
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-responsive liquid crystalline epoxy networks with exchangeable disulfide bonds†
Yuzhan Lia,
Yuehong Zhanga,
Orlando Riosb,
Jong K. Keumc and
Michael R. Kessler *ad
*ad
aSchool of Mechanical and Materials Engineering, Washington State University, PO Box 642920, Pullman, WA 99164-2920, USA. E-mail: MichaelR.Kessler@wsu.edu; Tel: +1-509-335-8654
bDeposition Sciences Group, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
cCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
dDepartment of Mechanical Engineering, North Dakota State University, Fargo, ND 58102, USA
First published on 27th July 2017
Abstract
The increasing demand for intelligent materials has driven the development of polymers with a variety of functionalities. However, combining multiple functionalities within one polymer is still challenging because of the difficulties encountered in coordinating different functional building blocks during fabrication. In this work, we demonstrate the fabrication of a multifunctional liquid crystalline epoxy network (LCEN) using the combination of thermotropic liquid crystals, photo-responsive azobenzene molecules, and exchangeable disulfide bonds. In addition to shape memory behavior enabled by the reversible liquid crystalline phase transition and photo-induced bending behavior resulting from the photo-responsive azobenzene molecules, the introduction of dynamic disulfide bonds into the LCEN resulted in a structurally dynamic network, allowing the reshaping, repairing, and recycling of the material.
1. Introduction
Shape memory polymers are smart materials that are able to change shapes when exposed to external stimuli, such as heat, light, electric fields, or magnetic fields.1–3 The unique properties and technological significance of shape memory polymers have led to broad development activities ranging from biomedical (e.g., biosensors, drug delivery systems, and intelligent implants) to aerospace applications (e.g., actuators, self-deployable devices, and morphing structures). Liquid crystalline networks (LCNs) are a special class of shape memory polymers defined by a reversible liquid crystalline (LC) phase transition and a unique coupling between the mesogen and the polymer network.4–6 They are able to undergo large dimensional changes owing to the macroscopic orientation of the LC domains.7 These unique properties make them excellent candidates for shape memory and actuation applications.8–16 In recent years, photo-responsive LCNs were developed by incorporating photo-responsive molecules, such as azobenzene, which allows the material to convert light energy into mechanical work.17–19 Here, exposure to light causes the transformation of azobenzene between two geometrically different isomers, resulting in a variety of shape changes of the bulk liquid crystalline network material, including bending, twisting, and oscillating.20–26 Gelebart et al. recently reported a photoresponsive polymer film that exhibited continuous and directional mechanical waves resulting from a fast isomerization process of the azobenzene molecules.27 The incorporation of azobenzene into polymers has been proven to be an effective way to create smart materials that can response to external light stimuli. However, in order to further increase the intelligence of these materials, additional functional building blocks need to be introduced to the materials.Recently, dynamic covalent chemistries were incorporated into LCNs, which allowed for the reversible breaking and reformation of the covalent bonds.28–31 The use of a catalytically/thermally controlled transesterification reaction resulted in a rearrangeable network structure that improved the reprocessability of the material. However, these LCNs were still not fully recyclable because the transesterification reaction relied on the exchange reaction between hydroxyl and ester groups. Although the chemical bonds in the network kept exchanging, their total number remained constant. Therefore, the recyclability of the material was still limited as it heavily relied on depolymerization induced by chemical bond reduction.
Disulfide chemistries have been used in physiological applications where the behavior of large protein molecules, including their conformational change, biological activity, and reduction, is controlled by reversible sulfur–sulfur bonds.32,33 In recent years, dynamic disulfide bonds were used to design structurally adaptive polymers with self-healing and shape memory properties.34–36 While the use of disulfide chemistry has the potential to produce reprocessable and recyclable LCNs, it remains largely unexplored. The introduction of dynamic disulfide bonds into LCNs is expected to result in rearrangeable networks, which will improve the reprocessability of LCNs and allow for practical shape memory applications.
In this work, we demonstrate that different functional building blocks (thermally responsive liquid crystals, photo-responsive azobenzene molecules, and dynamic disulfide bonds) can be integrated into a liquid crystalline epoxy network (LCEN), and that the resulting material exhibits adaptive performance, including shape memory and photomechanical behavior, when exposed to different external stimuli (light or heat). In addition, we demonstrate that the introduction of dynamic disulfide bonds facilitates the reshaping, repairing, and recycling of LCEN by taking advantage of the disulfide exchange and thiol–disulfide interchange reactions.
2. Experimental section
2.1 Materials
Epichlorohydrin was obtained from Acros Organics. Benzyltrimethylammonium bromide, sebacic acid (SA), 4,4′-dithiodibutryic acid (DSA), and imidazole (ID) were purchased from Sigma-Aldrich. 4,4′-(1,2-Diazenediyl)bisphenol was purchased from Synthon Chemicals. All chemicals were used as received without further purification. The epoxy monomer (4,4′-diglycidyloxyazobenzene, AE) was synthesized according to a procedure reported in an earlier work.372.2 Preparation of LCEN films
The LCEN was synthesized by curing of AE, SA, DSA, and ID at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.9:0.1:0.1. The four chemicals were dissolved in acetone and sonicated for 10 min. After evaporation of the solvent, the powder mixture was grinded using a pestle and mortar. LCEN films were prepared using the parallel plate fixture of a strain-controlled rheometer (ARES G2, TA Instruments). The parallel plates were preheated to 170 °C before the powder mixture was loaded. After the material was fully melted, the gap between the plates was reduced to 0.2 mm and maintained constant throughout the curing process. The material was cured at 170 °C for 1 h in ARES G2, then moved to a convection oven and cured at 170 °C for 3 h and 200 °C for 1 h.
:0.9:0.1:0.1. The four chemicals were dissolved in acetone and sonicated for 10 min. After evaporation of the solvent, the powder mixture was grinded using a pestle and mortar. LCEN films were prepared using the parallel plate fixture of a strain-controlled rheometer (ARES G2, TA Instruments). The parallel plates were preheated to 170 °C before the powder mixture was loaded. After the material was fully melted, the gap between the plates was reduced to 0.2 mm and maintained constant throughout the curing process. The material was cured at 170 °C for 1 h in ARES G2, then moved to a convection oven and cured at 170 °C for 3 h and 200 °C for 1 h.
2.3 Characterizations
The thermal and LC transitions of the LCEN were determined using a differential scanning calorimeter (Discovery DSC, TA Instruments) under a nitrogen purge of 20 mL min−1. A heat–cool–heat cycle with a ramp rate of 20 °C min−1 was used. The glass transition (Tg) and LC transition (Tlc) of the LCEN were determined from the second heating scan.The structure of the LCEN was characterized using two-dimensional wide-angle X-ray scattering (Anton Paar SAXSess mc2). The scattering beam was recorded on an imaging plate (multi-sensitive storage phosphor) and read using an imaging plate reader. The X-rays were generated at 40 kV/50 mA and the wavelength of the used X-ray beam was λ = 1.541 Å (Cu-Kα radiation). The structure of LCEN films at strain values of 0%, 100%, and 200% was examined using wide angle X-ray scattering (WAXS) configurations.
Static tensile tests were carried out using the ARES G2 rheometer in a film tension geometry. The LCEN film was heated to 80 °C (between Tg and Tlc) and the axial force was measured under a strain ramp rate of 0.002 s−1. Cyclic thermomechanical tensile tests were carried out using the ARES G2 in a film tension geometry. The LCEN film was heated to 160 °C (above Tlc), and then a constant force of 0.1 N was applied. Then the temperature was decreased to 80 °C at a cooling rate of 3 °C min−1 and then increased to 160 °C again at a heating rate of 3 °C min−1. The strain of the LCEN film was measured during the temperature cycling. Triple shape memory tests were also performed using ARES G2 in a film tension geometry and consisted of the following steps: (1) heating the sample to 160 °C, (2) applying a constant axial force of 0.2 N, (3) decreasing the temperature to 80 °C at a cooling rate of 3 °C min−1, (4) increasing the axial force to 0.5 N, (5) equilibrating the sample for 2 min, (6) decreasing the temperature to 40 °C at a cooling rate of 3 °C min−1, (7) decreasing the axial force to 0.2 N, (8) increasing the temperature to 80 °C at a heating rate of 3 °C min−1, (9) equilibrating the sample for 2 min, and (10) increasing the temperature to 160 °C at a heating rate of 3 °C min−1. Creep and creep recovery tests were also carried out using ARES G2 in a film tension geometry. A constant force was applied for 20 min, followed by a 20 min recovery period.
The light source used for the photomechanical characterization was generated by a UV curing lamp system with a 320–500 nm internal filter (OmniCure S2000, Excelitas Technologies). The light was guided into an adjustable collimating adapter (Lumen Dynamics) and then passed through a bandpass filter (355 nm or 442 nm) and a polarized beamsplitter cube (Thorlabs). The final intensity of the light was measured using either a blue light radiometer (Solartech) or a UV radiometer (UV Process Supply).
3. Results and discussion
3.1 Thermal and LC properties
Fig. 1a shows the chemical structures of monomer, curing agents, and catalyst used to synthesize the LCEN. The curing reaction started with the ring-opening of epoxy monomer. Adding flexible aliphatic chains increased the mobility of the mesogens and facilitated their self-organization into an ordered LC phase. However, hydroxyl groups were also created by the opening of the epoxy rings, which acted as crosslinking sites and disrupted the LC order. Therefore, imidazole was used as a catalyst to promote the ring-opening reaction and facilitate formation of the LC phase. The formed LC phase was retained in the material through the reaction of hydroxyl groups with either epoxy through etherification or carboxylic acid through esterification, which created a lightly crosslinked network with LC structure (ESI Fig. S1†). In order to create structurally dynamic LCENs, disulfide bonds were introduced into the LCEN by using DSA as a co-curing agent. It was determined that when DSA was used as the only curing agent, the resulting LCEN was overly dynamic, affecting the stability of the formed LC phase. Therefore, the molar ratio of DSA was limited to 0.1 to control the amount of disulfide bonds in the LCEN. | ||
| Fig. 1 Synthesis and thermal properties of LCEN. (a) Chemical structures of monomer, curing agents, and catalyst. (b) Thermal and LC properties of LCEN determined by DSC. | ||
Fully cured LCEN films exhibited a Tg of 59.0 °C and a Tlc of 114.6 °C (Fig. 1b). Two-dimensional wide-angle X-ray scattering (2D WAXS) confirmed that the prepared LCEN films exhibited smectic LC polydomain structure (Fig. 2a). The quantified 2D WAXS patterns are shown in Fig. 2b. The sharp peak (q = 3.5 nm−1, d = 18.0 Å) was the result of scattering from the periodic smectic layer, while the broad peak (q = 13.4 nm−1, d = 4.7 Å) was caused by the scattering of the less regulated neighboring mesogens in the smectic layers. The liquid crystallinity of the LCEN was also examined using polarized optical microscopy, in which a highly birefringent texture was observed (ESI Fig. S2†). Fig. 2c shows the static tensile behavior of the LCEN films. After the initial elastic response, the force–strain curve exhibited a plateau region, which was attributed to the macroscopic orientation of the LCEN, where the LC domains were aligned in the direction of the applied force. The increase in the degree of orientation of the LC domains upon mechanical stretching was confirmed by ex situ 2D WAXS experiments. Fig. 2d shows the azimuthal intensity distribution of LCEN films at different strain levels, which were used to determine the degree of orientation see Fig. 2a. Owing to the macroscopic orientation of the LC domains, the synthesized LCEN was able to exhibit large dimensional change, which would be beneficial for shape memory applications.
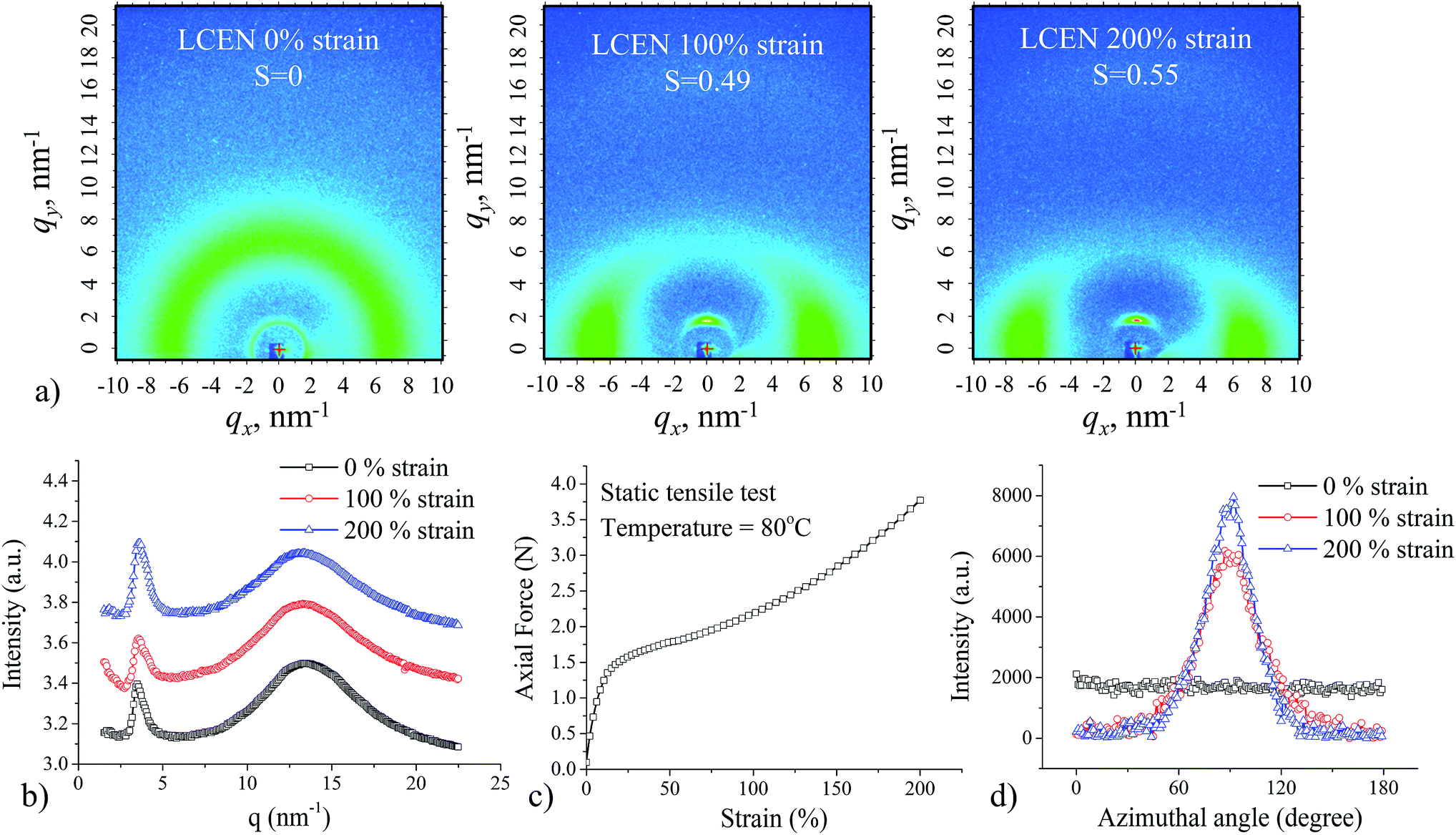 | ||
| Fig. 2 Structure characterization of LCEN films. (a) 2D WAXS patterns of LCEN films at different strain levels. (b) Q integration of the 2D WAXS data of LCEN films with different strain levels. (c) Static tensile behavior of LCEN film. (d) Azimuthal intensity distribution determined from 2D WAXS patterns for LCEN films with different strain levels. | ||
3.2 Shape memory properties
Fig. 3a shows reversible shape change of the LCEN films in a cyclic thermomechanical tensile test. The LCEN films exhibited elongation behavior during the cooling process. The rate of the elongation reached a maximum at 116.4 °C (ESI Fig. S3†), which was close to the Tlc determined by differential scanning calorimetry (DSC) experiments and indicated that the elongation was caused by the formation of the LC domains and their macroscopic orientation in response to the applied axial force. During the heating process, however, the LCEN films contracted because the LC phase transitioned to an isotropic phase, where the material lost liquid crystallinity and returned to a random conformation. Because of the reversible nature of the LC phase transition, the elongation and contraction behavior of the LCEN in the cooling and heating process, respectively, can be repeated several times. It is worth mentioning that a creep behavior of the LCEN was observed during the cyclic thermomechanical tensile test, where the deformation was not able to be fully recovered. This was attributed to the relatively low crosslink density of the material. Fig. 3b shows triple shape memory behavior of the LCEN films, which was facilitated by the two reversible transitions (Tg and Tlc). A two-stage shape programming process was used to induce two consecutive shape changes caused by the orientation of the LC domains and the deformation of the elastomeric network, respectively. During the cooling process, the LCEN first transformed from an amorphous rubber to an LC rubber. The LC formation and orientation allowed for the first shape change observed in the triple shape memory experiment. Then, the axial force applied on the LCEN film was increased from 0.1 N to 0.5 N to induce the second shape change, which was caused by the deformation of the rubbery network. Further cooling led to a transformation of the LCEN from an LC rubber state to an LC glass state, which allowed the deformation to be fixed even when the axial force was removed. Subsequently, the changes in shape of the LCEN films were reversed when the material was heated to 80 °C and 160 °C, respectively. Fig. 3c demonstrates the shape memory capacity of the LCEN material. Here, an LCEN box was flattened and folded backward at 160 °C to adopt a temporary shape. After cooling to room temperature, the temporary shape was retained as a result of the restriction of the LC domains and the glassy polymer network. The permanent shape was recovered by heating the material back to 160 °C to remove the restriction. In addition, the incorporation of the azobenzene molecules allowed to initiate the shape recovery process by UV irradiation because of a photo-thermal effect of the azobenzene molecules (Video S1†).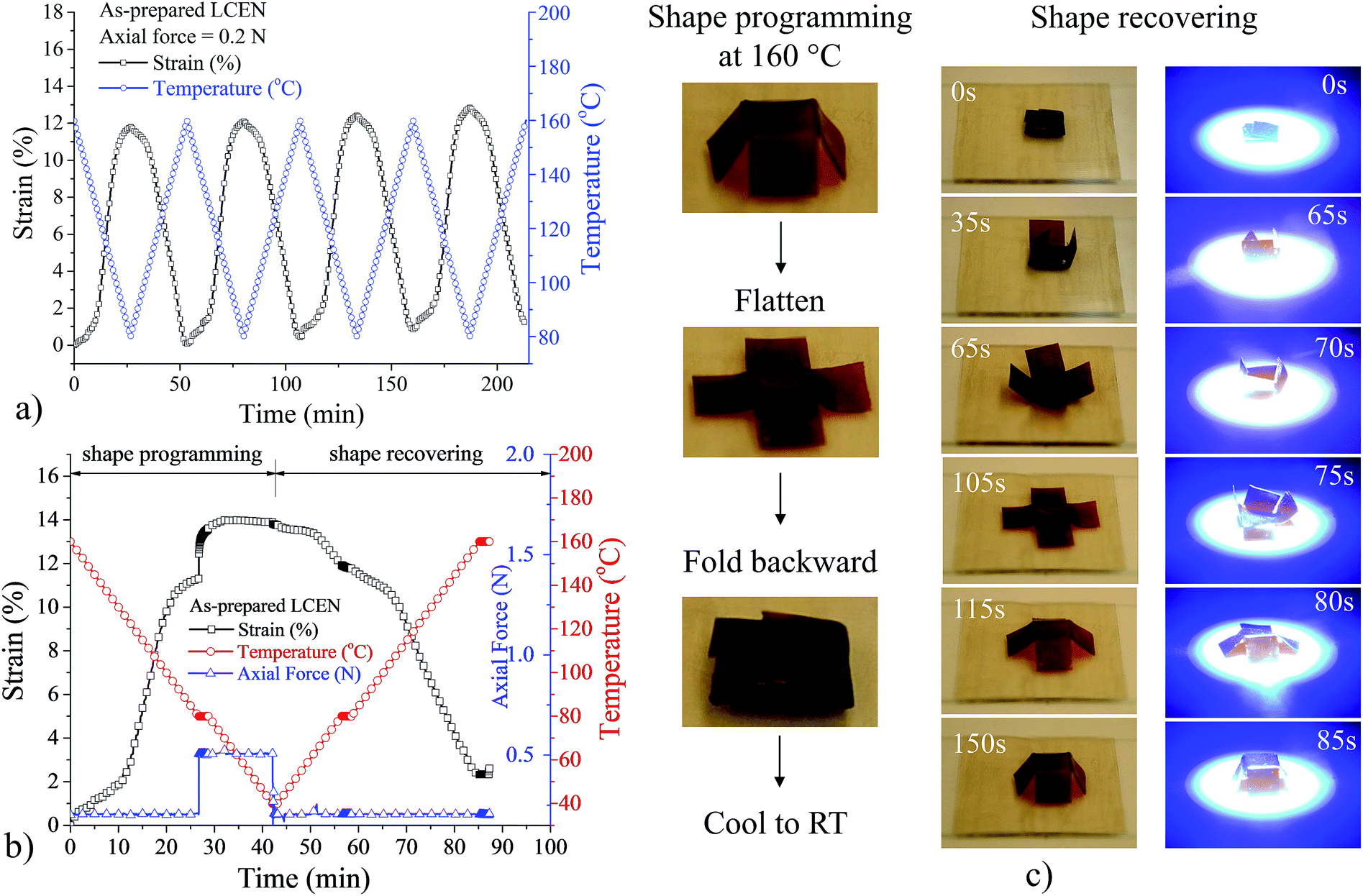 | ||
| Fig. 3 Thermomechanical and shape memory behavior of LCEN. (a) Cyclic thermomechanical tensile test of LCEN films. (b) Triple shape memory test of LCEN films. (c) Demonstration of thermal and UV-induced shape memory behavior of LCEN. | ||
3.3 Photomechanical properties
The photo-chemical properties of azobenzene molecules, such as their blue light induced photo-alignment38 and UV light induced photo-isomerization,39 facilitated control of the photomechanical behavior of the LCEN films using light with different wavelengths. Fig. 4a shows the bidirectional bending behavior of the LCEN films triggered by polarized blue light. When the polarization direction of the blue light was parallel to the long axis of the LCEN film, the film bent toward the light source (Video S2†). In contrast, when the polarization direction was perpendicular to the long axis of the LCEN, the film bent away from the light source (Video S3†). In both cases, the direction of the azobenzene molecules on the light-fronting surface was changed in response to the incident polarized blue light, leading to either a contraction or an expansion of the surface layer, which ultimately resulted in bending of the LCEN films. Fig. 4b shows the controlled bending direction of the LCEN films initiated by polarized UV light. Although in both case, the bending behavior of the LCEN films was caused by the shape change of the material on the light-fronting surface, the underlying mechanisms were different. The polarized blue light caused alignment of the azobenzene molecules perpendicular to the polarized light through a trans–cis–trans transformation,40 whereas exposure to polarized UV light led to a bending of selective azobenzene molecules through a trans–cis transformation.41 The change in direction or shape of the azobenzene molecules on the light-fronting surface led to expansion or contraction of the surface layer, resulting in bending of the LCEN films. | ||
| Fig. 4 Photomechanical properties of LCEN films (arrows indicate polarization direction of the incident light). (a) Blue light induced bidirectional bending at a light intensity of 40 mW cm−2. (b) UV light induced bending of LCEN film with controlled direction at a light intensity of 80 mW cm−2. | ||
3.4 Reprocessability and recyclability
In order to add new functionalities to the material, dynamic disulfide bonds were incorporated to create structurally dynamic LCEN. The effect of exchangeable disulfide bonds on viscoelastic properties of the LCEN was characterized using creep tests (Fig. 5a). The creep experiments were performed at temperatures higher than Tlc to eliminate any possible influence of LC orientation on creep behavior of the LCEN films. In contrast to the creep behavior of traditional crosslinked polymer networks, where the deformations can be fully recovered, the synthesized LCEN films exhibited permanent deformations at all the investigated temperatures. | ||
| Fig. 5 Reprocessability and recyclability of LCEN. (a) Creep and creep recovery behavior of LCEN at different temperatures. (b) Reshaping of LCEN. (c) Repairing of LCEN using UV light. (d) Effect of disulfide bonds on recycling of LCEN using 2-mercaptoethanol. (e) Recycling of LCEN using 2-mercaptoethanol under reflux. | ||
The creep test results are summarized in Table 1. Instantaneous deformation of the LCEN films was determined by the initial strain of the material upon application of the axial force, which reflected the elastic response of the material. Creep deformation of the LCEN films was determined as the accumulated strain of the LCEN throughout the creep process, which reflected the viscoelastic behavior of the material. When the applied force was removed, only instantaneous deformation was recovered, and creep deformation became permanent deformation. This effect was attributed to the disulfide exchange reaction, where the disulfide bonds incorporated into the material underwent an equilibrium of breaking and reformation, resulting in a dynamic LCEN with rearrangeable network structure. In addition, the rate of the disulfide exchange reaction was affected by temperature, resulting in a strong temperature dependence of the permanent deformation of the LCEN films. This unique functionality allowed for the change of the permanent shape of the LCEN, which is a crucial factor limiting the applications of shape memory polymers. As shown in Fig. 5b, the as-prepared LCEN film was reshaped into a box by remolding at 200 °C for 1 h. The breaking and reformation of the disulfide bonds also allowed the LCEN films to be repaired. Fig. 5c shows UV-induced self-healing behavior of the LCEN films. Here, the trace of the carved letters “WSU” was significantly reduced after UV irradiation for only 3 min. The disulfide exchange reaction also allowed for reprocessing of the material, where as-prepared LCEN film was chopped into small pieces and hot-pressed at 200 °C for 4 h. The reprocessed LCEN films showed similar thermal, liquid crystalline, thermomechanical, and shape memory behavior to those of as-prepared LCEN films (ESI Fig. S4–S6†).
| Creep temperature (°C) | Instantaneous deformation (%) | Creep deformation (%) | Instantaneous recovery (%) | Permanent deformation (%) |
|---|---|---|---|---|
| 160 | 1.88 | 4.12 | 1.99 | 4.01 |
| 180 | 3.76 | 9.75 | 3.82 | 9.58 |
| 200 | 6.03 | 20.41 | 7.13 | 19.31 |
In addition to the self-exchange reaction, disulfide bonds are able to exchange with thiol groups. The thiol–disulfide interchange is a reaction between a monothiol (RSH) and a disulfide (R′SSR′) with the formation of a new disulfide (RSSR′) and a thiol (R′SH). The interchange reaction allows for the recycling of polymers containing disulfide bonds using low-molecular thiol compounds.42,43 In order to investigate the recyclability of LCEN films, 2-mercaptoethanol was used as a monothiol to initiate the thiol–disulfide interchange reaction. Fig. 5d shows the effect of disulfide bonds on recyclability of the LCEN. Compared to the thiol solution of the control LCEN (without disulfide bonds), significant coloration was observed for the solution containing LCEN with disulfide bonds after 16 h at room temperature, indicating partial dissolution of the LCEN through the interchange reaction between thiol groups and disulfide bonds. However, the glassy network structure of the LCEN at room temperature impeded penetration of the thiol compound into the network. In order to further reduce crosslink density of the LCEN to fully recycle the material, the solution was heated to 150 °C. After refluxing for 1 h, the LCEN containing disulfide bonds was fully dissolved in the thiol solution (Fig. 5e), indicating significant breakage of the network structure because of the reduction of crosslink density.
4. Conclusions
In summary, a multifunctional polymer was synthesized by integrating different functional building blocks into a LCEN, including thermally responsive liquid crystals, photo-responsive azobenzene molecules, and dynamic disulfide bonds. The functional groups showed good compatibility and the resulting material exhibited adaptive performance when exposed to different external stimuli. The macroscopic liquid crystalline orientation and the reversible liquid crystalline phase transition allowed for shape memory behavior, whereas the photo-alignment and photo-isomerization of the azobenzene molecules allowed for the control of photomechanical behavior of the LCEN. The incorporation of dynamic disulfide bonds added new functionalities to the LCEN through disulfide–disulfide exchange and thiol–disulfide interchange reactions, which resulted in LCEN that could be reshape, repaired, and recycled.Acknowledgements
The majority of this work was supported by the Air Force Office of Scientific Research (Award FA-9550-12-1-0108). In addition, a portion of the research was conducted at the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Division of Scientific User Facilities, U.S. Department of Energy, managed by UT-Battelle, LLC, for the U.S. Department of Energy. Also, some of the research was sponsored by the Critical Materials Institute, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, and Advanced Manufacturing Office, under contract DE-AC05-00OR22725 with UT-Battelle, LLC.References
- M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed.
- G. J. Berg, M. K. McBride, C. Wang and C. N. Bowman, Polymer, 2014, 55, 5849–5872 CrossRef CAS.
- Q. Zhao, H. J. Qi and T. Xie, Prog. Polym. Sci., 2015, 49–50, 79–120 CrossRef CAS.
- L. T. de Haan, A. P. H. J. Schenning and D. J. Broer, Polymer, 2014, 55, 5885–5896 CrossRef CAS.
- T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087–1098 CrossRef CAS PubMed.
- C. Ohm, M. Brehmer and R. Zentel, Adv. Mater., 2010, 22, 3366–3387 CrossRef CAS PubMed.
- S. Krause, F. Zander, G. Bergmann, H. Brandt, H. Wertmer and H. Finkelmann, C. R. Chim., 2009, 12, 85–104 CrossRef CAS.
- J. E. Marshall, S. Gallagher, E. M. Terentjev and S. K. Smoukov, J. Am. Chem. Soc., 2013, 136, 474–479 CrossRef PubMed.
- C. Ohm, C. Serra and R. Zentel, Adv. Mater., 2009, 21, 4859–4862 CrossRef CAS PubMed.
- H. Qin and P. T. Mather, Macromolecules, 2009, 42, 273–280 CrossRef CAS.
- S.-k. Ahn and R. M. Kasi, Adv. Funct. Mater., 2011, 21, 4543–4549 CrossRef CAS.
- H. Yang, M.-X. Liu, Y.-W. Yao, P.-Y. Tao, B.-P. Lin, P. Keller, X.-Q. Zhang, Y. Sun and L.-X. Guo, Macromolecules, 2013, 46, 3406–3416 CrossRef CAS.
- I. A. Rousseau and P. T. Mather, J. Am. Chem. Soc., 2003, 125, 15300–15301 CrossRef CAS PubMed.
- K. A. Burke, I. A. Rousseau and P. T. Mather, Polymer, 2014, 55, 5897–5907 CrossRef CAS.
- Y. Yu and T. Ikeda, Angew. Chem., Int. Ed., 2006, 45, 5416–5418 CrossRef CAS PubMed.
- J. Cui, D.-M. Drotlef, I. Larraza, J. P. Fernández-Blázquez, L. F. Boesel, C. Ohm, M. Mezger, R. Zentel and A. del Campo, Adv. Mater., 2012, 24, 4601–4604 CrossRef CAS PubMed.
- H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149–2180 CrossRef CAS PubMed.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- T. J. White, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 877–880 CrossRef CAS.
- C. L. van Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677–682 CrossRef CAS PubMed.
- K. M. Lee, M. L. Smith, H. Koerner, N. Tabiryan, R. A. Vaia, T. J. Bunning and T. J. White, Adv. Funct. Mater., 2011, 21, 2913–2918 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- K. M. Lee and T. J. White, Macromolecules, 2012, 45, 7163–7170 CrossRef CAS.
- J. J. Wie, K. M. Lee, T. H. Ware and T. J. White, Macromolecules, 2015, 48, 1087–1092 CrossRef CAS.
- J. J. Wie, K. M. Lee, M. L. Smith, R. A. Vaia and T. J. White, Soft Matter, 2013, 9, 9303–9310 RSC.
- T. H. Ware and T. J. White, Polym. Chem., 2015, 6, 4835–4844 RSC.
- A. H. Gelebart, D. Jan Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS PubMed.
- P. J. Hogg, Trends Biochem. Sci., 2003, 28, 210–214 CrossRef CAS PubMed.
- W. J. Wedemeyer, E. Welker, M. Narayan and H. A. Scheraga, Biochemistry, 2000, 39, 4207–4216 CrossRef CAS PubMed.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS.
- Y. Li, O. Rios, J. K. Keum, J. Chen and M. R. Kessler, ACS Appl. Mater. Interfaces, 2016, 8, 15750–15757 CAS.
- K. M. Lee, N. V. Tabiryan, T. J. Bunning and T. J. White, J. Mater. Chem., 2012, 22, 691–698 RSC.
- T. Ikeda, J.-i. Mamiya and Y. Yu, Angew. Chem., Int. Ed., 2007, 46, 506–528 CrossRef CAS PubMed.
- T. J. White, S. V. Serak, N. V. Tabiryan, R. A. Vaia and T. J. Bunning, J. Mater. Chem., 2009, 19, 1080–1085 RSC.
- K. M. Lee, H. Koerner, R. A. Vaia, T. J. Bunning and T. J. White, Soft Matter, 2011, 7, 4318–4324 RSC.
- L. M. Johnson, E. Ledet, N. D. Huffman, S. L. Swarner, S. D. Shepherd, P. G. Durham and G. D. Rothrock, Polymer, 2015, 64, 84–92 CrossRef CAS.
- A. Takahashi, T. Ohishi, R. Goseki and H. Otsuka, Polymer, 2016, 82, 319–326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06343a |
| This journal is © The Royal Society of Chemistry 2017 |