
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polycaprolactone-templated reduced-graphene oxide liquid crystal nanofibers towards biomedical applications†
Sasan Jalili-Firoozinezhad‡§
 ab,
Mohamad Hasan Mohamadzadeh Moghadam‡c,
Mohammad Hossein Ghanianb,
Mohammad Kazemi Ashtianib,
Hossein Alimadadid,
Hossein Baharvand*be,
Ivan Martin*a and
Arnaud Scherbericha
ab,
Mohamad Hasan Mohamadzadeh Moghadam‡c,
Mohammad Hossein Ghanianb,
Mohammad Kazemi Ashtianib,
Hossein Alimadadid,
Hossein Baharvand*be,
Ivan Martin*a and
Arnaud Scherbericha
aDepartment of Biomedicine, University Hospital Basel, University of Basel, Hebelstrasse 20, CH-4031 Basel, Switzerland. E-mail: ivan.martin@usb.ch
bDepartment of Stem Cells and Developmental Biology, Cell Science Research Center, Royan Institute for Stem Cell Biology and Technology, ACECR, Tehran, Iran. E-mail: baharvand@royaninstitute.org
cCondensed Matter National Laboratory, Institute for Research in Fundamental Sciences, Tehran, 19395-5531, Iran
dCenter for Electron Nanoscopy, Technical University of Denmark, Fysikvej, Building 307, DK-2800 Kongens Lyngby, Denmark
eDepartment of Developmental Biology, University of Science and Culture, Tehran, Iran
First published on 14th August 2017
Abstract
Here, we report a facile method to generate electrically conductive nanofibers by coating and subsequently chemically reducing graphene oxide (GO) liquid crystals on a polycaprolactone (PCL) mat. Ultra large GO sheets obtained are in favor of charge carrier mobility and oriented morphology of the GO coating. We showed that coating the reduced GO (rGO) not only retains the three-dimensional topography, fiber orientation and size of the template PCL, but also makes it electroconductive. Our preliminary in vitro assessments using mesenchymal stem cells revealed no induced cytotoxicity yet increased cellular metabolism on PCL-templated rGO fibers.
Introduction
To date, electroactive nanofibrous substrates have been widely employed in various applications such as fuel cells, sensors and batteries.1 High surface-to-volume ratio, providing electrical cues and the capability to recapitulate the native extracellular matrix (ECM) architecture make electroconductive nanofibers an ideal platform for cell culture, tissue engineering, drug delivery and biosensor applications.2,3 Among different fabrication strategies, electrospinning serves as a facile, inexpensive, high-adaptable and scalable method to generate nanoscale fibrous structures from a wide variety of polymers.4–7Graphene, a single layer carbon crystal with a packed honeycomb lattice structure, shows exceptional physicochemical, thermal and electrical properties as compared to other carbon materials.8–10 Thanks to its biocompatibility and potential to regulate cellular behaviors, graphene has been employed in various biomedical fields.11,12 Although graphene can be formed in different shapes and architectures, efforts to make graphene nanofibers have been hampered by irregular shape and size of graphene sheets, and lack of efficient assembly methods. However, a new strategy has recently emerged to facilitate the generation of graphene oxide (GO) sheets and their coating on top of electrospun poly-L-lactide or poly(vinyl chloride) fiber meshes.13,14 Adapting this approach, we immobilized GO liquid crystal phases onto polycaprolactone (PCL) nanofibers, used as a template, and then reduced the GO (rGO), to form PCL-templated graphene nanofibers.
Results and discussion
In the current study, we took benefit of GO dispersion in the nematic phase, where large GO sheets could undergo liquid crystalline phase transition at very low concentration. While inter-sheet contacts and inter-layer gaps between small GO sheets compromise 2D conductivity,15 large GO liquid crystals obtained in the current work deliver excellent electrical properties. Having proper electrical conductivity and nanofibrous morphology at the same time would make our construct suitable for various biomedical applications.GO was synthesized using the modified Hummers' method16 (Fig. 1A) in which, graphite oxide can be swelled in water (and polar organic compounds),17,18 followed by a spontaneous exfoliation as ions and impurities are being removed.17 This process was observed in our experiment as well (Fig. S1A†). Chemical treatment of graphite introduces oxygenated functional groups on its surface and edges. Using Fourier transform infrared (FTIR) spectrum (Fig. 2A), the main functional groups were recognized: epoxy (1072 cm−1, –O– stretching), hydroxyl (1412 cm−1, O–H deformation), carboxyl or carbonyl (1620 cm−1, O–H stretching and 1710 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching).19–21 Ultraviolet-visible (UV-vis) spectroscopy showed a distinct peak at 230 nm (due to π–π* transition of CC bond in sp2-portion of GO) along with a shoulder at 300 nm (due to n–π* transition of C–O bond in oxidized-portion of GO) (Fig. 2B, S1B and C†).
O stretching).19–21 Ultraviolet-visible (UV-vis) spectroscopy showed a distinct peak at 230 nm (due to π–π* transition of CC bond in sp2-portion of GO) along with a shoulder at 300 nm (due to n–π* transition of C–O bond in oxidized-portion of GO) (Fig. 2B, S1B and C†).
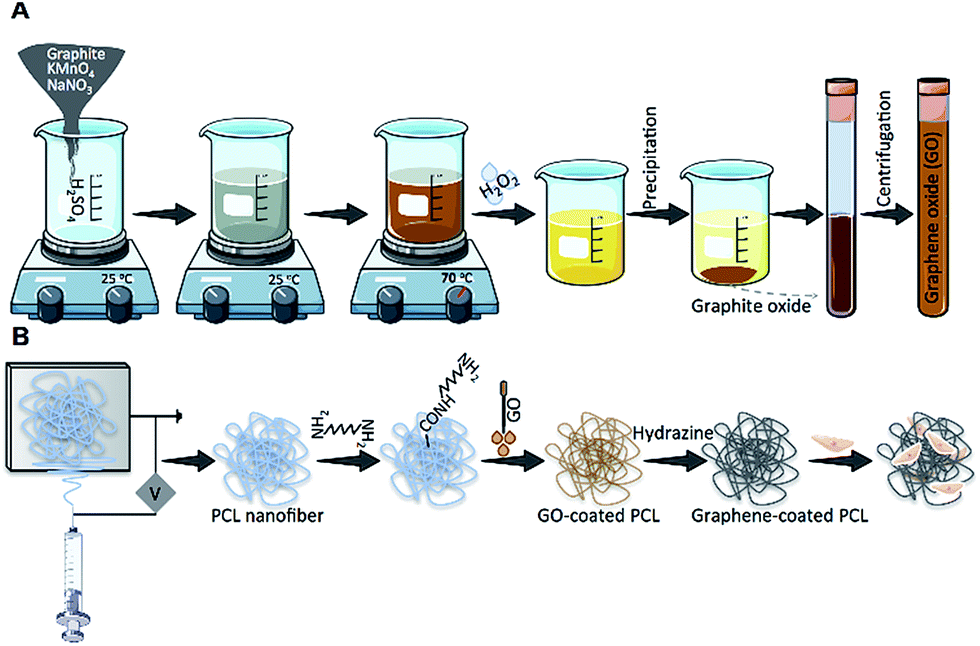 | ||
| Fig. 1 Schematic illustration of (A) graphene oxide (GO) synthesis, (B) polycaprolactone (PCL) electrospinning, fabrication of rGO-coated PCL nanofibers (PCL–rGO) and in vitro cell seeding. | ||
 | ||
| Fig. 2 Graphene oxide (GO) characterization. (A) FTIR spectrum of GO. The main peaks attributed to the functional groups on GO sheets were indicated by arrows. (B) UV-vis absorption spectrum of stable GO aqueous dispersion. (C) X-ray diffraction (XRD) pattern of GO film. (D) Representative atomic force microscopy (AFM) micrograph and (E) height profile of an exfoliated GO sheet (the cursor pair) showing single-layered structures with approximately 0.8 nm thickness. (F) Statistical analysis of sheet thickness by AFM performed on 50 randomly selected sheets. | ||
The presence of oxygen moieties results in increment in interlayer distance between GO sheets. Based on XRD measurement, one peak centred at 11° was found which is typical for GO sheets (Fig. 2C). By applying Bragg's law (nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ), the d-spacing in GO was calculated around 0.8 nm which was in accordance with previous studies.18,22–24 The exfoliation of GO into single layer sheets has significant importance in the case of processing and eventual application. In this regard, atomic force microscopy (AFM) measurement was used to study the thickness of GO sheets. From images shown in Fig. 2D–F, a thickness of ∼0.8 nm was obtained which is in line with X-ray diffraction (XRD) result.
sinθ), the d-spacing in GO was calculated around 0.8 nm which was in accordance with previous studies.18,22–24 The exfoliation of GO into single layer sheets has significant importance in the case of processing and eventual application. In this regard, atomic force microscopy (AFM) measurement was used to study the thickness of GO sheets. From images shown in Fig. 2D–F, a thickness of ∼0.8 nm was obtained which is in line with X-ray diffraction (XRD) result.
Electrical properties of GO coatings depend on its lateral size.25–27 Bigger lateral sizes of GO results in the higher electrical conductivity.28,29 Therefore, preserving lateral size of GO sheet during synthesis and fabrication is a key factor. Applying several centrifuging steps, small sheets were removed, hence, GO dispersion containing large sheets was obtained. Scanning electron microscopy (SEM) images of GO sheets alongside optical microscopy indicated the presence of large GO sheets (Fig. S2; 3A and C; S1D and E†). The size distribution of GO sheets was calculated as 21 ± 12 μm (Fig. 3B). Aqueous dispersion of large GO possesses a unique property in which it can undergo liquid crystalline phase transition at very low concentration.30,31 In other words, stacks of GO sheets in liquid phase will be oriented in a special direction which enables more facile fabrication process.30 As shown in Fig. 3D, GO dispersion is in the nematic phase at concentration of 0.25 mg ml−1. It should be taken into account that orientation of these sheets is favorable when achieving high electrical conductivity.
 | ||
| Fig. 3 (A) SEM image of large area of GO sheet on Si/SiO2 substrate. Wrinkling can also be seen on basal plane of GO. Scale bar: 10 μm. (B) Size distribution of GO sheets calculated from 108 images. The average size is 21 μm and the standard deviation is 12 μm. Minimum and maximum size was measured 2 and 51 μm, respectively. (C) Optical micrograph of large area GO sheet on coverslip. Scale bar: 20 μm. (D) Polarized optical microscopy (POM) micrograph of GO sheet at 0.25 mg ml−1. The bright part is due to orientation of stacks of GO sheet against incident light. | ||
Electrospinning of graphene is quite challenging due to the irregular size/shape of chemically derived graphenes and the movable layer-by-layer stacking of graphenes, which could obstruct direct assembly of 2D microscopic graphene sheets into macroscopic nanofiber. Although many strategies have been devised to generate composite nanofiber substrates based on graphene,32,33 which fail to provide pure graphene interface, an alternative strategy is to use a polymeric nanofiber mat template, e.g. PCL, to immobilize GO on the surface (Fig. 1B). However, introduction of –NH2 groups onto the PCL mat template is essential for even and stable coating of GO sheets. The ninhydrin assay was used to determine the efficiency of hexamethylenediamine (HMD) in introducing amino groups on the surface of the PCL fibrous substrate. GO could be easily immobilized on surface through electrostatic attractions or hydrogen bonding between amino moieties of PCL on one side and hydroxyl, carboxyl and ether groups of GO on the other side.13,34 HMD, as an aminolyzing reagent, has been previously reported for inducing amino groups on PCL nanofibrous scaffolds for subsequent immobilization with biomolecules. HMD-aminolyzation enhances the cytocompatibility and hydrophilicity of PCL substrates without cross-reacting with, adsorbing to or changing diameter of PCL fibers.35,36 As described previously,35 UV-vis spectrophotometry was used to evaluate amination success. The presence of amino groups was confirmed by recording a more intense absorption band at 560 nm (32.5% increase) on aminolyzed PCL (Fig. 4A). To obtain rGO-coated fibers, GO was first tightly immobilized onto clean PCL mat, which gave a brownish shade to the fibers' color. Through chemical reduction of GO-coated fibers by hydrazine vapor, rGO-coated substrates were obtained. Addition of hydrazine hydrate altered the GO color from brownish to dark gray, confirming the reduction of GO to rGO (Fig. 4B). To evaluate morphological features, substrates were investigated by SEM. As shown in Fig. 5A, PCL nanofibers appeared to have a uniform randomly oriented bead-free morphology. As depicted in Fig. 5C–D, more than 50% of fibers were distributed in the size range of 400–1000 nm, with average fiber diameter of 852.2 ± 205.4 nm. Graphene coating did not change the topography of PCL nanofibers (Fig. 5B), so that the average fiber diameter (921.5 ± 270.6 nm) and distribution of PCL–rGO resembled those of PCL (Fig. 5C and D).
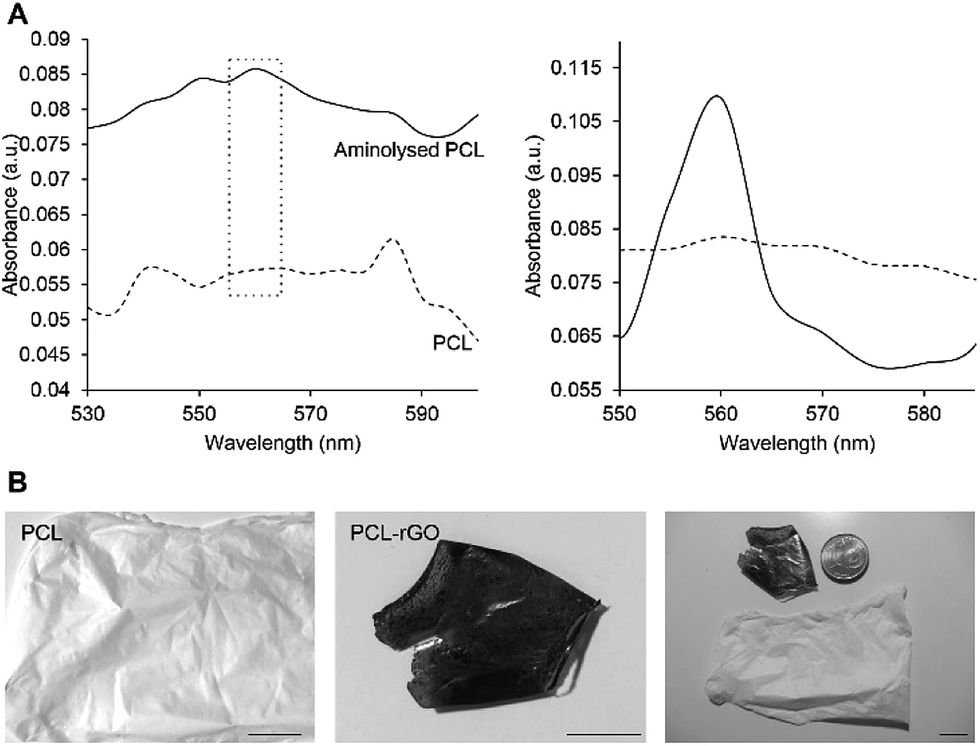 | ||
| Fig. 4 (A) UV-vis spectra of polycaprolactone (PCL) and aminolysed PCL samples. Right panel represents the difference between absorbance patterns of two substrates. (B) Digital images of electrospun polycaprolactone (PCL) and graphene-coated PCL (PCL–rGO) substrates. Scale bars: 10 mm. | ||
 | ||
| Fig. 5 Morphological and physical characterization of samples. (A) Representative field emission-scanning electron microscopy (FE-SEM) micrographs of (A) polycaprolactone (PCL) nanofibers and (B) graphene-coated PCL nanofibers (PCL–rGO). (C) Fiber diameter distribution and (D) average fiber diameter in intact and graphene coated nanofibers. (E) Electrical conductivity measurements of PCL and PCL–rGO. Scale bar A – left panel: 100 μm, B – left panel: 50 μm, A – middle panel, B – middle panel: 30 μm, A – right panel, B – right panel: 5 μm. ns: not significant. | ||
Fiber diameter plays an important role in modulating the adhesion, migration, proliferation and differentiation of cells. We used fibers with average diameter of 700–1000 nm which is known in neural engineering context to enhance elongation of neural stem cells and directing their differentiation towards neural lineages, compared to small (200–400 nm) and large (1000–1500 nm) dimensions.37,38 However, given the versatility of electrospinning to fabricate different sizes of PCL nanofiber platforms, our approach could be employed to fabricate rGO-coated nanofibers with various diameters to target specific biological responses. Although in previous studies fabrication of nanofiber structures based on GO has been reported,13,39 the above-described approach to employ GO liquid crystal phase has not been used so far to generate electro-conductive rGO-coated nanofibers. Comparing the in vitro behavior of stem cells on GO- versus rGO-based PCL fibers is beyond the scopes of this research; however, thanks to different degrees of π–π stacking, hydrogen and electrostatic bonding, rGO has been shown to accelerate the differentiation of mesenchymal stromal/stem cells (MSCs) towards the osteogenic lineages12 or favor the maintenance of stem cells pluripotency40 as compared to GO or inert substrates.
As illustrated in Fig. 5E, a four-point probe conductivity assay revealed that introduction of rGO onto the neat PCL fibers, resulted in induction of conductivity up to 3.9 ± 0.4 S cm−1 in PCL–rGO. The relatively high electrical conductivity obtained in this work could be explained by the harnessing of two unique features of synthesized GO. First, the presence of ultra large GO sheets ensures the presence of conductive pathways within the coated layer, providing less physical junctions between neighboring sheets.41 Second, oriented GO sheets, adhered on the PCL substrate, are in favor of charge carrier mobility. Size of GO sheets affects their orientation on the PCL template. At very dilute GO dispersion with small lateral size, the isotropic phase, in which no orientation is achievable, is expected to be dominant (0.1 mg ml−1, Fig. 6A). This trend is observed even at a relatively higher concentration (1 mg ml−1, Fig. 6B). However, ultra large sheets enable nematic liquid crystalline phase to be dominant even at very dilute solutions (0.1–0.2 mg ml−1) used for dip coating process (Fig. 6C and D). This, in turn, favors adhesion of ordered GO sheet onto the PCL substrate, which is schematically shown in Fig. 7. Given the high electrical conductivity of PCL–rGO, this substrate could be harnessed for certain biological applications as compared to PCL per se. Electrically conductive nanofibers have been previously shown to increase cell attachment, sustain cell recovery and enhance neuronal/cardiomyogenic differentiation with or without external electrical stimulations as compared to non-conductive fibers.42
 | ||
| Fig. 6 Polarized optical microscopy (POM) images of aqueous GO dispersion with small lateral size (0.6 ± 0.5 μm) at concentration of (A) 0.1 mg ml−1, and (B) 1 mg ml−1. POM images of aqueous GO dispersion with large lateral size (21 ± 12 μm) at concentration of (C) 0.1 mg ml−1, and (D) 0.2 mg ml−1. | ||
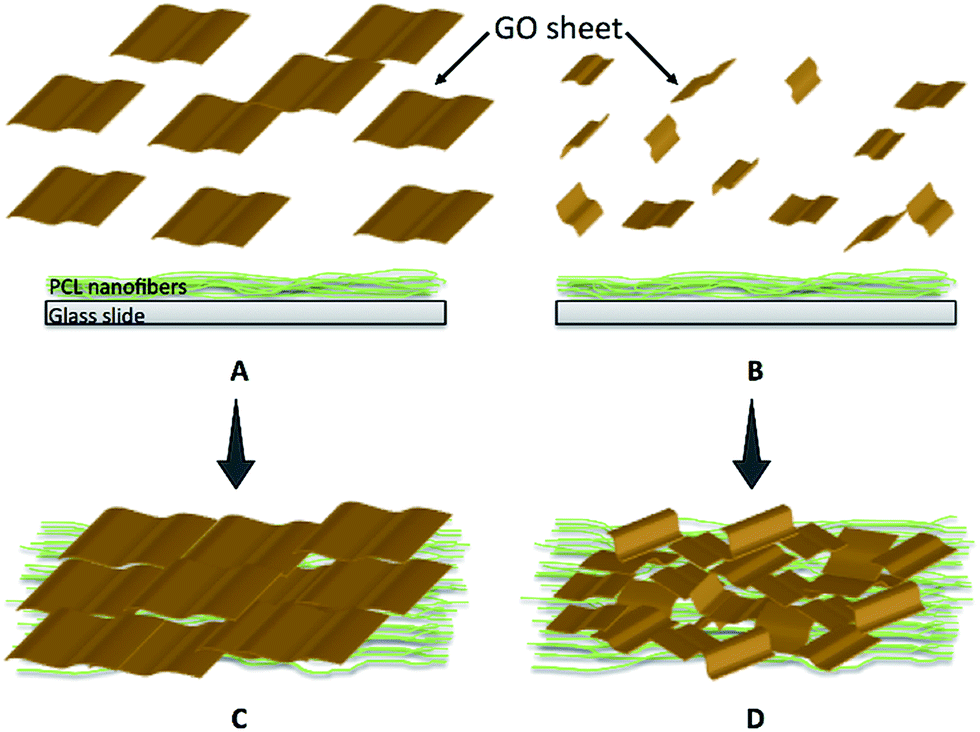 | ||
| Fig. 7 Schematic representation of orientation of GO sheets on the PCL substrate: (A) nematic liquid crystalline phase containing large GO sheets in dilute solution which is in favor of oriented morphology of coating, and (B) isotropic phase of GO sheets in dilute solution when small GO sheets are used. The latter will result in disoriented film morphology in which GO sheets are randomly distributed in the coating. Schemes (C) and (D) depict the resultant films coated on the PCL substrate from (A) and (B), respectively. | ||
As initial steps to check whether introduction of rGO could affect the cytotoxicity of PCL fibers, analyses of viability and metabolic activity of MSCs on PCL and PCL–rGO substrates were performed at days 3, 7, and 14 using the MTS assay. Cell viability assessment confirmed that both samples were cytocompatible, with more than 80% viability in all culture intervals (Fig. 8A). However, as depicted in Fig. 8B, PCL–rGO showed significant increase (P < 0.05) in absorbance at 490 nm from day 7 to 14 of culture. Moreover, the absorbance value of PCL–rGO at day 14 was considerably higher (P < 0.05) than other samples, suggesting greater levels of cell metabolic activity and proliferation following rGO deposition. The extent of cellular metabolism and proliferation of MSCs have been previously reported to improve on graphene and other electro-conductive scaffolds in vitro.42–44
 | ||
| Fig. 8 In vitro biocompatibility of samples. MTS assay of mesenchymal stem cells (MSCs) at different intervals showing (A) viability and (B) metabolic activity of PCL and PCL–rGO substrates. Both samples are biocompatible, while the metabolism of cells on PCL–rGO at day 14 is higher than PCL and standard tissue culture polystyrene (TCPS) samples. (n = 4, *P < 0.05, **P < 0.01). | ||
Overall, we aimed to develop a graphene-based nanofibrous substrate via facile approach, i.e. using the commonly used electrospun PCL template. Thorough in vitro investigations were beyond the prospects of the current work; however, having the fibrous and electro-conductive features, combined, this construct could be later employed to study the differentiation behavior of MSCs in absence or presence of external electrical stimulations. Electroconductive nanofibers show great promise in tissue engineering area, since they provide ECM-like topographical cues and deliver electrical signals to cells simultaneously and thus, promote electroactive tissue repair.45
Conclusions
In this study, we fabricated electroconductive rGO nanofibers via a simple process by immobilization of GO liquid crystals on electrospun PCL nanofibers. We used PCL nanofibers as a template to coat and reduce GO liquid crystals. Ultra large GO liquid crystals obtained in the current study are in favour of charge carrier mobility and affect the orientation of GO sheets on the PCL template. Moreover, the large GO liquid crystals enable conductive pathways within the coated layers and reduce physical junctions between adjacent sheets, thus providing high electrical conductivity in the resulting PCL-templated rGO fibers. Preliminary in vitro analyses confirmed that rGO nanofibers support active metabolism of human MSCs with enhanced cellular proliferation as compared to standard cell culture plates and PCL nanofibers. Our future studies will aim to assess functional properties of the PCL–rGO towards regeneration of electro-responsive tissues such as muscle and myocardium.Experimental
Graphene oxide (GO) synthesis
To generate GO aqueous dispersion, graphite flakes (1 g, Merck) were added to H2SO4 solution (80 ml, 98% w/v, Merck) and remained under constant stirring for 2 h. Following the addition of KMnO4 (4–6 g, Sigma) to the mixture and stirring for an overnight, the resultant solution first turned to dark green during the oxidation process of graphite, and then changed to dark brown. Next, double distilled water (d.d. H2O, 300 ml) was added to quench the oxidation reaction. To inactivate residual (un/non-reacted) KMnO4, aqueous solution of H2O2 (10 ml, 10% w/v, Merck) was added, which gave yellowish color to the mixture (Fig. S1A†). To remove excess ions and acids from the suspension, the mixture was centrifuged at least three times with d.d. H2O and HCl (1 M, Merck) at 8000 rpm. GO formed after spontaneous exfoliation process in graphite oxide. Finally, to remove unexfoliated particles, GO dispersion was purified through dialysis (12 kDa tube) process in d.d. H2O for 72 h at 25 °C. Schematic representation of GO synthesis was depicted in Fig. 1A.Fabrication of polycaprolactone (PCL) nanofibers
To generate electrospun nanofibrous substrate, a polymer solution with overall concentration of 12 wt% was obtained by dissolving PCL (80 kDa, Sigma) in chloroform:methanol (3:1 v/v, Merck). The solution was electrospun via a 10 ml syringe with a 20 gauge blunt needle at a flow rate of 1 ml h−1. After applying a high voltage to the needle (14 kV), nanofibers were collected on a grounded drum placed at a distance of 17 cm from the needle tip and rotating at 250 rpm. Electrospun fibers were then vacuum dried at 25 °C for 24 h to remove any residual solvent remained in the construct and kept in a desiccator for further investigations.
Surface modification of PCL electrospun fibers mat
Prior to perform aminolysis of PCL fibrous substrates, they were rinsed in ethanolic aqueous solution (50% v/v) for 2–3 h followed by several washes in d.d. H2O. Aminolysis procedure was carried out by immersing the fiber mats in a hexamethylenediamine (HMD)/2-propanol (Sigma) solution (12% w/v) for 24 h at 37 °C in a shaker incubator. The aminolyzed fibers were then rinsed several times with d.d. H2O for 24 h at 25 °C and subsequently vacuum dried at 30 °C for 24 h until of a constant weight.Graphene-coated on PCL nanofibers
To cover the PCL nanofibers with reduced graphene (rGO), fiber mats were immersed into the GO solution (1 mg ml−1) for 2 h. rGO-Coated fibers were obtained by keeping the samples under hydrazine monohydrate vapor (20% aqueous solution, Merck) in a desiccator for 24 h at 40 °C, after which the color of PCL fibers transformed from white to greyish black due to the reduction of GO. The samples were then vacuum dried for 24 h at 40 °C and rinsed several times for 24 h with d.d. H2O (Fig. 1B).Characterizations
To analyze the topology profiles of exfoliated GO sheets, following the deposition of a GO solution on a freshly cleaved mica surface, atomic force microscopy (AFM) images were obtained by a Dual scope 95-200 (DME, Denmark) working under tapping mode. X-ray diffraction (XRD) pattern of GO was obtained using a Philips X-ray spectrometer (Netherlands) after drying the GO dispersion in a lyophilizer (Christ, Alpha 1-2 LD, Germany). Ultraviolet-visible (UV-vis) spectrum of GO was obtained using a Nicolet Evolution spectrophotometer (Thermo Scientific, USA) in the wavelength range of 200–800 nm. To evaluate the heterogeneity and birefringence of GO dispersion polarized optical microscopy (POM) was performed by Leica DMR microscope in transmission mode. Optical images were recorded by placing a drop of dispersion on a glass coverslip and observed under an inverted microscope (BX51, Olympus) equipped with an Olympus DP72 digital camera. To characterize the size and size distribution of GO flakes, secondary electron (SE) imaging was applied in an FEI Helios NanoLab™ 600 dual beam scanning electron microscope. To ensure high quality imaging, GO flakes were dispersed on SiO2 substrate and imaged by electron probe current of 11 pA at an acceleration voltage of 2 kV. To evaluate the morphology and architecture of GO, PCL and PCL–rGO substrates, samples were observed by a field emission scanning electron microscopy (FE-SEM, FEI Nova NanoSEM 230, USA). Average fiber diameter and distribution were determined using Image Analyzer software (Image J 1.44p). The electrical conductivity of PCL and PCL–rGO substrates was measured by a four-point probe method using Keithley instrument (Model 196 System DMM, USA). The resistivity of substrates with determined thickness (t) was obtained using the following equation: ρ = (π/ln2) × (V/I) × t, where V is the measured voltage and I is the applied current. The conductivity of samples was obtained as the inverse of resistivity.
Determination of the amino groups
To detect the presence of NH2 groups on the aminolyzed PCL fiber mats, ninhydrin assay was performed. The substrates were first immersed in ninhydrin/ethanol solution (1 M, Sigma) for 1 min and then heated at 80 °C for 10 min to facilitate ninhydrin-amino reaction, which in turn made the surface of nanofibers blue. To dissolve the PCL mat, 1,4-dioxane (Merck) was added and to stabilize the blue compound, isopropanol (Merck) was introduced into the mixture. The UV-vis spectra of the modified and neat nanofibers were recorded in the range of 450–650 nm to determine the presence of NH2 groups on the surface of modified PLC nanofibers.In vitro studies
Statistical analysis
Data were expressed as mean ± standard deviation (SD). Statistical differenced in the measured properties between groups were analyzed by one-way analysis of variance (ANOVA) and the independent Student's t-test using SPSS 16.0 software. Post hoc analysis was performed using Tukey's HSD tests. P values of less than 0.05 were considered significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant provided by a Swiss excellence scholarship from the Swiss National Science Foundation to S. J.-F. (SNF grant #2013.0022) and by a grant from Royan Institute to S. J.-F. and M. H. M. M. We are grateful to Ms Evi Bieler (Zentrum fur Mikroskopie, University of Basel) for her excellent technical assistance with scanning electron microscopy.Notes and references
- L. Persano, A. Camposeo and D. Pisignano, Prog. Polym. Sci., 2015, 43, 48–95 CrossRef CAS.
- S. Jalili-Firoozinezhad, F. Mirakhori and H. Baharvand, in Stem-Cell Nanoengineering, ed. H. Baharvand and N. Aghdami, John Wiley & Sons, Inc, 2015, pp. 265–283 Search PubMed.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, RSC Adv., 2015, 5, 37553–37567 RSC.
- S. Tavakol, S. Jalili-Firoozinezhad, O. Mashinchian and M. Mahmoudi, in Nanoscience in Dermatology, ed. M. R. Hamblin, P. Avci and T. W. Prow, Academic Press, Boston, 2016, pp. 337–352 Search PubMed.
- S. H. Lim and H.-Q. Mao, Adv. Drug Delivery Rev., 2009, 61, 1084–1096 CrossRef CAS PubMed.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- Z. Su, J. Ding and G. Wei, RSC Adv., 2014, 4, 52598–52610 RSC.
- A. Savchenko, Science, 2009, 323, 589–590 CrossRef CAS PubMed.
- Editorial article, Nat. Nanotechnol., 2008, 3, 517 Search PubMed.
- Y. Li, P. Zhang, Z. Ouyang, M. Zhang, Z. Lin, J. Li, Z. Su and G. Wei, Adv. Funct. Mater., 2016, 26, 2122–2134 CrossRef CAS.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- W. C. Lee, C. H. Y. X. Lim, H. Shi, L. A. L. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 CrossRef CAS PubMed.
- K. Zhang, H. Zheng, S. Liang and C. Gao, Acta Biomater., 2016, 37, 131–142 CrossRef CAS PubMed.
- Z.-Q. Feng, T. Wang, B. Zhao, J. Li and L. Jin, Adv. Mater., 2015, 27, 6462–6468 CrossRef CAS PubMed.
- X. Lin, J. Jia, N. Yousefi, X. Shen and J.-K. Kim, J. Mater. Chem. C, 2013, 1, 6869–6877 RSC.
- S. H. Aboutalebi, M. M. Gudarzi, Q. B. Zheng and J.-K. Kim, Adv. Funct. Mater., 2011, 21, 2978–2988 CrossRef CAS.
- M. M. Gudarzi, M. H. M. Moghadam and F. Sharif, Carbon, 2013, 64, 403–415 CrossRef CAS.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, S. E. Moulton, J. M. Razal and G. G. Wallace, ACS Nano, 2013, 7, 3981–3990 CrossRef CAS PubMed.
- M. H. Mohamadzadeh Moghadam, S. Sabury, M. M. Gudarzi and F. Sharif, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1545–1554 CrossRef CAS.
- C.-M. Chen, J.-Q. Huang, Q. Zhang, W.-Z. Gong, Q.-H. Yang, M.-Z. Wang and Y.-G. Yang, Carbon, 2012, 50, 659–667 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- T. Szabó, E. Tombácz, E. Illés and I. Dékány, Carbon, 2006, 44, 537–545 CrossRef.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- X. Wang, H. Bai and G. Shi, J. Am. Chem. Soc., 2011, 133, 6338–6342 CrossRef CAS PubMed.
- J. Chen, Y. Li, L. Huang, N. Jia, C. Li and G. Shi, Adv. Mater., 2015, 27, 3654–3660 CrossRef CAS PubMed.
- X. Lin, X. Shen, Q. Zheng, N. Yousefi, L. Ye, Y.-W. Mai and J.-K. Kim, ACS Nano, 2012, 6, 10708–10719 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- J. Zhao, S. Pei, W. Ren, L. Gao and H.-M. Cheng, ACS Nano, 2010, 4, 5245–5252 CrossRef CAS PubMed.
- S. Naficy, R. Jalili, S. H. Aboutalebi, R. A. G. Iii, K. Konstantinov, P. C. Innis, G. M. Spinks, P. Poulin and G. G. Wallace, Mater. Horiz., 2014, 1, 326–331 RSC.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, J. M. Razal, S. E. Moulton and G. G. Wallace, Mater. Horiz., 2013, 1, 87–91 RSC.
- W. Guo, X. Zhang, X. Yu, S. Wang, J. Qiu, W. Tang, L. Li, H. Liu and Z. L. Wang, ACS Nano, 2016, 10, 5086–5095 CrossRef CAS PubMed.
- C. Chen, T. Zhang, Q. Zhang, X. Chen, C. Zhu, Y. Xu, J. Yang, J. Liu and D. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 10183–10192 CAS.
- Y. Zhu, C. Gao, X. Liu and J. Shen, Biomacromolecules, 2002, 3, 1312–1319 CrossRef CAS PubMed.
- W. Mattanavee, O. Suwantong, S. Puthong, T. Bunaprasert, V. P. Hoven and P. Supaphol, ACS Appl. Mater. Interfaces, 2009, 1, 1076–1085 CAS.
- S. Regis, S. Youssefian, M. Jassal, M. D. Phaneuf, N. Rahbar and S. Bhowmick, J. Biomed. Mater. Res., Part A, 2014, 102, 1697–1706 CrossRef PubMed.
- G. T. Christopherson, H. Song and H.-Q. Mao, Biomaterials, 2009, 30, 556–564 CrossRef CAS PubMed.
- H. B. Wang, M. E. Mullins, J. M. Cregg, C. W. McCarthy and R. J. Gilbert, Acta Biomater., 2010, 6, 2970–2978 CrossRef CAS PubMed.
- S. Shah, P. T. Yin, T. M. Uehara, S.-T. D. Chueng, L. Yang and K.-B. Lee, Adv. Mater., 2014, 26, 3673–3680 CrossRef CAS PubMed.
- G.-Y. Chen, D. W.-P. Pang, S.-M. Hwang, H.-Y. Tuan and Y.-C. Hu, Biomaterials, 2012, 33, 418–427 CrossRef CAS PubMed.
- H. Kim, J. I. Jang, H. H. Kim, G.-W. Lee, J. A. Lim, J. T. Han and K. Cho, ACS Appl. Mater. Interfaces, 2016, 8, 3193–3199 CAS.
- A. Orza, O. Soritau, L. Olenic, M. Diudea, A. Florea, D. Rus Ciuca, C. Mihu, D. Casciano and A. S. Biris, ACS Nano, 2011, 5, 4490–4503 CrossRef CAS PubMed.
- P. Baei, S. Jalili-Firoozinezhad, S. Rajabi-Zeleti, M. Tafazzoli-Shadpour, H. Baharvand and N. Aghdami, Mater. Sci. Eng., C, 2016, 63, 131–141 CrossRef CAS PubMed.
- M. Kalbacova, A. Broz, J. Kong and M. Kalbac, Carbon, 2010, 48, 4323–4329 CrossRef CAS.
- J. Y. Lee, Polym. Rev., 2013, 53, 443–459 CrossRef CAS.
- A. Papadimitropoulos, E. Piccinini, S. Brachat, A. Braccini, D. Wendt, A. Barbero, C. Jacobi and I. Martin, PLoS One, 2014, 9, e102359 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06178a |
| ‡ These authors contributed equally. |
| § Current address: Wyss Institute for Biologically Inspired Engineering at Harvard University, Boston, MA 02115, USA. E-mail: E-mail: sasan.jalili@wyss.harvard.edu. |
| This journal is © The Royal Society of Chemistry 2017 |