
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted isolation of sulfur-containing metabolites from Lsr2-deletion mutant strain of Streptomyces roseosporus†
Lina Denga,
Rui Wanga,
Guowei Wang*a,
Mingxu Liua,
Guojian Liaoa,
Zhihua Liaob and
Min Chen *a
*a
aCollege of Pharmaceutical Sciences, Key Laboratory of Luminescent and Real-Time Analytical Chemistry, Ministry of Education, Southwest University, Chongqing 400715, P. R. China. E-mail: mminchen@swu.edu.cn; wangguowei1987@163.com
bSchool of Life Sciences, Southwest University, Chongqing 400715, P. R. China
First published on 1st August 2017
Abstract
Deletion of the Lsr2 gene in Streptomyces roseosporus up-regulated silent gene clusters and produced new secondary metabolites. An ultra-performance liquid chromatography quadrupole time of flight mass spectrometry (UPLC-QTOF-MS/MS) method was used to analyze metabolites of the mutant and wild-type strains, and recognize previously unreported sulfur-containing compounds based on their molecular formulas and fragmentation ions. The targeted isolation of unidentified compounds afforded six new sulfur-containing compounds, pyrismycins A–F (1–6), together with seven known analogues 7–13. Their cytotoxic effects were evaluated using four clinically relevant human cancer cell lines, gastric carcinoma SGC7901, breast carcinoma MDA-MB-231, lung carcinoma A549 and hepatocellular carcinoma HepG2. Compound 7 exhibited the most potent cytotoxicity with IC50 values of 1.7, 5.8 and 6.3 μM against the SGC7901, HepG2 and MDA-MB-231, respectively.
Introduction
Streptomyces produce a wealth of chemically diverse secondary metabolites with a wide range of biological activities. These have been used as antibacterial, antifungal, anticancer, antiparasitic and immunosuppressive agents.1 Sulfur-containing metabolites are common across the Streptomyces kingdom and constitute a family of natural products which play an important role in the health care for humans.2,3 Among the top 200 best-selling drugs in the current drug market, about 86% of them are sulfur and/or nitrogen containing compounds.4Microbial genomics and bioinformatics analysis has revealed that many microorganisms have far greater potential to produce specialized metabolites than have been discovered by classic screening-based methods. Bioinformatics analysis of the genome of S. roseosporus disclosed 25 gene clusters, which include 14 NRPS or PKSs, three for terpene biosynthesis, three for siderophore, two for bacteriocin, one ectoine, and two other gene clusters.5 Relative to the limited number of secondary metabolites isolated from S. roseosporus, many gene clusters in this bacteria seemingly are not expressed, implying that there is additional metabolic potential in this species.
A number of approaches have been developed to activate and up-regulate silent or cryptic gene clusters to produce new compounds.6,7 Lsr2 is a small nucleoid-associated protein present in S. roseosporus that regulates the expression of many biosynthetic gene cluster and influences the organization of bacterial chromatin, but examples of influence the metabolites are less common.8,9 Thus, we speculated that deletion of the Lsr2 in S. roseosporus might active or repress gene expression and produce novel compounds.
Herein, we report the isolation and structure elucidation of six new sulfur-containing compounds, pyrismycins A–F (1–6), with seven known analogues (7–13), from the Lsr2-deletion (ΔSrlsr2) mutant strain of S. roseosporus. In this research, UPLC-QTOF-MS/MS, a successful tool for dereplication purposes,10 was used to identify sulfur-containing metabolites from the ΔSrlsr2 strain, with present in the wild-type (WT) strain, and then used to guide the isolation work. The cytotoxic effects of isolated compounds on four clinically relevant human cancer cell lines, SGC7901 gastric carcinoma, MDA-MB-231 breast carcinoma, A549 lung carcinoma and HepG2 hepatocellular carcinoma, were evaluated (Fig. 1).
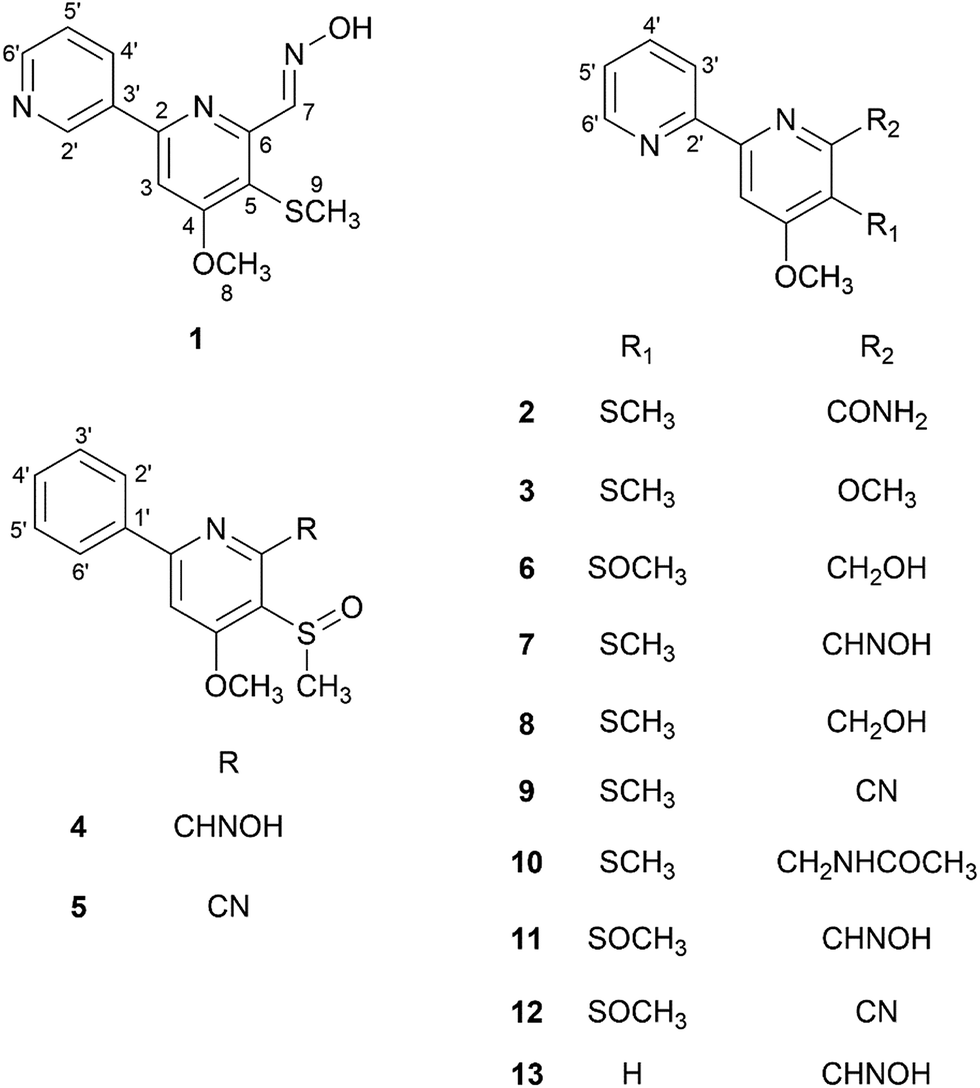 | ||
| Fig. 1 Chemical structures of compounds 1–13. | ||
Results and discussion
UPLC-QTOF-MS analysis revealed 22 new peaks in the ethyl acetate extract of ΔSrlsr2 than were present in a similar extract of the WT strain (Fig. 2). This result indicated that deletion of the Lsr2 gene increased the number of secondary metabolites produced by this mutant strain. 14 peaks were identified as potential sulfur-containing derivatives, as supported by the molecular formula and fragment ions, and compared with the Streptomyces compounds previously indexed in the SciFinder database (Table S1†).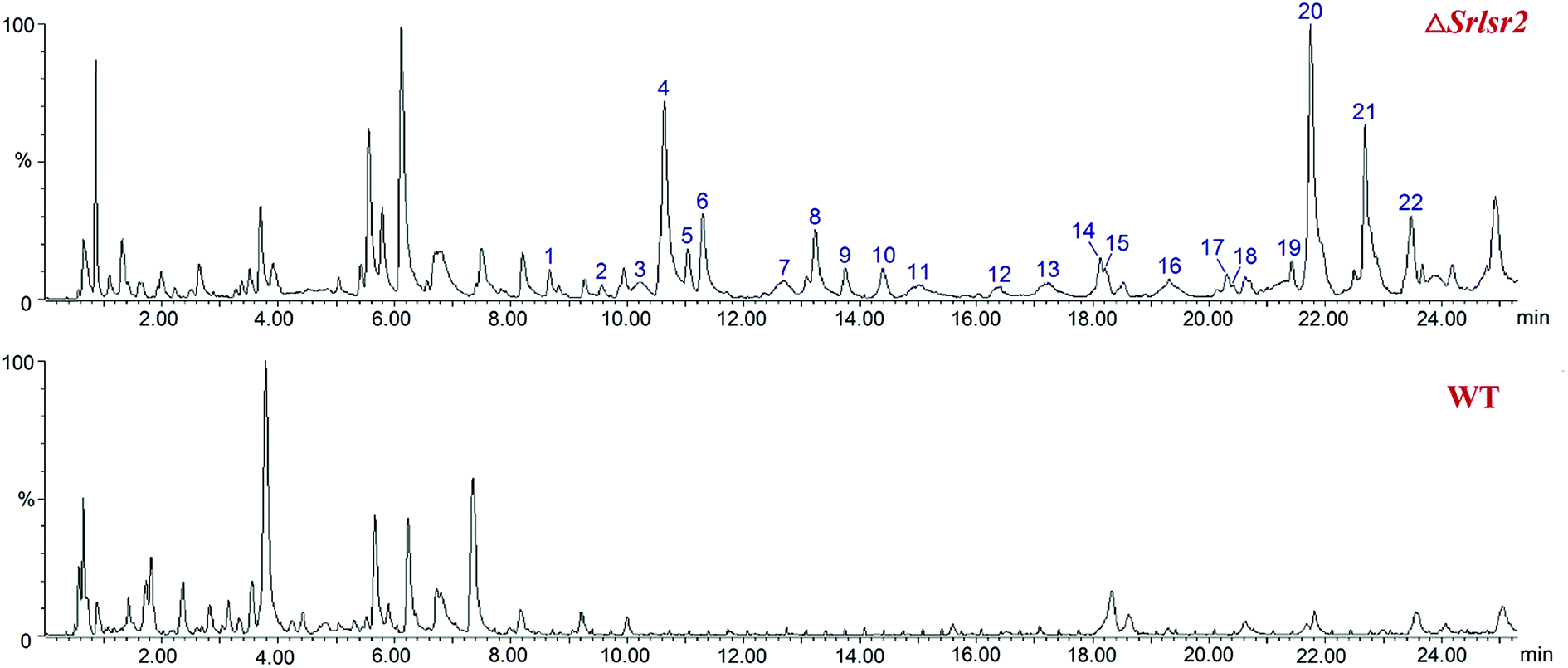 | ||
| Fig. 2 UPLC-QTOF-MS profiles of the acetate extracts from ΔSrlsr2 (the upper diagram) and WT (the lower diagram) strains of S. roseosporus. | ||
Peak 9 (tR = 13.75 min) showed molecular ions at m/z 291.0794 [M + H]+ and its molecular formula was determined to be C14H14N2O3S (Fig. 3). Tandem mass showed that the fragmentation of [M + H]+ precursor ion generated [M − OH]+ ion (m/z 273.0695), [M − OH − CH2]+ ion (m/z 259.0539), [M − OH − OCH3]+ ion (m/z 243.0593) and fragment at m/z 229.0446. The fragment ion m/z 229.0446 further fragmented into fragment ion m/z 204.0485 and 181.0759. The transition from fragment ion m/z 156.0802 to 130.0660 indicated that it was a phenylpyridine derivative.11,12 Such a molecular formula and the basic structure units have not been found previously in Streptomyces metabolites. Similar fragmentation was also observed for peak 15. Thus, both peaks 9 and 15 were targeted as potentially new metabolites.
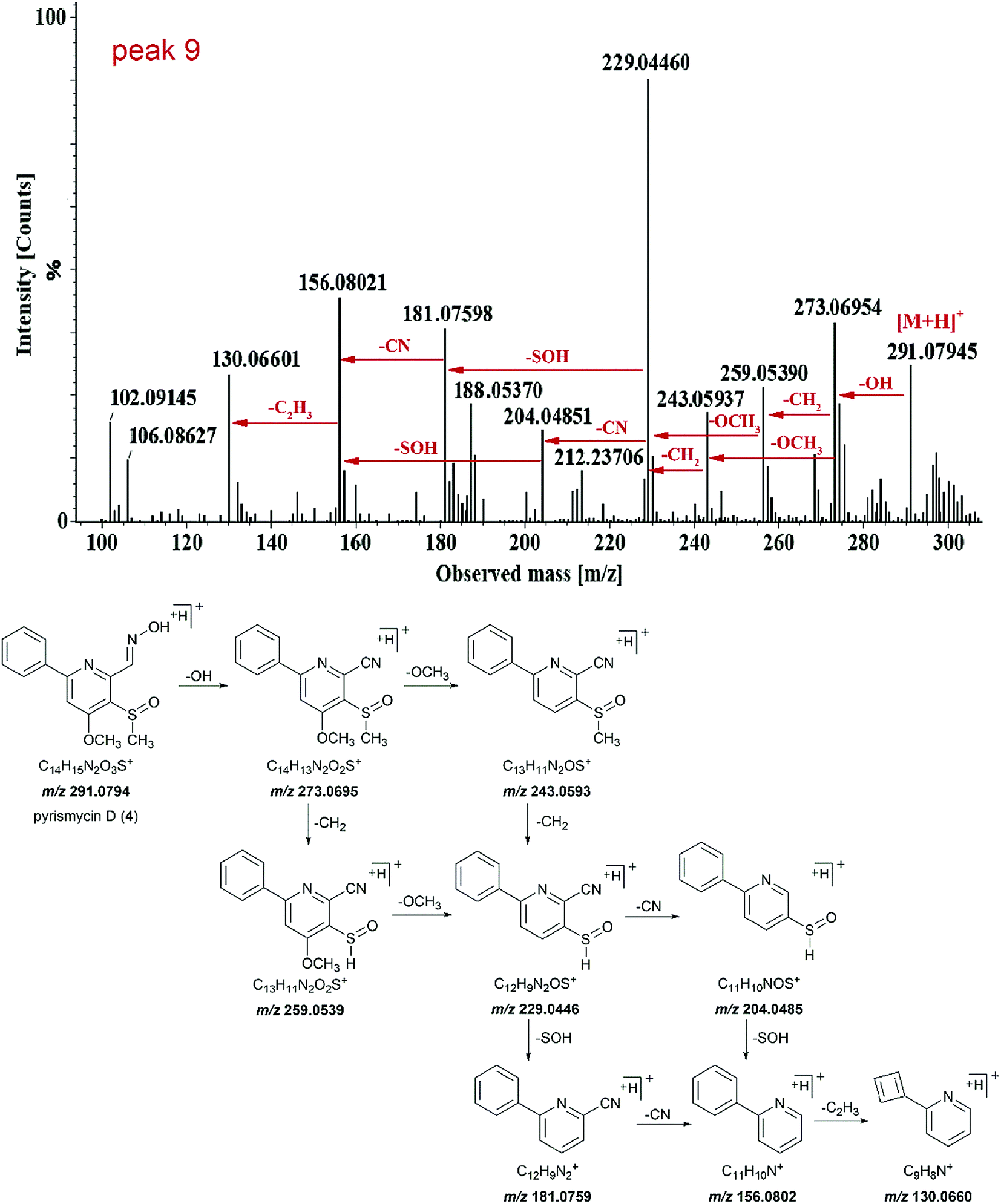 | ||
| Fig. 3 HR-MS/MS spectra and proposed fragmentation pathway of peak 9 (4). | ||
Peak 17 (tR = 20.31 min), peak 18 (tR = 20.43 min) and peak 19 (tR = 21.44 min) shared a same molecular formula of C13H14N3O2S with nearly same molecular ions at m/z 276.0797, 276.0794, and 276.0795 [M + H]+. They could be identified as isomeric compounds since their fragment pattern were different (Table S1†). The MS/MS spectra of peaks 18 and 17 are shown in Fig. S1,† with the molecular ions and the main fragment ions obtained under the high energy mode. The MS/MS spectra of peak 17 and peak 19 were nearly the same. The neutral loss of 33, 31, 28 and 26 mass units could account for the elimination of –SH, –OCH3, –CO and –CN, respectively. The product ions at m/z 157.0761 and 130.0652 were attributed to the characteristic fragment of dipyridyl derivatives,12 and the mass difference between peaks 17 and 18 was m/z 182.0712 and 185.0707 units for cyano-substituted dipyridyl and aldehyde-substituted dipyridyl, respectively, suggesting the existence of new sulfur-containing compounds.12
The similar investigation was applied to all 22 new peaks. As a result, eight target peaks (5, 9, 15, 16, 17, 18, 20a and 21) were considered as unknowns of interest. Combined with the HPLC analysis on the subfractions (Fig. S2†) of every separation process, the extract was separated by column chromatography and semi-preparative HPLC to obtain six new sulfur-containing compounds, pyrismycins A–F (1–6), together with seven known analogues 7–13. Their structures were elucidated by the means of HRESIMS, NMR data and single-crystal X-ray diffraction experiment.
Compound 1 (peak 17) was isolated as a yellow amorphous powder. Its molecular formula was assigned as C13H13N3O2S from HRESIMS peak at m/z 276.0799 [M + H]+ (calcd for 276.0807), which requires nine degrees of unsaturation. The 1H NMR spectrum showed signals for a proton spin system that included δH 9.33 (1H, d, J = 1.8 Hz, H-2′), 8.66 (1H, dd, J = 4.8, 1.6 Hz, H-6′), 8.49 (1H, dd, J = 6.9, 4.8 Hz, H-5′) and 7.56 (1H, ddd, J = 6.9, 1.8, 1.6 Hz, H-4′) (Table 1). The above data along with the observation of the 13C NMR and DEPT resonances of one olefinic nonprotonated carbons at δC 133.6 (C-3′) and four olefinic methines carbons at δC 150.1 (C-6′), 148.0 (C-2′), 134.2 (C-5′), 128.9 (C-4′) indicated the presence of a mono-substituted pyridine ring (Table 2).13 Similarly, the 13C NMR and DEPT data further displayed four olefinic nonprotonated carbons at δC 166.8 (C-4), 154.6 (C-2), 153.0 (C-6), 120.0 (C-5) and one olefinic methine carbons at 103.9 (C-3), along with a singlet at δH 7.70 (1H, s, H-3), suggesting a tetra-substituted pyridine ring in 1.14 The HMBC correlation between H-3 with C-3′ was detected and confirmed the existence of 2,3′-dipyridyl moiety (Fig. 4). The 1H NMR data also showed signals for one methoxyl group and one methyl group at δH 4.08 (3H, s, H-8) and 2.33 (3H, s, H-9), respectively. The HMBC correlation between δH 4.08 with δC 166.8 indicated that the methoxyl group was substituted at C-4. Taking into consideration the presence of one sulfur atom in the molecular formula of 1 and the chemical shifts of H-9, C-9 (δC 17.7), and C-5, the existence of a methylthio group was established. The HMBC correlation between δH 2.33 with δC 120.0 indicated that the methylthio group was located at C-5 position. Eight degrees of unsaturation were occupied by two pyridyl groups. Thus, the remaining unsaturation suggested that 1 may possess a double bond, which was consistent with the NMR data of the downfield carbon chemical shift at δC 146.7 (C-7) and one N–OH proton signal at δH 11.78 (1H, brs). HMBC correlations of δH 8.72 (H-7) with δC 153.0 and 120.0 confirmed the presence of an oxime group at C-6. The geometry of the oxime group was established by the comparison of the chemical shift of the N–OH signal with those in the known bipyridine analogue, SF2738 A (7).14,15 Thus, compound 1 was characterized as (E)-4-methoxy-5-(methylthio)-[2,3′-bipyridine]-6-carboxaldehyde oxime and named pyrismycin A.
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 3 | 7.70, s | 8.03, s | 8.10, s | 7.68, s | 7.93, s | 8.03, s |
| 7 | 8.72, s | 3.91, s | 8.77, s | 4.87, d (8.8) | ||
| 8 | 4.08, s | 4.06, s | 4.10, s | 4.08, s | 4.13, s | 4.04, s |
| 9 | 2.33, s | 2.33, s | 2.35, s | 3.00, s | 3.06, s | 3.01, s |
| 2′ | 9.33, d (1.8) | 8.18, dd (7.7, 1.8) | 8.19, dd (7.2, 2.4) | |||
| 3′ | 8.40, ddd (8.0, 1.2, 1.0) | 8.31, ddd (8.0, 1.3, 1.2) | 7.52, m | 7.57, m | 8.47, ddd (7.8, 1.3, 1.2) | |
| 4′ | 7.56, ddd (6.9, 1.8, 1.6) | 7.94, ddd (8.0, 6.8, 1.6) | 7.96, ddd (8.0, 6.0, 2.0) | 7.52, m | 7.57, m | 7.96, ddd (7.8, 6.8, 1.6) |
| 5′ | 8.49, dd (6.9, 4.8) | 7.48, ddd (6.8, 4.8, 1.2) | 7.51, ddd (6.0, 4.8, 1.3) | 7.52, m | 7.57, m | 7.50, ddd (6.8, 4.9, 1.3) |
| 6′ | 8.66, dd (4.8, 1.6) | 8.70, ddd (4.8, 1.6, 1.0) | 8.73, ddd (4.8, 2.0, 1.2) | 8.18, dd (7.7, 1.8) | 8.19, dd (7.2, 2.4) | 8.71, ddd (4.9, 1.6, 1.2) |
| NOH | 11.78, brs | 11.93, brs | ||||
| –CONH2 | 7.98, d (1.77) 7.57, s | |||||
| 7-OH | 5.35, brs |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 2 | 154.6, C | 158.0, C | 156.2, C | 159.4, C | 161.2, C | 157.7, C |
| 3 | 103.9, CH | 103.2, CH | 104.3, CH | 104.1, CH | 107.5, CH | 103.3, CH |
| 4 | 166.8, C | 166.5, C | 167.0, C | 165.0, C | 164.1, C | 164.6, C |
| 5 | 120.0, C | 118.5, C | 117.7, C | 126.2, C | 132.3, C | 126.3, C |
| 6 | 153.0, C | 154.2, C | 167.0, C | 150.4, C | 136.3, C | 159.8, C |
| 7 | 146.7, CH | 168.5, C | 52.5, CH3 | 146.1, CH | 115.0, C | 62.8, CH2 |
| 8 | 56.6, CH3 | 56.2, CH3 | 56.5, CH3 | 56.7, CH3 | 57.5, CH3 | 56.5, CH3 |
| 9 | 17.7, CH3 | 17.4, CH3 | 17.4, CH3 | 39.5, CH3 | 39.5, CH3 | 39.0, CH3 |
| 1′ | 137.5, C | 130.5, C | ||||
| 2′ | 148.0, CH | 155.3, C | 153.6, C | 127.1, CH | 127.4, CH | 153.9, C |
| 3′ | 133.6, C | 120.9, CH | 120.7, CH | 128.6, CH | 128.9, CH | 121.2, CH |
| 4′ | 128.9, CH | 137.2, CH | 137.8, CH | 130.0, CH | 130.7, CH | 137.3, CH |
| 5′ | 134.2, CH | 124.5, CH | 125.1, CH | 128.6, CH | 128.9, CH | 124.9, CH |
| 6′ | 150.1, CH | 149.1, CH | 149.6, CH | 127.1, CH | 127.4, CH | 149.2, CH |
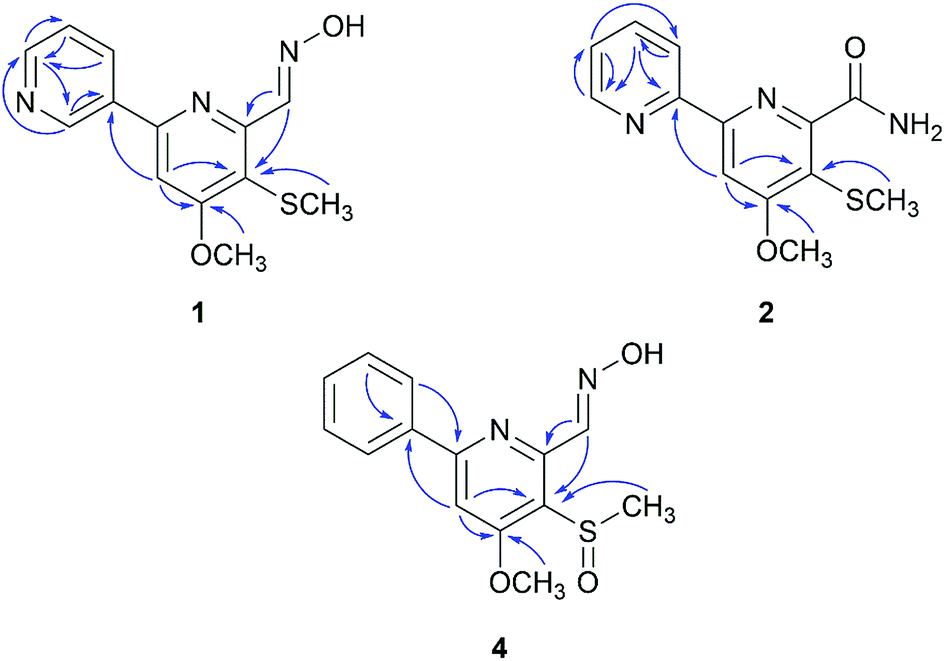 | ||
| Fig. 4 Key HMBC correlations of 1, 2 and 4. | ||
Compound 2 (peak 18) was isolated as yellow amorphous powder. Its molecular formula was determined to be C13H13N3O2S based on the observation of an [M + Na]+ ion at m/z 298.0619 (calcd for 298.0626) in its positive ion mode HRESIMS spectrum. Similar to the known compound 10,15 whose structure was elucidated by a single-crystal X-ray diffraction experiment (Fig. S3†), 2 is also a 2,2′-dipyridyl derivative, as shown by the presence of a proton spin system including δH 8.70 (1H, ddd, J = 4.8, 1.6, 1.0, H-6′), 8.40 (1H, ddd, J = 8.0, 1.2, 1.0, H-3′), 7.94 (1H, ddd, J = 8.0, 6.8, 1.6, H-4′) and 7.48 (1H, ddd, J = 6.8, 4.8, 1.2, H-5′), a singlet signal at δH 8.03 (1H, s, H-3) (Table 1), five olefinic nonprotonated carbons at δC 166.5 (C-4), 158.0 (C-2), 155.3 (C-2′), 154.2 (C-6) and 118.5 (C-5), and five olefinic methine carbons at δC 149.1 (C-6′), 137.2 (C-4′), 124.5 (C-5′), 120.9 (C-3′) and 103.2 (C-3) (Table 2). The HMBC correlation of δH 8.03 with δC 155.3 further confirmed the existence of a 2,2′-dipyridyl skeleton (Fig. 4). A methoxyl group at δH 4.06 (3H, s, H-8) and δC 56.2(C-8), a methylthio group at δH 2.33 (3H, s, H-9) and δC 17.4 (C-9) and an amide group at δH7.98 (1H, d, J = 1.77 Hz), 7.57 (1H, s) and δC 168.5 (C-7) were observed in NMR data of 2. Cross peaks of δH 4.06 with δC 166.5 and δH 2.33 with δC 118.5 in HMBC indicated that the methoxyl group and methylthio group were located at C-4 and C-5, respectively. Thus, the amide group was substituted at C-6. 2 was characterized as 4-methoxy-5-(methylthio)-[2,2′-bipyridine]-6-carboxamide and named pyrismycin B.
Compound 3 (peak 5) was isolated as yellow amorphous powder. Its molecular formula was determined as C13H14N2O2S according to an [M + Na]+ ion at m/z 285.0666 (calcd for 285.0674) in its positive ion mode HRESIMS spectrum. Comparison of the 13C and 1H NMR data of 3 with 2 revealed extensive similarities, except that the methoxy signals at δH 3.91 (3H, s, H-7) and δC 52.5 (C-7) in 3 was replaced by an amide group (δH 7.98 and 7.57, δC 168.5) in 2. The HMBC correlation of δH 3.91 with δC 167.0 (C-6) indicated that the methoxyl group was substituted at C-6. Thus, compound 3 was characterized as 4,6-dimethoxy-5-(methylthio)-2,2′-bipyridine and named pyrismycin C.
Compound 4 (peak 9) was isolated as yellow amorphous powder. The molecular formula was determined to be C14H14N2O3S from the [M + H]+ peak at m/z 291.0793 (calcd for 291.0803) in its positive ion mode HRESIMS spectrum. The NMR data of 4 were similar to pyrisulfoxin A (11), previously isolated from Streptomyces californicus BS-75.16 Both compounds shared the same tetra-substituted pyridine ring as revealed by the presence at δC 165.0 (C-4), 159.4 (C-2), 150.4 (C-6), 126.2 (C-5), 104.1 (C-3) (Table 2). Moreover, a methoxyl group at δH 4.08 (3H, s, H-8), a methylsulfoxide group at δH 3.00 (3H, s, H-9) and an oxime group at δH 8.77 (1H, s, H-7) (Table 1) were also analogous of 11 and substituted at C-4, C-5 and C-6 based on the HMBC correlations of δH 4.08 with δC 165.0, δH 3.00 with δC 126.2 and δH 8.77 (1H, s, H-7) with δC 150.4, respectively (Fig. 4). The difference lies on the mono-substituted pyridine ring in 11 and the mono-substituted phenyl ring in 4, as shown by the protons at δH 8.18 (each 1H, dd, J = 7.7, 1.8 Hz, H-2′, 6′) and 7.51–7.53 (3H, m, H-3′, 4′, 5′), as well as the aromatic carbon signals at δC 137.5 (C-1′), 130.0 (C-4′), 128.6 × 2 (C-3′, 5′) and 127.1 × 2 (C-2′, 6′). The HMBC correlations of δH 7.68 (1H, s, H-3) and 7.52 (each 1H, m, H-3′/4′/5′) with C-1′ and 8.18 (H-2′) with C-2 confirmed the pyridine ring was connected at C-2 (Fig. 4). Thus, 4 was characterized as (E)-4-methoxy-5-(methylsulfinyl)-6-phenylpicolinaldehyde oxime and named pyrismycin D.
Compound 5 (peak 15) was isolated as yellow amorphous powder. Its HRESIMS data exhibited a [M + Na]+ ion at m/z 295.0511 (calcd for 295.0517) corresponded to a molecular formula of C14H12N2O2S and revealed eleven degrees of unsaturation. The UV and NMR data features of compound 5 were very similar to those of 4, except for the substitution of a cyano group (δC 115.0) at C-6 in 5, which was also supported by the existence of two nitrogen atom in its molecular formula, instead of an oxime group in 4. Thus, compound 5 was characterized as 4-methoxy-5-(methylsulfinyl)-6-phenylpicolinonitrile and named pyrismycin E.
Compound 6 (peak 16) was isolated as yellow amorphous powder. Its molecular formula was assigned as C13H14N2O3S on the basis of a [M + Na]+ ion at m/z 301.0620 (calcd for 301.0623) in its positive ion mode HRESIMS spectrum. The 1H and 13C NMR data of 6 indicated it is an analogue of 11.14 Comparison of the NMR data of 6 with those of 11 showed the absence of the oxime group, and the presence of a methylol group at δC 62.8 (C-7), δH 4.87 (2H, d, J = 8.8 Hz, H-7) and 5.35 (1H, brs) in 6. This methylol group was positioned at C-6 based on the HMBC correlation of δH 4.87 with δC 159.8 (C-6). Thus, 6 was characterized as (4-methoxy-5-(methylsulfinyl)-[2,2′-bipyridin]-6-yl) methanol and named pyrismycin F.
The structures of compounds 7–13 were identified as the previously reported natural compounds, SF2738A (7, peak 19),14 SF2738C (8, peak 6),13 SF2738D (9, peak 4),15 SF2738E (10, peak 11),15 pyrisulfoxin A (11, peak 7),16 pyrisulfoxin B (12, peak 14)16 and caerulomycin A (13, peak 12),11 by a comparison of their 1H and 13C NMR data with the literature.
All isolates (1–13) were evaluated for cytotoxicity against the SGC7901, MDA-MB-231, A549 and HepG2 cell lines (ESI Experimental section†). Compounds 7, 9 and 11 displayed measurable IC50 values, but others showed no cytotoxicity (Table S2†). The most potent one, SF2738A (7), showed cytotoxic activities against SGC7901 with IC50 value of 1.7 μM, HepG2 with IC50 value of 5.8 μM and MDA-MB-231 with IC50 value of 6.3 μM, while pyrisulfoxin A (11) showed distinct cytotoxic activity for HepG2 with IC50 value of 7.2 μM. A gross structure–activity relationships (SAR) for this class of compounds was discussed as follows: (1) the 2,2′-dipyridyl moiety may be crucial for the cytotoxicity since pyrismycin A (1), with a greatly similar structures with its isomer 7 except for the 2,3′-dipyridyl moiety, has no displayed deemed cytotoxic for any of the cell lines used. (2) The absence of oxime group in C-7 may remarkably reduce the cytotoxicity. Compound 3 with the methoxyl group in place of the oxime group of 7 significantly decreased the inhibitory activities, which also can be observed in the other analogues of 2 and 8–10. (3) As compared with 7, the presence of a methylsulfoxide group at C-5 in 11 may obviously improve the selectivity of the types of cancer cell lines.
Experimental
General experimental procedures
UV spectra were recorded on a Shimadzu UV-260 spectrophotometer (Shimadzu Corp., Japan) in absolute methanol (MeOH). IR spectra were record as KBr pellets on the Avatar 360 E.S.P spectrophotometer (Thermo Nicolet Co. Boston, MA, USA). NMR data were acquired with a Bruker Avance 400 spectrometer using solvent signals (DMSO; δH 2.50/δC 39.52) as reference. UPLC-QTOF-MS was operated using an Acquity UPLC system (Waters Corp., Milfoed, USA) with a QTOF-MS (XEVO-G2 QTOF, Waters MS Technologies, Manchester, UK), controlled by MassLynx V4.1 software. Semipreparative reversed-phase HPLC separation was performed on a Shimadzu LC-6AD instrument packed with a YMS-pack ODS-A column (5 μm, 250 × 10 mm). X-ray diffraction was realized on a Bruker D8 QUEST X-ray single crystal diffractometer using Mo Kα radiation (λ = 0.71073). Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), MCI Gel CHP 20/P120 (Mitsubishi Chemical Corp., Japan) and ODS-A 12 nm S-50 μm (YMC Co., Ltd, Japan) were used for column chromatography, respectively. Human gastric carcinoma SGC7901, breast carcinoma MDA-MB-231, lung carcinoma A549 and hepatocellular carcinoma HepG2 were obtained from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.Bacteria material
S. roseosporus Lsr2-deletion mutant strain (ΔSrlsr2) and WT strain were obtained from Dr Guojian Liao, College of Pharmaceutical Sciences, Southwest University, China. The voucher specimens (no. 2016102901 and 2016102902) were deposited in the College of Pharmaceutical Sciences, Southwest University, China.Fermentation and isolation
The ΔSrlsr2 strain was grown on PDA plates at 28 °C for 6 days. Then the agar plate were cut into small pieces and inoculated into sterilized liquid potato dextrose broth medium in Erlenmeyer flasks for further incubation at 28 °C on a rotary shaker at 170 rpm for 6 days. The fermented whole broth was filtered to remove the mycelia, and the broth was then extracted four times with ethyl acetate to get about 60 L of extract solution. Then solvent was evaporated under reduced pressure to afford a crude residue (20.9 g). The extract was applied to an ODS column eluted with water, 30%, 40%, 50%, 60%, 70%, 80%, 100% MeOH and concentrated, affording 4 combined fractions (Fr.1–Fr.4). Fr.3 was then separately isolated on MCI gel column and eluted with MeOH–H2O to obtain 3 subfractions Fr.3.1 to Fr.3.3. Fr.3.1 was separated by semipreparative HPLC, using a gradient of 75–92% MeOH in H2O over 47 min, to give 3 (2.5 mg, tR = 36 min) and 9 (1.6 mg, tR = 23 min). Fr.3.2 was separated over Sephadex LH-20 column chromatography, using CH2Cl2–MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as an eluent, to yield two subfractions (Fr.3.2.1 and Fr.3.2.2). Fr.3.2.1 was further purified using semipreparative HPLC, eluting with a MeOH–H2O (2:3) over 88 min, to give 4 (1.8 mg, tR = 75 min), 8 (3.5 mg, tR = 32 min) and 11 (2.5 mg, tR = 36 min). Similarly, Fr.3.2.2 was subjected to semipreparative HPLC, eluted with 33% MeOH in H2O over 63 min, to provide 5 (1.0 mg, tR = 54 min), 10 (2.7 mg, tR = 27 min), 12 (2.2 mg, tR = 58 min) and 13 (1.2 mg, tR = 33 min). Fr.3.3 was chromatographed by semipreparative HPLC, eluting with a step gradient of 32–60% MeOH in H2O over 93 min, giving 1 (2.0 mg, tR = 64 min), 2 (1.8 mg, tR = 80 min), 6 (2.1 mg, tR = 39 min) and 7 (3.0 mg, tR = 85 min).
:3) as an eluent, to yield two subfractions (Fr.3.2.1 and Fr.3.2.2). Fr.3.2.1 was further purified using semipreparative HPLC, eluting with a MeOH–H2O (2:3) over 88 min, to give 4 (1.8 mg, tR = 75 min), 8 (3.5 mg, tR = 32 min) and 11 (2.5 mg, tR = 36 min). Similarly, Fr.3.2.2 was subjected to semipreparative HPLC, eluted with 33% MeOH in H2O over 63 min, to provide 5 (1.0 mg, tR = 54 min), 10 (2.7 mg, tR = 27 min), 12 (2.2 mg, tR = 58 min) and 13 (1.2 mg, tR = 33 min). Fr.3.3 was chromatographed by semipreparative HPLC, eluting with a step gradient of 32–60% MeOH in H2O over 93 min, giving 1 (2.0 mg, tR = 64 min), 2 (1.8 mg, tR = 80 min), 6 (2.1 mg, tR = 39 min) and 7 (3.0 mg, tR = 85 min).
Structure characterization
ε) 239 (3.9) nm; IR (KBr) νmax 3431, 2929, 2872, 2364, 1631, 1568, 1377, 1300, 1232, 1128, 1018, 871, 790, 758, 692 cm−1; 1H and 13C NMR data see Tables 1 and 2; HRESI (+) MS m/z 276.0799 (calcd for 276.0807).ε) 285 (3.8) nm; IR (KBr) νmax 3441, 3361, 2926, 2858, 1662, 1570, 1452, 1375, 1217, 1124, 1045, 854, 786, 738, 661 cm−1; 1H and 13C NMR data see Tables 1 and 2; HRESI (+) MS m/z 298.0619 (calcd for 298.0626).ε) 284 (3.6) nm; IR (KBr) νmax 3433, 2933, 1724, 1647, 1635, 1598, 1438, 1369, 1224, 1033, 781, 692 cm−1; 1H and 13C NMR data see Tables 1 and 2; HRESI (+) MS m/z 285.0666 (calcd for 285.0674).ε) 247 (3.8) nm; 1H and 13C NMR data see Tables 1 and 2; HRESI (+) MS m/z 291.0793 (calcd for 291.0803).ε) 243 (3.8) nm, 279 (3.7) nm; 1H and 13C NMR data see Tables 1 and 2; HRESI (+) MS m/z 295.0511 (calcd for 295.0517).ε) 281 (4.1) nm; IR (KBr) νmax 3441, 3346, 2987, 2926, 1564, 1421, 1271, 1217, 1028, 945, 881, 788, 686, 621, 540 cm−1; 1H and 13C NMR data see Tables 1 and 2; HRESI (+) MS m/z 301.0620 (calcd for 301.0623).243 reflections measured, 13668 independent reflections (Rint = 0.0224). The final R1 values were 0.0536 (I > 2σ (I)), and the final wR (F2) values were 0.1460 (all data). The goodness of fit on F2 was 1.057. Crystallographic data were deposited with the Cambridge Crystallographic Data Centre (deposition code: CCDC 1533762).†UPLC-QTOF-MS conditions
Chromatographic separation was performed with a 2.1 × 50 mm i.d., 1.7 μm UPLC BEH C18 reversed-phase column and kept at a temperature of 40 °C. The mobile phase consisted of water (A) and acetonitrile (B). The linear gradient elution was performed as follows: 0–0.4 min, 5% B; 0.4–20 min, 5–45% B; 20–24 min, 45–65% B; 24–26 min, 65–76% B; 26–35 min, 76–100% B; 35–36 min, 100–0.5% B; 36–38 min, 0.5–0.5% B. A flow rate of 0.4 mL min−1 was employed for elution, and the injected sample volume was set at 2 μL. Mass spectrometry was recorded using a Xevo G2 QTOF equipped with an ESI source and MS full scanning was conducted in positive ion modes over the range m/z 100–1000 Da, with a scan time of 0.2 s. The capillary voltages were set at 3000 V and the cone voltage was 40 V. Nitrogen gas was used both for the nebulizer and in desolvation. The desolvation and cone gas flow rates were 800 and 50 L h−1, respectively. The desolvation temperature was 450 °C, and the source temperature was 120 °C. The tandem MS data were acquired in the positive mode with a range of m/z 100–1000 Da, and the low energy was set as 6 V, while the high energy was ramped from 20 to 40 V. The cone voltage was set as 40 V.Conclusions
The combination of UPLC-QTOF-MS/MS and HPLC analysis is an effective chemical dereplication method for structural analysis of compounds and rapid isolation of unknowns of interest in ΔSrlsr2 strain, whereas the conventional separation often requires a great deal of work to obtain the desired compounds. Pyrisulfoxins A–F (1–6), six sulfur-containing metabolites have not yet been described in the literature, and seven known analogues (7–13) were isolated from the ΔSrlsr2 strain. Among these compounds tested for cytotoxicity, SF2738A (7) showed the highest activity for SGC7901 cell lines, and pyrisulfoxin A (11) showed moderate cytotoxic for HepG2 cell lines. It was suggested that the 2,2′-dipyridyl moiety may be crucial for the cytotoxicity, and the absence of oxime group in C-7 may remarkably reduce the cytotoxicity.This is the first report that deletion of the global regulator Lsr2 in S. roseosporus upregulated the expression of biosynthetic gene cluster and produced new compounds, pyrisulfoxins A–F (1–6). And this further demonstrated that deletion of the global regulator Lsr2 is an effective approach to produce new secondary metabolites.
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National High Technology Research and Development Program of China (2011AA100607), Fundamental Research Funds for the Central Universities (XDJK2014A008), and National Natural Science Foundation of China (81602987).References
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- J. A. Worrall and E. Vijgenboom, Nat. Prod. Rep., 2010, 27, 742–756 RSC.
- C. Christophersen and U. Anthoni, J. Sulfur Chem., 1986, 4, 365–442 CAS.
- A. L. Demain and S. Sanchez, J. Antibiot., 2009, 62, 5–16 CrossRef CAS PubMed.
- L. Shen, master thesis, Southwest University, 2016.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988–1005 RSC.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509–522 CrossRef CAS PubMed.
- B. R. Gordon, R. Imperial, L. Wang, W. W. Navarre and J. Liu, J. Bacteriol., 2008, 190, 7052–7059 CrossRef CAS PubMed.
- H. M. Ashmead, L. Negron, K. Webster, V. Arcus and J. A. Gerrard, Biopolymers, 2014, 103, 261–270 Search PubMed.
- S. P. Gaudencio and F. Pereira, Nat. Prod. Rep., 2015, 32, 779–810 RSC.
- S. H. Kim, H. Ko, H. S. Bang, S. H. Park, D. G. Kim, H. C. Kwon, S. Y. Kim, J. Shin and D. C. Oh, Bioorg. Med. Chem. Lett., 2011, 21, 5715–5718 CrossRef CAS PubMed.
- J. Regueiro, A. Giri and T. Wenzl, Anal. Chem., 2016, 88, 6500–6508 CrossRef CAS PubMed.
- F. Trecourt, B. Gervais, O. Mongin, C. L. Gal, F. Mongin and G. Queguiner, J. Org. Chem., 1998, 63, 2892–2897 CrossRef CAS.
- K. Shindo, Y. Yamagishi, Y. Okada and H. Kawai, J. Antibiot., 1994, 47, 1072–1074 CrossRef CAS PubMed.
- S. Gomi, S. Amano, E. Sato, S. Miyadoh and Y. Kodama, J. Antibiot., 1994, 47, 1385–1394 CrossRef CAS PubMed.
- N. Tsuge, K. Furihata, K. Shin-Ya, Y. Hayakawa and H. Seto, J. Antibiot., 1999, 52, 505–507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI Experimental section; Table S1 (sulfur-containing metabolites detected and characterized in ΔSrlsr2 strain by UPLC-QTOF-MS and HR-MS/MS analysis); Table S2 (cytotoxic activities of 1–13 isolated from the ΔSrlsr2 strain of S. roseosporus); Fig. S1 (HR-MS/MS spectra and proposed fragmentation pathway of peaks 17 and 18 (1 and 2)); Fig. S2 (HPLC guided isolation of sulfur-containing metabolites); Fig. S3 (X-ray crystal structure of 10); NMR and HRMS spectra of compounds 1–6. CCDC 1533762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06482a |
| This journal is © The Royal Society of Chemistry 2017 |