
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic formation of C(sp3)–F bonds via decarboxylative fluorination with mechanochemically-prepared Ag2O/TiO2 heterogeneous catalysts†
G. Tarantino,
L. Botti,
N. Dimitratos and
C. Hammond *
*
Cardiff Catalysis Institute, Cardiff University School of Chemistry, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hammondc4@cardiff.ac.uk; Web: http://www.blogs.cardiff.ac.uk/hammond
First published on 12th June 2017
Abstract
Mechanochemically-prepared, Ag2O-containing solid materials, are shown to be efficient heterogeneous catalysts for the synthesis of C(sp3)–F bonds via decarboxylative fluorination. Five catalytic cycles without loss of intrinsic activity could be performed with the optimal catalyst, composed of 1 wt% Ag2O supported on TiO2 (P25), despite the challenging conditions. The catalyst is easily prepared from the corresponding oxides in 20 minutes by simple mechanical mixing methods. In addition to ease of separation and re-use, the turnover numbers obtained over the solid catalyst are over one order of magnitude higher than those obtained with the state-of-the-art homogeneous catalyst, AgNO3, under otherwise identical conditions. To the best of our knowledge, this represents the first true heterogeneous catalyst for the selective formation of C(sp3)–F bonds with electrophilic fluorine donors, representing a major breakthrough in the field of catalytic fluorination.
Introduction
Fluorinated compounds are widely employed in several industrial fields as agrochemicals, pharmaceuticals and materials.1 Furthermore, 18F compounds are widely used as tracers for Positron Emission Tomography.2 Unfortunately, the synthesis of fluorinated molecules is very difficult and whilst several breakthroughs3 have been achieved, the synthesis of C(sp3)–F bonds via catalytic fluorination remains an immense challenge. Whilst several recent studies have demonstrated that transition metals such as iron, copper, silver and palladium react with electrophilic fluorinating reagents such as F-TEDA (Selectfluor®), NFSI or DAST,1a to yield new C(sp3)–F and C(sp2)–F bonds,4 several of these methods require stoichiometric (i.e. 100 mol%) metal loadings (i.e. metal use is non-catalytic, hence TON < 1). Moreover, heterogeneous catalysis – preferred for several process intensification reasons – has not yet been achieved, likely due to the extremely challenging conditions, which can easily induce restructuring, leaching and hence, deactivation, of solid catalysts.The decarboxylative fluorination of aliphatic carboxylic acids, catalysed by homogeneous AgNO3, represents a powerful way to selectively synthesise new C(sp3)–F bonds from readily available substrates under mild reaction conditions (Scheme 1).5 However, despite the potential of this process, an active and reusable heterogeneous catalyst has not yet been developed, and catalyst activity remains extremely low, with typical turnover numbers being below 5 even after extended periods i.e. <24 h. Accordingly, the development of a highly active, stable and truly heterogeneous catalyst for this process is the focus of this study.
 | ||
| Scheme 1 Decarboxylative fluorination of aliphatic carboxylic acids to yield new C(sp3)–F bonds. | ||
Results and discussion
Amongst a range of metal oxide-supported, Ag-containing catalysts, we first identified 1 wt% Ag/TiO2, prepared by sol immobilisation6 (henceforth denoted 1AgTiO2(SI)), as being a suitable catalyst for decarboxylative fluorination. The performance of the material after various heat treatment procedures was first evaluated for the decarboxylative fluorination of 2,2-dimethylglutaric acid (DMGA, (1a)), a useful model substrate possessing both a primary and a tertiary carboxylic group (Scheme 2). Under conditions optimized from homogeneous AgNO3 (20 mol% Ag, 1.0 equivalent K2CO3, 25 °C) higher levels of activity were observed with the heterogeneous catalyst following calcination at 350 °C (Fig. 1, left). The only product observed over the timescale of the reaction by NMR and HPLC was 4-fluoro-4-methylpentanoic acid (4F4MPA (1b), selectivity > 95%) confirming the higher reactivity of the tertiary carboxylic group in accordance with previous studies.5 | ||
| Scheme 2 Decarboxylative fluorination of 1a to yield 1b with Selectfluor® as fluorine donor. | ||
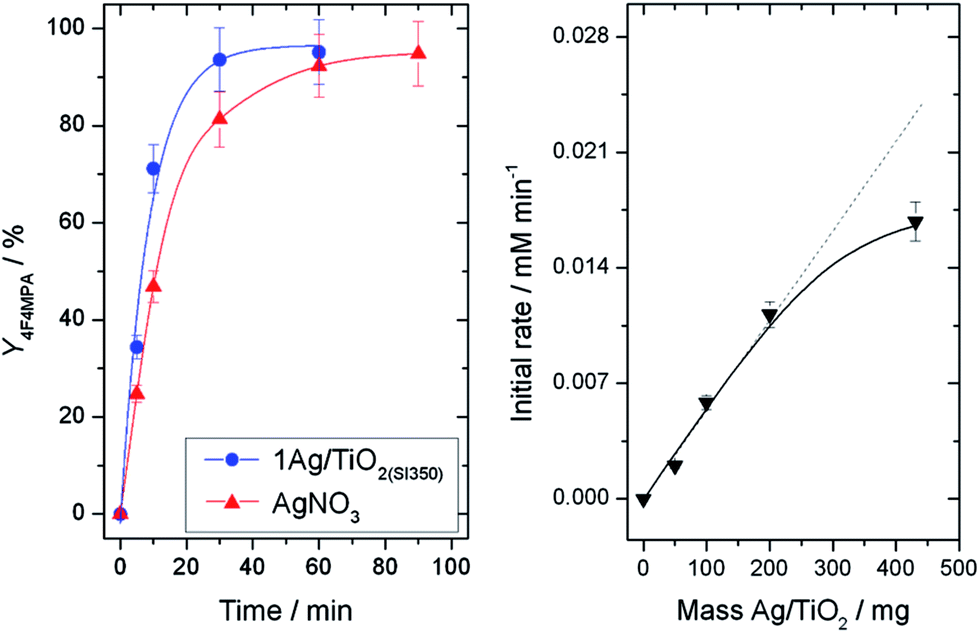 | ||
| Fig. 1 (Left) Yield of 1b with time over (blue/circles) 1Ag/TiO2(SI350) and (red/triangles) AgNO3. Reaction conditions: 0.2 mmol of 1a, 0.4 mmol of Selectfluor®, 0.04 mmol of Ag, 4 mL H2O, 0.2 mmol K2CO3, N2 atmosphere, 25 °C. (Right) Initial rate with different amounts of 1Ag/TiO2(SI350). Reaction conditions otherwise identical to left. | ||
Whilst the system exhibited good kinetic behaviour between 0–200 mg loading of 1Ag/TiO2(SI350) (Fig. 1, right) the rate deviated from linear above this loading, demonstrating transport limitations to be present at the highest catalyst loading. Accordingly, all further studies were performed with 100 mg of 1Ag/TiO2(SI350) (4.64 mol% Ag), which represents the best compromise between intrinsic activity and overall performance. To further optimise reaction conditions, the impact of the loading of base (in a range 0–2 equivalents) on the catalytic performance of 1Ag/TiO2(SI350) was investigated (ESI Fig. S1†). An optimal base loading of 1.16 equivalents (K2CO3/DMGA ratio of 1.16) was identified. Interestingly substantially lower activity was found in the absence of K2CO3.
The system under study represents a formidable challenge from a catalyst stability perspective, given that various amounts of carboxylic groups, fluorine sources and also HF (as possible reaction by-product)7 are present to some extent. Each of these species are known to cause extensive reorganisation, leaching and hence, permanent deactivation, of solid catalysts.8 To rule out any homogeneous contribution to the reaction rate, a hot filtration experiment at the optimal base loading was performed (Fig. 2, left).8d The removal of the solid catalyst from the reaction mixture clearly terminated the catalytic reaction, demonstrating catalysis to be truly heterogeneous, and confirming that potential traces of Ag in solution did not contribute to the reaction rate. Full termination of the reaction also implies Ag to be a true catalyst, and not solely a reaction initiator in the same manner as Cu(I) as observed by Pitts et al.7 TiO2-supported Ag is, therefore, the first truly heterogeneous catalysts for the formation of C(sp3)–F bonds with electrophilic fluorine donors.
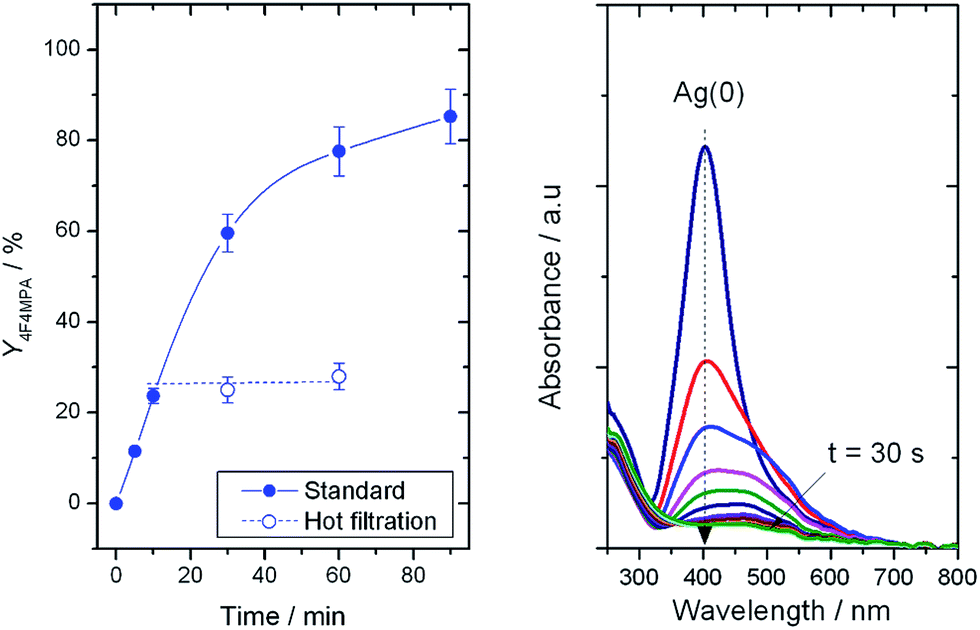 | ||
| Fig. 2 (Left) Solid line: yield of 1b with time without filtering the catalyst. Dashed line, catalytic activity of the supernatant solution following filtration of the catalyst at 10 min. Reaction conditions identical to Fig. 1, left, albeit with 0.23 mmol of K2CO3 and 100 mg of 1Ag/TiO2(SI350). (Right) UV-Vis spectra of colloidal Ag(0) after addition of Selectfluor® and K2CO3. | ||
According to previous studies, decarboxylative fluorination requires cationic Ag species for efficient catalysis to be observed.5 However, sol immobilisation catalysts are known to contain a heterogeneous distribution of cationic and metallic species following heat treatment.8,9 Thus, to understand the catalytic roles of Ag(0) and Ag(I), we investigated the interaction between Selectfluor® and Ag. Accordingly, a colloidal solution of Ag(0) was placed into a UV-Vis cuvette, and the Plasmon resonance10 of Ag(0) at 400 nm monitored in situ following addition of Selectfluor® and K2CO3 (Fig. 2, right). This treatment resulted in the disappearance of the Plasmon peak in <30 s. Clearly, any residual Ag(0) species in the solid catalysts are immediately oxidised to Ag(I) upon interaction with Selectfluor®, and cationic Ag is responsible for catalytic performance.
Given the requirement for cationic Ag active sites, the preparation of materials directly containing Ag(I), and not Ag(0), was targeted. Mechanochemistry, whereby mechanical energy is employed to facilitate chemical processes, has recently received considerable attention as a sustainable and scalable method of catalyst synthesis.11 In addition, it allows metal oxides such as Ag2O to be directly employed as solid precursors of the desired active phase.11c We first investigated the mechanochemical synthesis of Ag2O/TiO2 catalysts by simple physical grinding (denoted 1Ag2O/TiO2(PM)). Physically grinding mixtures of Ag2O and TiO2 in a pestle and mortar for 20 min led to a catalyst demonstrating almost identical behaviour to heat-treated 1Ag/TiO2(SI350), despite the rapidity of the method and the complete absence of high temperature heat treatments (Fig. 3, left). Notably, in the absence of the TiO2 support i.e. when Ag2O is ground alone, and in the absence of the grinding stage i.e. when Ag2O and TiO2 were placed into the reaction mixture together without prior physical contact, high levels of leaching occurred (Fig. 3, right). Moreover, homogeneous contributions to the reaction rate were observed (ESI Fig. S2†). Clearly, obtaining strong interaction between Ag2O and TiO2 is essential. We note that homogeneous catalytic activity with Ag2O has previously been reported by the Ritter group.12
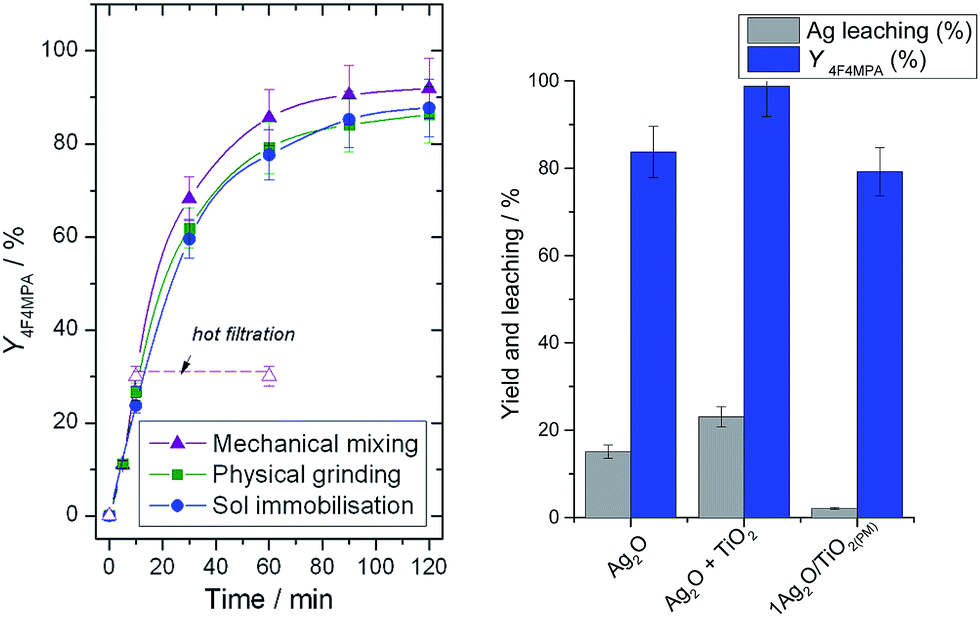 | ||
| Fig. 3 (Left) Yield of 1b against time for 1Ag2O/TiO2(PM) (green squares), 1Ag/TiO2(SI350) (blue circles) and 1Ag2O/TiO2(MM) (purple triangles). Reaction conditions as Fig. 2, left. (Right) Ag leaching (grey bars) at 10 min and yield of 1b (blue bars) at 60 min over neat Ag2O, not supported Ag2O + TiO2, and 1Ag2O/TiO2(PM). | ||
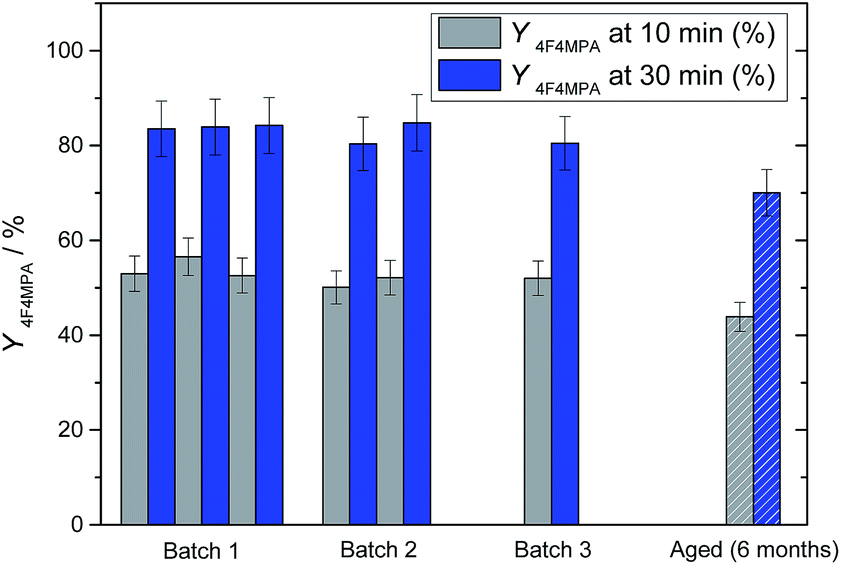 | ||
| Fig. 4 Activity and reproducibility of three different batches of 1Ag2O/TiO2(MM). Grey bars are percentage yield of 1b at 10 min, blue bars conversion at 30 min. White and blue/grey bars correspond to the activity of 1Ag2O/TiO2(MM) aged for 6 months. Reaction conditions as Fig. 2, right, 30 °C. | ||
In view of this, and to allow a greater control of the preparation conditions, thus improving the reproducibility of the preparation method (ESI, Fig. S3†), the optimal catalyst composition was re-made by mechanochemical mixing in a high frequency milling machine. Mixing Ag2O and TiO2 for 20 minutes at 15 Hz led to the formation of a catalyst that was both more active and more stable than the sol immobilisation and physical grinding analogues (the catalyst is denoted 1Ag2O/TiO2(MM)). For 1Ag2O/TiO2(MM), catalysis was also totally heterogeneous (Fig. 3 (left)), and fully reproducible and stable over long periods of time (Fig. 4). Preparing an effective and reproducible catalyst directly from Ag2O so rapidly, without the presence of solvents or other additives, and in readily available experimental apparatus, presents several synthetic advantages over other catalyst preparation methodologies, such as sol immobilisation (Scheme 3).
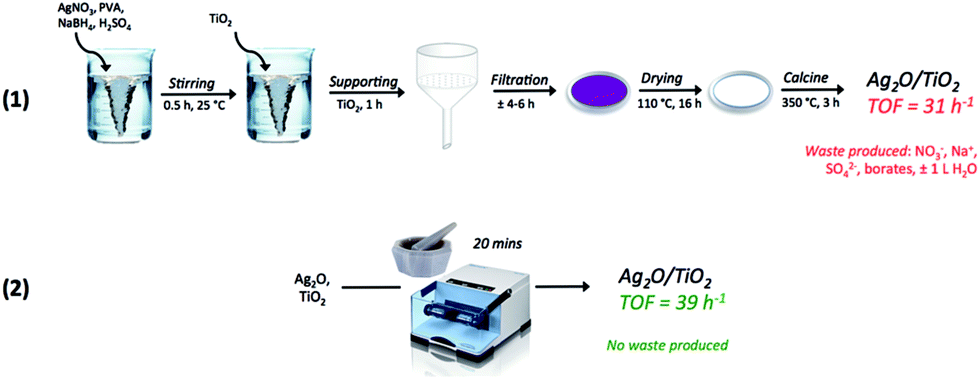 | ||
| Scheme 3 Graphical representation of classical “wet” preparation methodologies e.g. sol immobilisation (1), and physical methods e.g. mechanochemical mixing (2). | ||
To further optimize the preparation of 1Ag2O/TiO2(MM), a series of different milling frequencies, in a range 3–30 Hz, was explored (Fig. 5). Interestingly, increasing the milling frequency results in a decrease in catalytic activity, with 1b yield values decreasing from 95% to around 10% over the frequency series. Nevertheless, while metal leaching values below the instrument detectability limits were detected by MP-AES analysis for 1Ag2O/TiO2(MM) milled at higher frequencies (15 Hz, 22.5 and 30 Hz), leaching values of 6% and 5% were found for the two lowest frequencies (3 Hz and 7.5 Hz, respectively). This demonstrates that although low frequencies result in active materials, they do not provide sufficient energy to result in strong metal-support interactions nor synthesise a truly heterogeneous catalysts. Accordingly, all further studies utilised 1Ag2O/TiO2(MM) milled at 15 Hz.
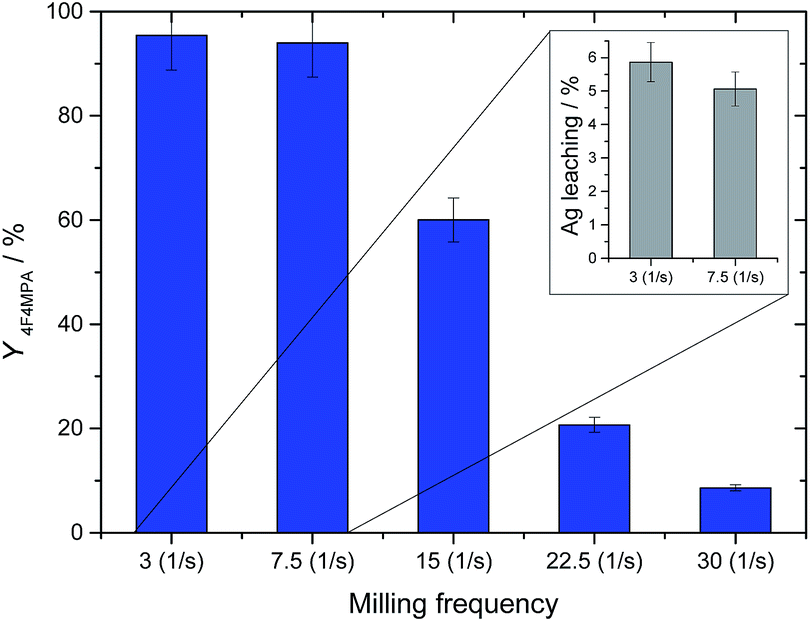 | ||
| Fig. 5 Yield of 1b (blue bars) at 30 min with 1% Ag2O/TiO2 milled at various frequencies. Inset on the top right: Ag leaching values at 10 min for 1% Ag2O/TiO2 milled at 3 Hz and 7.5 Hz. Reaction conditions as Fig. 2, right, room temperature. | ||
To fully understand the effect of the different frequencies on the Ag speciation and the resultant performances of the catalysts, detailed characterization of the mechanochemically-prepared catalysts with Powder X-ray Diffraction (pXRD), Temperature Programmed Reduction (TPR) and X-ray Photoelectron Spectroscopy (XPS) was performed. XRD analysis (Fig. 6, left) reveals that upon increasing the frequency from 3 to 30 Hz, the reflection at 32.7°, corresponding to Ag2O, decreases in intensity, and a new reflection at 32.2°, characteristic of AgO, appears. As reported in literature, AgO is known to be a mixture of Ag(III) and Ag(I) oxides,13 and will therefore be denoted as Ag2O2 herein. In agreement with the trend seen in the XRD patterns, the presence of multiple Ag species in the higher frequency materials was also observed by TPR (Fig. 6, right). The TPR profile of 1Ag2O/TiO2(MM) prepared at 3 Hz clearly shows only one intense reduction step at 240 °C, at temperatures higher than that found for unsupported Ag2O (196 °C). We attribute this reduction peak to Ag2O interacting with the support material. Upon increasing the milling frequency from 3 Hz to 22.5 Hz, the reduction step at 240 °C gradually decreases, whilst a new reduction feature appears around 160–170 °C. This new feature perfectly overlaps with the reduction temperature measured for unsupported Ag2O2. The presence of two features in these samples indicates the presence of two distinct Ag species in these materials. At the highest frequency, only Ag2O2 was observed, indicating full transformation of Ag2O to Ag2O2. Although several of the Ag signals overlap, XPS analysis also revealed the presence of multiple AgOx species in the optimal material prepared at 15 Hz (Fig. 7, left). Deconvolution of the broad asymmetrical peak observed for 1Ag2O/TiO2(MM) into two different components; firstly, a signal at 368.0 eV, perfectly overlapping with the signal obtained for Ag2O; secondly, a signal at lower binding energy (367 eV), assigned in the literature to Ag(III).14
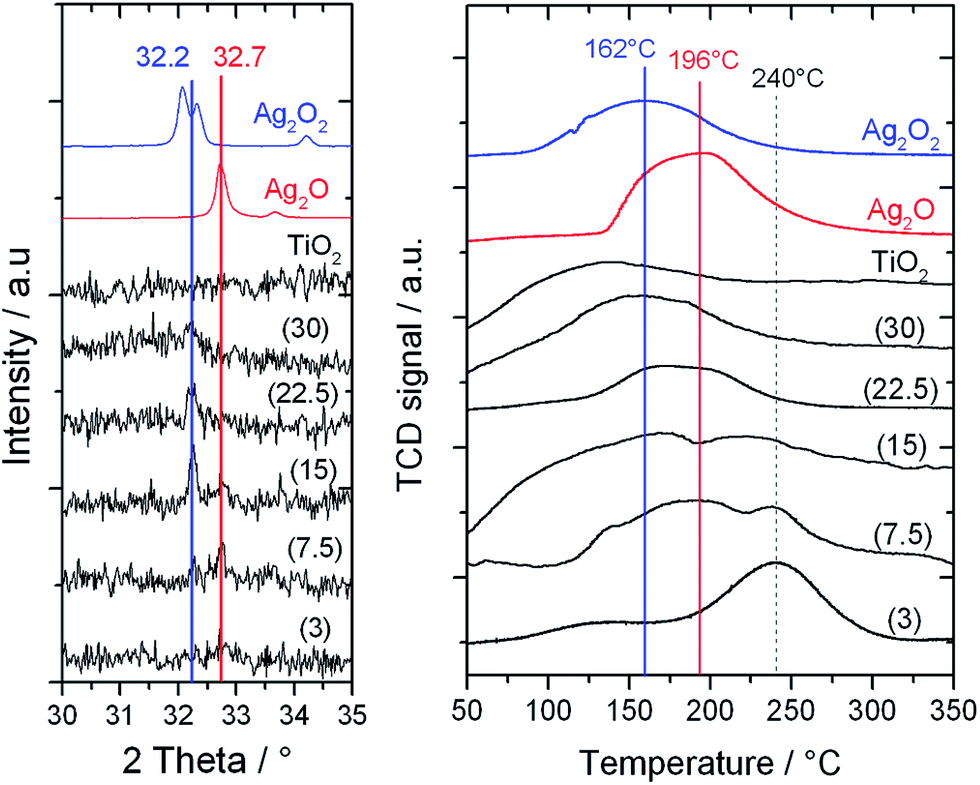 | ||
| Fig. 6 (Left) XRD patterns of 1Ag2O/TiO2(MM) prepared at different milling frequencies (3–30 Hz). (Right) TPR profiles of 1Ag2O/TiO2(MM) prepared at different milling frequencies (3–30 Hz). | ||
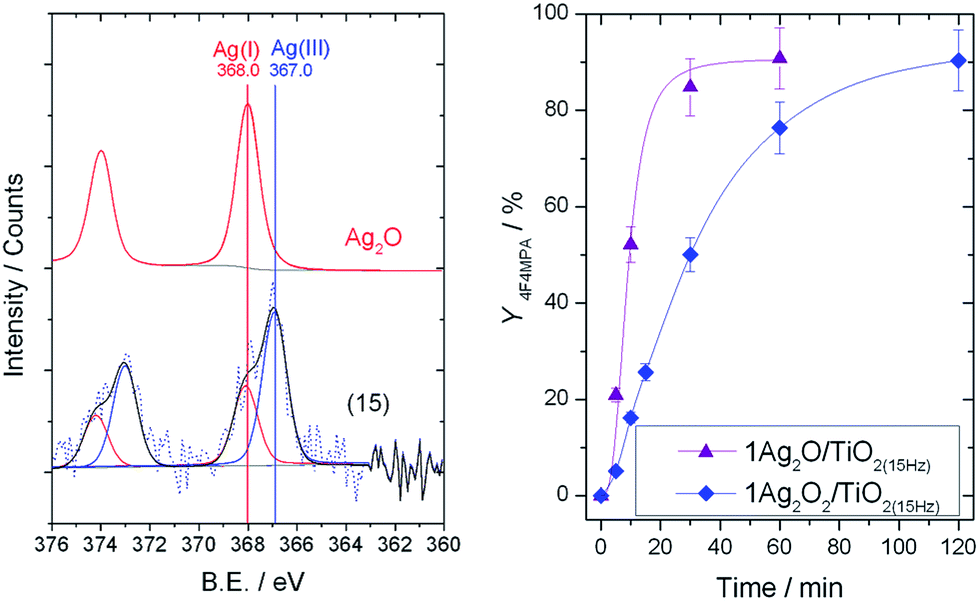 | ||
| Fig. 7 (Left) XPS analysis of commercial Ag2O and 1Ag2O/TiO2(MM) prepared at 15 Hz. (Right) Yield of 1b against time over 1Ag2O/TiO2(MM) (purple triangles), and 1Ag2O2/TiO2(MM) (blue rhomboids) both prepared at 15 Hz. Reaction conditions as Fig. 2, right, 30 °C. | ||
Clearly, increasing the energy of the mechanochemical preparation by increasing the milling frequency results in a transformation of Ag2O to Ag2O2, with a distribution of both oxides being observed at the optimal frequency (15 Hz).
An increase in Ag2O2 formation at higher frequencies appears to correlate with decreased catalytic performance (vide supra). To verify that the loss in activity observed upon increasing the milling frequency from 3–15 Hz correlates to the partial conversion of Ag2O into Ag2O2, 1Ag2O2/TiO2(MM) was prepared by milling Ag2O2 with TiO2 at 15 Hz. As expected, substantially decreased performance was observed (Fig. 7). Powder X-ray Diffraction (pXRD) analysis of 1Ag2O2/TiO2(MM) demonstrates that only Ag2O2 is present in this material (ESI Fig. S4†).
The loss of activity observed for 1Ag2O2/TiO2(MM) agrees well with the reaction mechanisms hypothesised in previous works.5 According to these studies, the key reaction step involves the oxidation of a Ag(I)-COOR species by Selectfluor®. It is likely that Ag(III) (present as approximately 50% of the Ag content in Ag2O2) cannot be further oxidized, thus stopping the reaction cycle and acting as a “trap”. According to this, it would be expected that catalysts milled at the highest frequencies (22.5 and 30 Hz) should still display some levels of catalytic performance, given that Ag2O2 contains approximately 50% Ag(I). We hypothesise that the greater than expected decrease in activity for these materials may be due to decreased crystallinity of the support, as evidenced by XRD and BET measurements (ESI Fig. S5 and Table S1†).
The general applicability of the heterogeneous catalytic system in terms of C(sp3)–F bond formation was also investigated with 1Ag2O/TiO2(MM) milled at 15 Hz as catalyst. These studies demonstrated the catalytic system to be versatile and able to convert even difficult primary carboxylic groups such as succinic acid (3a, Table 1 entry 3). In all cases, catalytic numbers of turnovers were observed within a short period of time (≤1 h). A strong influence of K2CO3 loading on both the activity and stability of the 1Ag2O/TiO2(MM) was also observed with these substrates, such as pivalic acid (2a, Table 1 entry 2) and 2,2-dimethyl-valeric acid (4a, Table 1 entry 4). In both cases, a two-fold increase in the TON values, 18.6 and 6.47 respectively, was observed when only 0.58 equivalents of K2CO3 were used (Table 1, entries 2 and 4). Optimizing the base![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carboxylic acid ratio for each individual substrate is critical for obtaining the highest levels of catalytic performance. Substrates not soluble in neat water, such as 2,2-dimethyl-3-phenylpropionic acid (5a), were also efficiently converted into the corresponding C(sp3)–F fluorinated compounds (5b, Table 1 entry 5). In this case, a water:acetone mixture (1:1) was employed as solvent, and TONs comparable or better to the ones obtained using AgNO3 were observed.5
:carboxylic acid ratio for each individual substrate is critical for obtaining the highest levels of catalytic performance. Substrates not soluble in neat water, such as 2,2-dimethyl-3-phenylpropionic acid (5a), were also efficiently converted into the corresponding C(sp3)–F fluorinated compounds (5b, Table 1 entry 5). In this case, a water:acetone mixture (1:1) was employed as solvent, and TONs comparable or better to the ones obtained using AgNO3 were observed.5
| Entry | Product | TONa | |
|---|---|---|---|
| a TON calculated as “mol (product)/mol (Ag)” for entries 1 and 3–5, and as “mol (2a converted)/mol(Ag)” for entry 2.b Reaction time 1 h.c 0.115 mmol K2CO3.d 200 mg 1Ag2O/TiO2(MM), 50 °C, 2 mL H2O.e 200 mg 1Ag/TiO2(MM), H2O/acetone (2 mL/2 mL), 60 °C. | |||
| 1 | 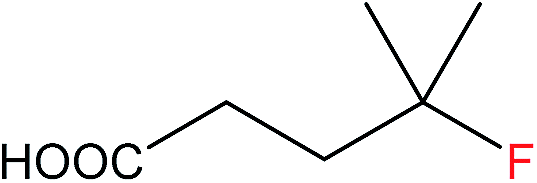 |
1b | 17.8 (34.2 after 2 cycles) |
| 2 |  |
2b | 9.6b, 18.6c |
| 3 | 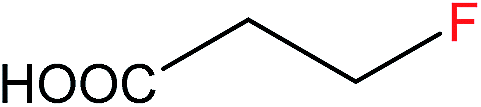 |
3b | 4.0d |
| 4 | 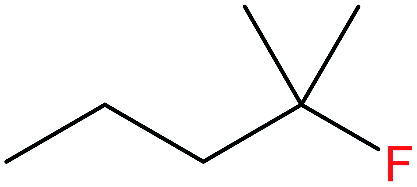 |
4b | 3.25b, 6.47b,c (14.6 after 5 cycles)b |
| 5 | 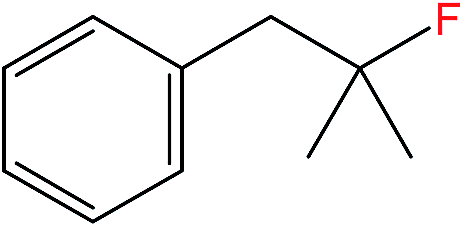 |
5b | 3.21c,e |
A major advantage of a heterogeneous catalyst over a homogeneous analogue is the ease in which it can be recovered and reused. Accordingly, we first explored the reusability of 1Ag2O/TiO2(MM) with 4a as substrate. To ensure that true kinetic comparisons were made, the activity of the catalyst in multiple cycles was examined at low conversion, so that the intrinsic activity and reusability of the catalyst was correctly examined.8b,d As can be seen (Fig. 8, left), 1Ag2O/TiO2(MM) exhibited good levels of reusability, even without any intermediate treatments being performed. Substrates containing additional functional groups, such as 1a, presented a greater challenge. Indeed, in the absence of secondary treatments, a large drop in the catalytic activity of 1Ag2O/TiO2(MM) was observed during the second cycle for 1a decarboyxlative fluorination (ESI Fig. S6†). However, even in these cases, the initial level of performance could be fully restored by suspending the catalyst in an aqueous K2CO3 solution for 60 min between successive cycles (Fig. 8, right). We hypothesize that removal of residual carboxylic species by K2CO3 is responsible for improved recyclability, as such species are well established poisons of metal-based catalysts.8a It is therefore clear that the heterogeneous catalyst possesses promising stability characteristics for intensified operation. For 4a and 1a, TONs over one order of magnitude higher than previously reported were obtained after 5 and 2 cycles, respectively.
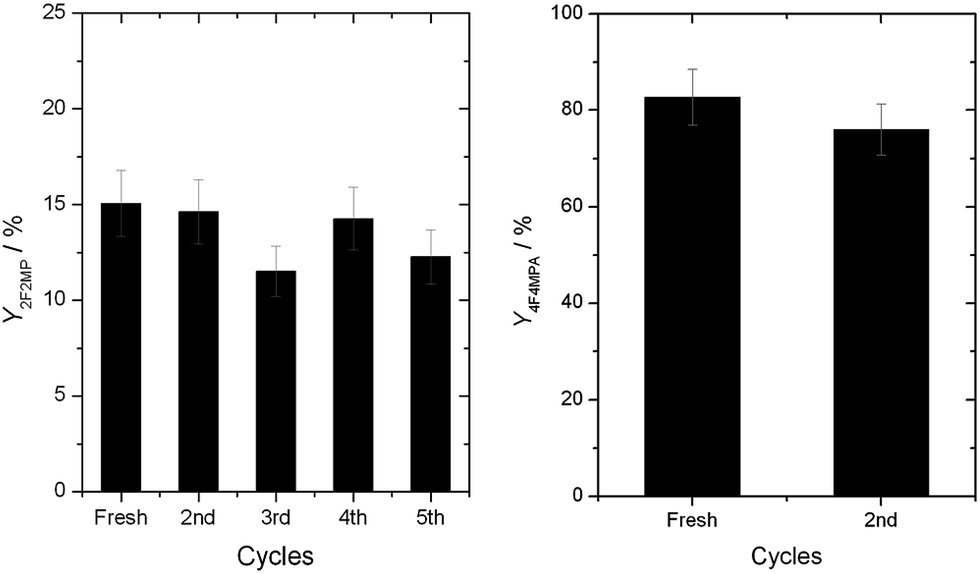 | ||
| Fig. 8 (Left) Reusability of 1Ag2O/TiO2(MM) at 30 °C with 4a as substrate. No intermediate treatments were performed. (Right) Reusability of 1Ag2O/TiO2(MM) at 30 °C with 1a as substrate. The catalyst was washed in an aqueous K2CO3 solution (0.06 M) for 1 h between cycles. Reaction conditions otherwise as Fig. 2, left. | ||
Conclusions
Mechanochemically-prepared 1 wt% Ag2O/TiO2 is found to be an efficient and reusable heterogeneous catalyst for the decarboxylative fluorination of various carboxylic acids in aqueous media at 25 °C. The mechanochemical mixture presents higher levels of catalytic activity than the state-of-the-art homogeneous catalyst, and is also easily recovered and reused over five successive catalytic cycles. Good general applicability of the catalytic system is also observed, with a variety of primary and tertiary carboxylic acid groups being selectively converted to the desired C(sp3)–F products. Spectroscopic characterisation of the mechanochemically-prepared materials indicates that phase transformations between Ag2O and Ag2O2 can occur, and that the optimal milling frequency in terms of activity and stability is 15 Hz.Acknowledgements
CH gratefully acknowledges The Royal Society for research funding and provision of a University Research Fellowship (UF140207).References
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) T. Ritter, Nature, 2010, 466, 447 CrossRef CAS PubMed.
- (a) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS PubMed; (b) R. Littich and P. J. H. Scott, Angew. Chem., Int. Ed., 2012, 51, 1106 CrossRef CAS PubMed; (c) A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545 RSC.
- (a) W. Liu, X. Huang, M. J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed; (b) M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570 CrossRef CAS PubMed; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (d) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024 CrossRef CAS PubMed; (e) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed; (f) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. Garcia-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed; (g) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed.
- (a) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588 CrossRef CAS PubMed; (b) T. Furuya, H. M. Kaiser and T. Ritter, Angew. Chem., Int. Ed., 2008, 47, 5993 CrossRef CAS PubMed; (c) S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722 CrossRef CAS PubMed; (d) P. S. Fier, J. Luo and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2552 CrossRef CAS PubMed; (e) A. R. Mazzotti, M. G. Campbell, P. Tang, J. M. Murphy and T. Ritter, J. Am. Chem. Soc., 2013, 135, 14012 CrossRef CAS PubMed; (f) T. Furuya, A. E. Strom and T. Ritter, J. Am. Chem. Soc., 2009, 131, 1662 CrossRef CAS PubMed; (g) N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224 CrossRef CAS PubMed; (h) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 4648 CrossRef CAS PubMed.
- (a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (b) N. R. Patel and R. A. Flowers, J. Org. Chem., 2015, 80, 5834 CrossRef CAS PubMed; (c) X. Zhang, Comput. Theor. Chem., 2016, 1082, 11 CrossRef CAS.
- J. Kennedy, J. Wilm, D. J. Morgan, M. Bowker, L. Lu, C. J. Kiely, P. P. Wells and N. Dimitratos, Catal., Struct. React., 2015, 1, 35 CrossRef.
- C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Leckta, J. Am. Chem. Soc., 2014, 136, 9780 CrossRef CAS PubMed.
- (a) S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242 CrossRef CAS; (b) I. Sadaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133 RSC; (c) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed; (d) C. Hammond, Green Chem., 2017 10.1039/C7GC00163K.
- R. C. Tiruvalam, J. C. Pritchard, N. Dimitratos, J. A. Lopez-Sanchez, J. K. Edwards, A. F. Carley, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2011, 152, 63 RSC.
- V. Amendola, O. M. Bakr and F. Stellacci, Plasmonics, 2010, 5, 85 CrossRef CAS.
- (a) C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736 CrossRef CAS PubMed; (b) C. Hammond, D. Padovan, A. Al-Nayili, E. K. Gibson, P. P. Wells and N. Dimitratos, ChemCatChem, 2015, 7, 3322 CrossRef CAS PubMed; (c) C. Hammond, J. Straus, M. Righetonni, S. E. Pratsinis and I. Hermans, ACS Catal., 2013, 3, 321 CrossRef CAS.
- (a) P. Tang and T. Ritter, Tetrahedron, 2011, 67, 4449 CrossRef CAS PubMed; (b) P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150 CrossRef CAS PubMed.
- D. Tudela, J. Chem. Educ., 2008, 85, 863 CrossRef CAS.
- (a) G. B. Hoflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B, 2000, 62, 11126 CrossRef CAS; (b) A. M. Ferraria, A. P. Carapeto and A. M. Botelho do Rego, Vacuum, 2012, 86, 1988 CrossRef CAS; (c) J. F. Weaver and G. B. Hoflund, J. Phys. Chem., 1994, 98, 8519 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06180c |
| This journal is © The Royal Society of Chemistry 2017 |