept in mind however, that the signal intensity in the MW spectra will depend on the flame temperature because of the temperature-dependent sampling rate through the quartz probe as described below.
It is shown next, that the same population differences can be achieved and that signal intensity from the MW spectra can easily be compared to mole fraction profiles. For this, we first followed the intensity of the rotational transitions of CH2O, CH2CO, and CH3CHO as gas samples were drawn from different positions (i.e., temperatures) in the flames. Exemplary results are shown in Fig. 3 for the 21,1–21,2 rotational transition of CH2O following sampling from the DME flame between 1085 and 2168 K (3.75–5.25 mm HAB). Although it is obvious from Fig. 3 that the signal intensity weakens when sampling further away from the burner surface, i.e., at higher temperatures, it is shown in the second step that this effect is unrelated to the efficiency of the cooling process in the supersonic jet and instead, can be traced back to the mole fraction profile and sampling rate through the quartz probe.
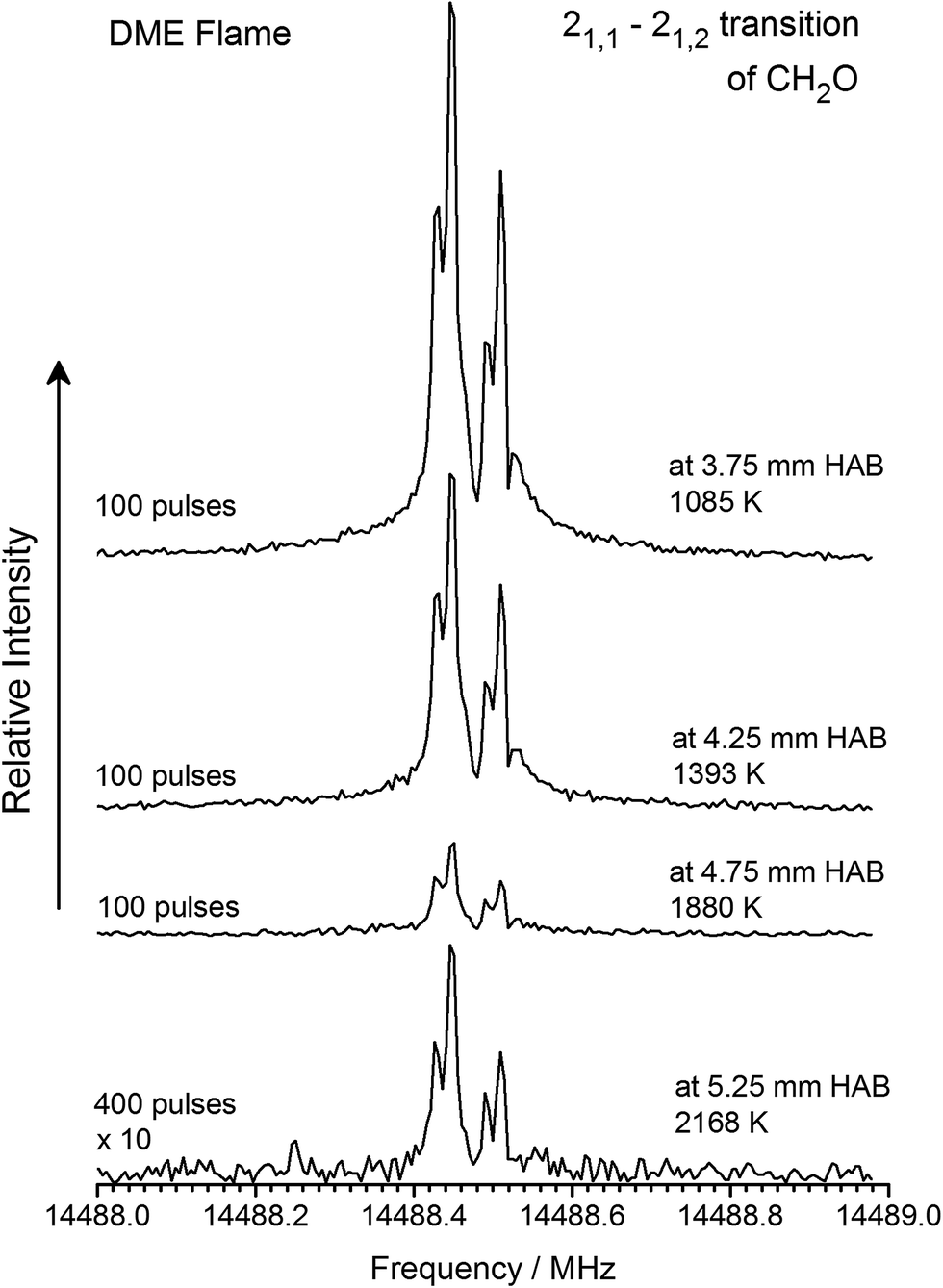 | ||
| Fig. 3 Signal intensity of the 21,1–21,2 rotational transition of CH2O as function of different sampling positions, i.e., temperatures, of the gases sampled. | ||
According to well-established relationships for flame-sampling experiments, the signal (Si) is proportional to the mole fraction of the sampled species xi and a sampling function FKT that is proportional to 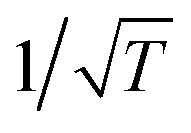 with T being the flame temperature at the sampling position:33,35
with T being the flame temperature at the sampling position:33,35
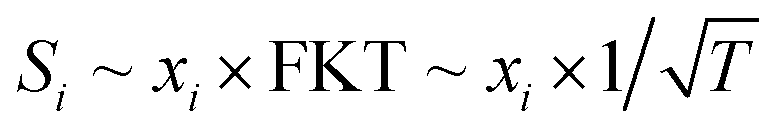 | (1) |
or
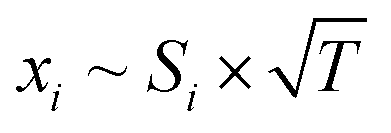 | (2) |
Following this procedure, we assembled the data to provide intensity profiles as function of height above the burner surface and compared the results with quantitative measurements from accompanying (but independent) flame-sampling molecular-beam mass spectrometry experiments. After correcting for the different temperatures at the various sampling positions according to eqn (2), jet FTMW and MBMS experiments compare quite favorable as shown in Fig. 4 for the profiles of formaldehyde and acetaldehyde in the DME and ethylene flames. As mentioned above, we have moved the FTMW profiles 0.25 mm further away from the burner surface to achieve this agreement. This shift is well within the combined measurement uncertainty of the zero-point determinations in both experiments.
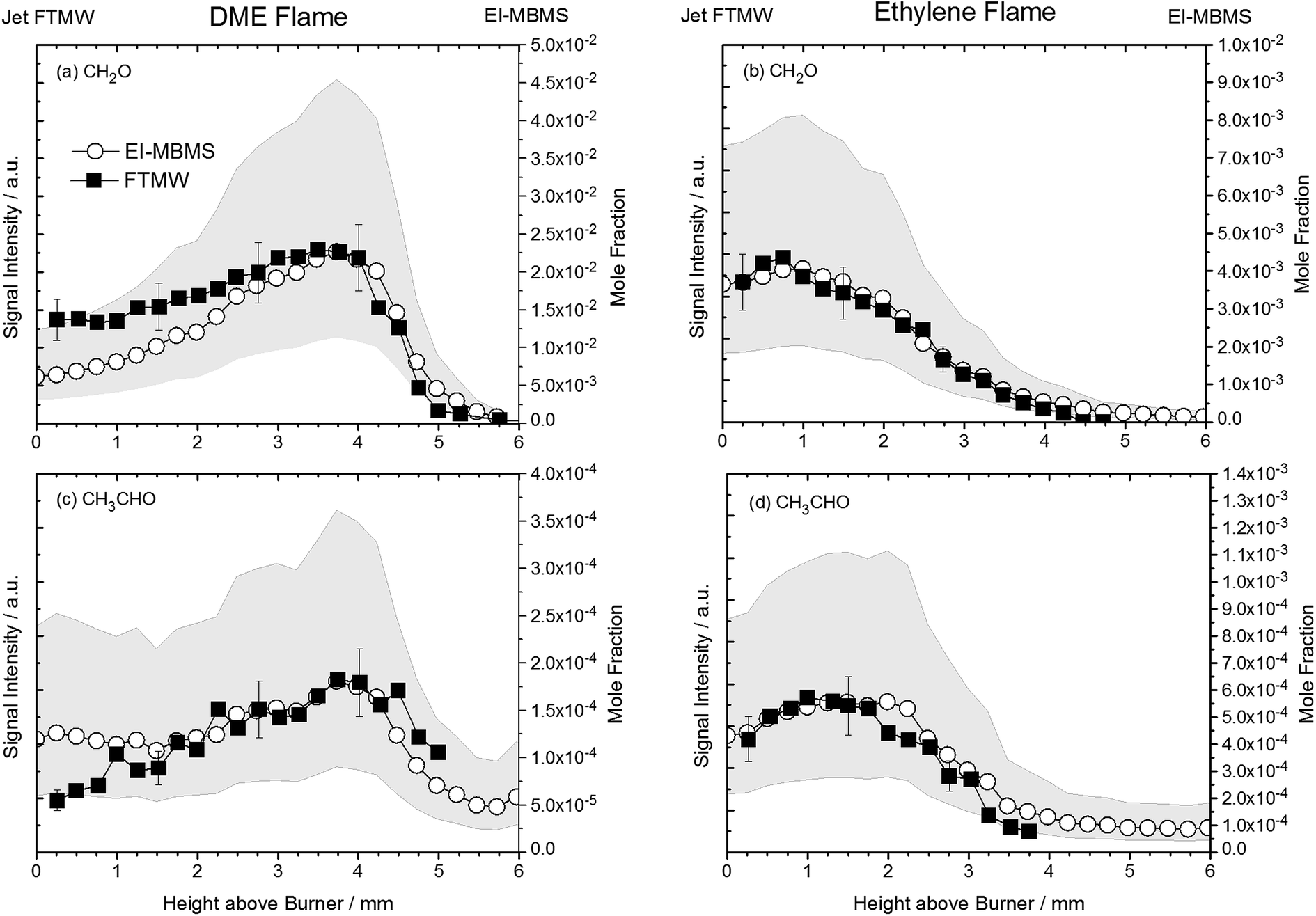 | ||
| Fig. 4 Comparison of temperature-corrected signal intensity profiles of the 21,1–21,2 rotational transition of CH2O and 10,1–00,0 of CH3CHO in the DME and ethylene flame with independent EI-MBMS measurements performed at Bielefeld University. Closed symbols are jet FTMW data, open symbols represent EI-MBMS data. Uncertainties of the EI-MBMS experiments are visualized as grey bands, uncertainties of the FTMW data are indicated as error bars. | ||
The uncertainty of quantitative flame-sampling experiments followed by mass spectrometry and the issues arising from comparing flame data obtained from different experiments set-ups have been addressed in ref. 10 and typically mole fraction profiles for intermediate species are determined to be accurate within a factor of two. These uncertainties for the EI-MBMS experiments are shown as grey bands in Fig. 4. For a meaningful comparison of the profiles in Fig. 4, pulse-to-pulse fluctuations in the MW spectroscopy experiments have to be considered. The reproducibility of the sample from the fast mixing nozzle may be the biggest factor but also the quality of the resonator, i.e. the mirror position may change during a so-called burner scan. Given these factors and the averaging time, we assume the signal intensity in the current experiments to be repeatable within 20%. These uncertainties are also included in Fig. 4. With this in mind, the agreement between the EI-MBMS and FTMW profiles can be considered very good, and it shows first that the rotational temperature in the molecular jet beam is indeed independent of the flame temperature at the sampling position and therefore the data can be converted into concentration profiles as function of height above the burner. And secondly, this agreement also implies a linear relationship between concentration and signal intensity.
We do not provide quantitative mole fraction profiles in this explorative study; however, the ultimate goal should be to perform quantitative experiments that can be used to provide validation targets for model purposes. With peak mole fractions of 2.3 × 10−2 (DME flame) and 4.1 × 10−3 (ethylene flame) determined for CH2O and 1.8 × 10−4 (DME flame) and 5.6 × 10−4 (ethylene flame) for CH3CHO from the EI-MBMS measurements, we estimate the detection limit of the current experimental setup to be of the order of ∼50 ppm, based on formaldehyde's dipole moment of 2.3 D. As a perspective, it should be kept in mind that the signal is linearly dependent on the molecule's dipole moment and consequently, larger detection limits (lower sensitivities) can be expected for molecules that have a weaker dipole moment. For this reason, the current work only focuses on oxygenated species (CH2O, CH2CO, and CH3CHO), because these species exhibit larger dipole moments compared to hydrocarbon intermediates from combustion processes.
A determination of the rotational temperature in the molecular jet was not possible, because in this explorative work we only traced low-mass molecules and only a few transitions were thus accessible given the frequency range of the existing MW spectrometer. The rotational temperature in the molecular jet is expected to be similar to other molecular jet experiments, i.e., of the order of ≤10 K. This estimate is based on the gas temperature before the expansion of 350–400 K as determined from the Doppler splittings and earlier measurements in flame-sampling experiments.26 Under these conditions, the expansion cooling should be comparable to complementary DC discharge and laser ablation experiments, in which routinely these low rotational temperatures are achieved.43,44 The fact that the sampled molecules before the molecular jet expansion are equilibrated at temperatures below 400 K also suggests that large populations of vibrationally excited states are not expected in the molecular jet, especially considering the low-mass molecules monitored in this study.
The achievement of such efficient cooling of the rotational temperature opens new avenues for combustion chemistry research and for FTMW spectroscopy to be developed into a broadly applicable diagnostic tool not just for general gas-phase physical chemistry problems39–42 but also for flame diagnostics, despite the high flame temperatures. Nevertheless, it should be kept in mind that protocols for quantitative applications still need to be developed for MW spectroscopy to be widely applicable to combustion systems. It is worth mentioning that Patterson and Doyle have used a He cell to cool molecules from a high flux room temperature beam to 8 K;45 a technique that might be useful for combustion research as well.
Conclusions
In this paper we demonstrate that molecular samples drawn from the hot environment of laboratory-scale model flames can be sufficiently cooled using a fast-mixing nozzle to allow for sensitive detection using microwave spectroscopy. The findings presented here show that microwave spectroscopy can be developed into a new diagnostic tool for combustion research and thus they open up an entire new field of research.Despite the fact that a permanent molecular dipole moment is needed in rotational spectroscopy, the FTMW diagnostic technique does not lack combustion targets. For example, in biofuel combustion46–48 as well as in low-temperature oxidation chemistry,49 many oxygenated intermediates are formed with their identity and formation pathways not necessarily well understood.50,51
Although the potential of this technique is already evident regarding its superb identification capacity, optimizing the coupling of the FTMW instrument with the flame and of the sampling conditions by measures such as eliminating the transfer line and aligning the flame coaxially with the resonator axis might provide at least an order of magnitude sensitivity increase over these first demonstration experiments with existing equipment. Combination of the broad-band chirp FTMW technology20,38 with various reactor sources shows promise for future work for observing previously undetected and identifying short-lived combustion intermediates.
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. The Bielefeld group acknowledges partial support by Deutsche Forschungsgemeinschaft (DFG) under contract KO 1363/31-1. The Hannover group is grateful for financial support from the DFG and the Land Niedersachsen.References
- F. Battin-Leclerc, A. Rodriguez, B. Husson, O. Herbinet, P. A. Glaude, Z. D. Wang, Z. J. Cheng and F. Qi, J. Phys. Chem. A, 2014, 118, 673–683 CrossRef CAS PubMed.
- C. Togbé, J. B. May-Carle, G. Dayma and P. Dagaut, J. Phys. Chem. A, 2010, 114, 3896–3908 CrossRef PubMed.
- A. Bohlin and C. J. Kliewer, J. Phys. Chem. Lett., 2014, 5, 1243–1248 CrossRef CAS PubMed.
- K. Kohse-Höinghaus, R. S. Barlow, M. Aldén and J. Wolfrum, Proc. Combust. Inst., 2005, 30, 89–123 CrossRef.
- T. A. Cool, K. Nakajima, T. A. Mostefaoui, F. Qi, A. McIlroy, P. R. Westmoreland, M. E. Law, L. Poisson, D. S. Peterka and M. Ahmed, J. Chem. Phys., 2003, 119, 8356–8365 CrossRef CAS.
- C. A. Taatjes, N. Hansen, D. L. Osborn, K. Kohse-Höinghaus, T. A. Cool and P. R. Westmoreland, Phys. Chem. Chem. Phys., 2008, 10, 20–34 RSC.
- R. K. Hanson and D. F. Davidson, Prog. Energy Combust. Sci., 2014, 44, 103–114 CrossRef.
- C.-J. Sung and H. J. Curran, Prog. Energy Combust. Sci., 2014, 44, 1–18 CrossRef.
- F. L. Dryer, F. M. Haas, J. Santner, T. I. Farouk and M. Chaos, Prog. Energy Combust. Sci., 2014, 44, 19–39 CrossRef.
- F. N. Egolfopoulos, N. Hansen, Y. Ju, K. Kohse-Höinghaus, C. K. Law and F. Qi, Prog. Energy Combust. Sci., 2014, 43, 36–67 CrossRef.
- N. Hansen, T. A. Cool, P. R. Westmoreland and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2009, 35, 168–191 CrossRef CAS.
- Y. Y. Li and F. Qi, Acc. Chem. Res., 2010, 43, 68–78 CrossRef CAS PubMed.
- S. R. Leone, M. Ahmed and K. R. Wilson, Phys. Chem. Chem. Phys., 2010, 12, 6564–6578 RSC.
- N. Hansen, S. J. Klippenstein, J. A. Miller, J. Wang, T. A. Cool, M. E. Law, P. R. Westmoreland, T. Kasper and K. Kohse-Höinghaus, J. Phys. Chem. A, 2006, 110, 4376–4388 CrossRef CAS PubMed.
- N. Hansen, S. J. Klippenstein, C. A. Taatjes, J. A. Miller, J. Wang, T. A. Cool, B. Yang, R. Yang, L. X. Wei, C. Q. Huang, F. Qi, M. E. Law and P. R. Westmoreland, J. Phys. Chem. A, 2006, 110, 3670–3678 CrossRef CAS PubMed.
- N. Hansen, S. J. Klippenstein, P. R. Westmoreland, T. Kasper, K. Kohse-Höinghaus, J. Wang and T. A. Cool, Phys. Chem. Chem. Phys., 2008, 10, 366–374 RSC.
- T. Baer and R. P. Tuckett, Phys. Chem. Chem. Phys., 2017, 19, 9698–9723 RSC.
- A. Bodi, P. Hemberger, D. L. Osborn and B. Sztaray, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS.
- J. Krüger, G. A. Garcia, D. Felsmann, K. Moshammer, A. Lackner, A. Brockhinke, L. Nahon and K. Kohse-Höinghaus, Phys. Chem. Chem. Phys., 2014, 16, 22791–22804 RSC.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, Rev. Sci. Instrum., 2008, 79, 053103 CrossRef PubMed.
- H. Dreizler, Ber. Bunsen-Ges., 1995, 99, 1451–1461 CrossRef CAS.
- G. B. Park and R. W. Field, J. Chem. Phys., 2016, 144, 200901 CrossRef PubMed.
- Y. J. Xu, J. Van Wijngaarden and W. Jäger, Int. Rev. Phys. Chem., 2005, 24, 301–338 CrossRef CAS.
- T. J. Balle and W. H. Flygare, Rev. Sci. Instrum., 1981, 52, 33–45 CrossRef CAS.
- J. U. Grabow, W. Stahl and H. Dreizler, Rev. Sci. Instrum., 1996, 67, 4072–4084 CrossRef CAS.
- M. Kamphus, N. N. Liu, B. Atakan, F. Qi and A. McIlroy, Proc. Combust. Inst., 2002, 29, 2627–2633 CrossRef CAS.
- M. Schnell, D. Banser and J.-U. Grabow, Rev. Sci. Instrum., 2004, 75, 2111–2115 CrossRef CAS.
- T. Emilsson, T. D. Klots, R. S. Ruoff and H. S. Gutowsky, J. Chem. Phys., 1990, 93, 6971–6976 CrossRef CAS.
- R. Bocquet, J. Demaison, L. Poteau, M. Liedtke, S. Belov, K. M. T. Yamada, G. Winnewisser, C. Gerke, J. Gripp and T. Köhler, J. Mol. Spectrosc., 1996, 177, 154–159 CrossRef CAS.
- R. D. Brown, P. D. Godfrey, D. McNaughton, A. P. Pierlot and W. H. Taylor, J. Mol. Spectrosc., 1990, 140, 340–352 CrossRef CAS.
- I. Kleiner, F. J. Lovas and M. Godefroid, J. Phys. Chem. Ref. Data, 1996, 25, 1113–1210 CrossRef CAS.
- F. J. Lovas, H. Lutz and H. Dreizler, J. Phys. Chem. Ref. Data, 1979, 8, 1051–1108 CrossRef CAS.
- P. Oßwald, K. Kohse-Höinghaus, U. Struckmeier, T. Zeuch, L. Seidel, L. Leon and F. Mauss, Z. Phys. Chem., 2011, 225, 1029–1054 CrossRef.
- D. Felsmann, H. Zhao, Q. Wang, I. Graf, T. Tan, X. Yang, E. A. Carter, Y. Ju and K. Kohse-Höinghaus, Proc. Combust. Inst., 2017, 36, 543–551 CrossRef CAS.
- U. Struckmeier, P. Oßwald, T. Kasper, L. Böhling, M. Heusing, M. Köhler, A. Brockhinke and K. Kohse-Höinghaus, Z. Phys. Chem., 2009, 223, 503–537 CrossRef CAS.
- A. Okaya, J. Phys. Soc. Jpn., 1956, 11, 258–263 CrossRef CAS.
- P. Thaddeus, L. C. Krisher and J. H. N. Loubser, J. Chem. Phys., 1964, 40, 257–273 CrossRef CAS.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer and B. H. Pate, J. Mol. Spectrosc., 2006, 238, 200–212 CrossRef CAS.
- C. Abeysekera, B. Joalland, N. Ariyasingha, L. N. Zack, I. R. Sims, R. W. Field and A. G. Suits, J. Phys. Chem. Lett., 2015, 6, 1599–1604 CrossRef CAS PubMed.
- C. Abeysekera, L. N. Zack, G. B. Park, B. Joalland, J. M. Oldham, K. Prozument, N. M. Ariyasingha, I. R. Sims, R. W. Field and A. G. Suits, J. Chem. Phys., 2014, 141, 214203 CrossRef PubMed.
- J. M. Oldham, C. Abeysekera, B. Joalland, L. N. Zack, K. Prozument, I. R. Sims, G. B. Park, R. W. Field and A. G. Suits, J. Chem. Phys., 2014, 141, 154202 CrossRef PubMed.
- K. Prozument, Y. V. Suleimanov, B. Buesser, J. M. Oldham, W. H. Green, A. G. Suits and R. W. Field, J. Phys. Chem. Lett., 2014, 5, 3641–3648 CrossRef CAS PubMed.
- D. P. Zaleski, D. P. Tew, N. R. Walker and A. C. Legon, J. Phys. Chem. A, 2015, 119, 2919–2925 CrossRef CAS PubMed.
- C. Karunatilaka, A. J. Shirar, G. L. Storck, K. M. Hotopp, E. B. Biddle, R. Crawley and B. C. Dian, J. Phys. Chem. Lett., 2010, 1, 1547–1551 CrossRef CAS.
- D. Patterson and J. M. Doyle, Mol. Phys., 2012, 110, 1757–1766 CrossRef CAS.
- K. Kohse-Höinghaus, P. Oßwald, T. A. Cool, T. Kasper, N. Hansen, F. Qi, C. K. Westbrook and P. R. Westmoreland, Angew. Chem., Int. Ed., 2010, 49, 3572–3597 CrossRef PubMed.
- S. M. Sarathy, P. Oßwald, N. Hansen and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2014, 44, 40–102 CrossRef.
- W. Leitner, J. Klankermayer, S. Pischinger, H. Pitsch and K. Kohse-Höinghaus, Angew. Chem., Int. Ed., 2017, 56, 5412–5452 CrossRef CAS PubMed.
- J. Zádor, C. A. Taatjes and R. X. Fernandes, Prog. Energy Combust. Sci., 2011, 37, 371–421 CrossRef.
- K. Moshammer, A. W. Jasper, D. M. Popolan-Vaida, Z. D. Wang, V. S. B. Shankar, L. Ruwe, C. A. Taatjes, P. Dagaut and N. Hansen, J. Phys. Chem. A, 2016, 120, 7890–7901 CrossRef CAS PubMed.
- Z. D. Wang, L. D. Zhang, K. Moshammer, D. M. Popolan-Vaida, V. S. B. Shankar, A. Lucassen, C. Hemken, C. A. Taatjes, S. R. Leone, K. Kohse-Höinghaus, N. Hansen, P. Dagaut and S. M. Sarathy, Combust. Flame, 2016, 164, 386–396 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |