
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe heterogeneous selective reduction of PHB as a useful method for preparation of oligodiols and surface modification
Paweł
Chaber
,
Michał
Kwiecień
 ,
Magdalena
Zięba
,
Michał
Sobota
and
Grazyna
Adamus
*
,
Magdalena
Zięba
,
Michał
Sobota
and
Grazyna
Adamus
*
Centre of Polymer and Carbon Materials, Polish Academy of Sciences, Zabrze 41-800, Poland. E-mail: gadamus@cmpw-pan.edu.pl
First published on 12th July 2017
Abstract
A selective heterogeneous reduction of natural PHB with lithium borohydride as a reducing agent has been described. Despite the method proceeding in a heterogeneous way, it allows obtaining hydroxyl terminated oligomers with low polydispersity, controlled average molecular weight, and defined end groups. The structure of resulting oligomers has been proven by 1H NMR and mass spectrometry. This method would constitute a source of PHB oligodiols useful in the synthesis of new tailor-made biomaterials. It has also been demonstrated that this simple method may be used to modify the surface chemistry of the polyester by generating free hydroxyl groups on the outermost face. The presence of hydroxyl groups on the polymer surface was confirmed by ATR-FTIR studies as well as contact angle measurement. As expected, the change in surface chemistry improved the hydrophilicity of the polymer surface. Since surface properties are critically important for cell–material interactions, this kind of modification may be a very useful tool in tissue engineering.
Introduction
Polyhydroxyalkanoates (PHAs) are a class of linear polymers that occur abundantly in nature. They are accumulated by a wide range of bacteria as the most important storage compounds for carbon and energy.1 They are produced inside the cells of bacteria, therefore they are often referred to as bacterial or microbial. From the chemical point of view, PHAs are polyesters formed by linking together a large number of hydroxyalkanoic acids. These hydroxyalkanoic acid monomers contain the hydroxyl and the carboxyl group separated by two to five carbon atoms. The sum of the carbon atoms in the whole molecule can be even higher than fourteen.2,3 Thus, PHAs are polymers in which the ester linkages occur quite often throughout the main chain, and in which an alkyl side chain is present. The general chemical structure of PHAs is given in Fig. 1. | ||
Fig. 1 General chemical structure of PHAs. In the figure, n = 1–4, x = 200–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and R is a hydrogen atom or an alkyl group. 000, and R is a hydrogen atom or an alkyl group. | ||
The bacterial polyesters have been drawing much attention because of their unique and promising properties such as superior biodegradability and biocompatibility.4,5 Owing to these properties, PHAs could be employed as biomaterials for tissue engineering and controlled drug delivery.4,6–8
PHAs certainly possess many interesting properties, but they are not without disadvantages. Their major shortcoming are their low mechanical properties. The difficulty of PHAs processing is another drawback which hinders their practical use. The most common and studied among PHAs polymer, poly(3-hydroxybutyrate) (PHB), is very stiff and brittle and has low elasticity.9 The stiffness of PHB is a result of its high crystallinity (about 80% (ref. 10)), which, in turn, arises from its regular structure. PHB is, namely, a stereoregular homopolyester with an R configuration at all the β-carbons, so the polymer is completely isotactic. Moreover, its processing is difficult as it readily undergoes thermal degradation at temperatures close to the melting point.11 Fortunately, all these difficulties may be overcome since the mechanical and thermal properties of PHAs are greatly affected by the monomer structure and copolymer composition.5,12,13 Therefore, by changing the chemical structure of the alkyl side chain or by incorporating other hydroxyalkanoic acid co-monomer units into the main chain of PHAs, it is possible to create new polymers with desired properties.
In general, biotechnological or chemical approach can be applied to modify the chemical composition of the bacterial polyesters. Since they are synthesised in nature by bacterial fermentation, the variation in the chemical structure can be achieved by using appropriate microorganism type and growth conditions.1,8 However, the bacterial enzymes simply accept only certain hydroxyalkanoic acids in the course of polymerisation. Thus, the biotechnological process leads to materials with structures similar only to those shown in Fig. 1, and hence to materials with limited thermomechanical properties. As a result, the microbial polyesters often do not meet the very specific requirements set to biomaterials in different medical applications.
Systematic research has been conducted on the development of new polymeric biomaterials based on structural fragments derived from microbial PHAs. Those structural fragments are very interesting building blocks for the synthesis of more sophisticated polymer architectures with tailored properties. They can be obtained using i.e. chemical reactions such as controlled degradation processes. In the case of structural segments, the end groups, which are usually ignored for long-chain PHAs, gain a significant importance since they determine the possibility of their further use in the synthesis of new materials.
Several attempts have been made to obtain structural fragments from PHAs.14–26 A molecular mass reduction of PHA may be achieved by various methods, such as an acid-catalysed alcoholysis (a transesterification reaction),14–18 an acid or a base hydrolysis,19 a saponification reaction,20,21 a thermal degradation,22,23 or a reduction reaction.24,25 All these methods lead to obtaining low-molecular-weight PHAs with well-defined end groups that can take part in further polymerisation. In the case of a saponification reaction and a thermal degradation, the formation of oligomers with olefinic and carboxylic end groups is observed.20,21 Those structural fragments could be polymerised to yield polymers via free-radical methods, but nevertheless, Nguyen et al. reported some difficulties in applying these chain-extending reactions. Much work has been done to obtain diol ended PHAs, particularly hydroxyl terminated PHB, which present some advantages over the other PHAs family members. Oligomers terminated by free hydroxyl groups are very useful in the chemical synthesis as they can react, for example, with diisocyanate monomers to form various polyurethanes, or with long-chain dicarboxylic acids to increase the distance between the ester linkages in the newly formed polymeric chains. For this purpose oligodiols of PHB are characterized with a lower steric hindrance, which results in higher reactivity in the above-mentioned reactions. PHB oligomers with reactive hydroxyl ends can be prepared by two methods: via transesterification reaction,14–18 and by borohydride reduction.24,25 In the first case 1,4-butanediol, ethylene glycol, and glycerol were employed, and in the second – sodium borohydride (NaBH4) and lithium borohydride (LiBH4) were used as reducing agents. Montoro compared both methods and concluded that the borohydride reduction was more efficient.26 A borohydride reduction of PHB has a significant advantage over a transesterification reaction because the latter is limited in terms of molar masses of the synthesised oligomers. The first could even result in the monomeric units.
Despite the fact that history of hydride reductions began over eighty years ago with a study of the reactions of diborane (B2H6),27 borohydride reductions of polyesters are not fully investigated. Bergamaschi et al. reported that the treatment of PHB (dissolved in chloroform) with sodium borohydride leads to the formation of oligomers terminated with hydroxyl groups, but these products were not the only ones present in the post-reaction mixture. Low-molecular-weight PHB with an unsaturated and a carboxylic end group were also observed, with the latter being dominant.24 Recently, we have developed a highly selective method for a controlled degradation of some PHAs, via a reduction reaction with lithium borohydride.25 However, despite all advantages the method has, it only applies to those PHAs which are soluble in solvents appropriate for lithium borohydride reduction (in tetrahydrofuran or diethyl ether). Since the natural PHB is insoluble in almost all common organic solvents, the reduction of PHB by using the abovementioned method has failed.
Here, we report a novel heterogeneous method of the reduction of sparingly soluble PHAs on the example of the most well-known among them, the PHB. The method allows obtaining low-molecular-weight PHB diols in a satisfactory yield at room temperature. In addition, the obtained oligomers have a high purity proved by 1H NMR and ESI-MSn analysis. Moreover, the elaborated method can be used to modify the chemistry of the surface layer of polyester devices fabricated, for example, by extrusion moulding. What is worth noting is that the modification of the surface proceeds under very mild conditions. It is also shown that elaborated method is useful to increase the hydrophilicity of the modified surface. The surface itself holds great importance in the cell–material interactions and to a great extent determines the biological performance of a biomaterial. It controls such important processes as cell adhesion and growth, processes which largely define success or failure of, for example, producing a scaffold.28,29
Experimental section
Materials
Poly(3-hydroxybutyrate) (PHB) was provided by the Biomer Company (Germany). In order to remove biological impurities the sample of raw PHB was purified by reprecipitation from its chloroform solution in hexane. Next, the obtained powder was dried in vacuum at room temperature and then it was ground in a mortar to average powder granulation. The sample of PHB was characterized by gel permeation chromatography (GPC) and 1H and 13C NMR. The number-average molar mass of PHB, determined by GPC, was 228000 g mol−1 with a polydispersity of 17.7.
Lithium borohydride, LiBH4, was purchased as a solution (as a 2.0 M solution in THF and a 0.5 M solution in diethyl ether) from Aldrich as well as phosphoric acid, H3PO4, (85 wt% in H2O, FG). Hydrochloric acid (HCl) solution in H2O (35–38%, pure p. a., Avantor), the hydrochloric acid solution in diethyl ether (2.0 M, Aldrich) and chloroform, CHCl3, (98.5%, pure p. a., Avantor) were used as received. Tetrahydrofuran, THF, (pure, Avantor), diethyl ether, DE, (pure, Avantor), toluene (pure, Avantor) were purified prior to use. THF and diethyl ether were distilled over potassium–sodium alloy and stored in Schlenk flasks under an argon atmosphere. Toluene was distilled from calcium hydride and stored over 4A molecular sieves.
Methods
In parallel to the functionalization of PHB discs' surfaces, a blind test was performed. The protocol of the test was the same as above except that herein lithium borohydride was not added. The discs were only just immersed in the solvent. After 24 hours they were treated with hydrochloric acid, washed with diethyl ether and finally dried over 72 hours.
000000 g mol−1; efficiency (1/2 ht) (plates per m) 56593] and a Shodex SE 61 refractive index detector. For the GPC analysis of the PHA oligodiols, a PLgel 3 µm Mixed-E column [Mw range up to 25000 g mol−1; efficiency (1/2 ht) (plates per m) 98869] and THF, as the solvent, were used. A sample solution volume of 10 µL (concentration of 1% w/v) was injected. Polystyrene standards with a narrow molecular mass distribution were used to generate calibration curves.
480 scans, a 9.40 ls pulse width, and 0.9088 s acquisition time.
ESI-MSn experiments
:1 v/v), and the solutions were introduced into the ESI source by continuous infusion using the instrument's syringe pump at a rate of 10 µL min−1. The LCQ ESI source was operated at 4.5 kV, and the capillary heater was set to 200 °C. Nitrogen was used as the nebulising gas. For ESI-MS/MS experiments, the ions of interest were isolated monoisotopically in the ion trap and were collisionally activated. The helium damping gas that was present in the mass analyser acted as a collision gas. The RF amplitude, which had a significant voltage range, was set to a value that caused the peak height of the molecular ion to decrease by at least 50%. The analysis was performed in the positive-ion mode.
The goniometer. Contact angle measurements (Θ) for PHB discs' surface were measured statically using a contact angle goniometer (CAM 101 from KSV instruments) equipped with a temperature control unit. The water contact angles were determined in air using the sessile drop method. A water drop of volume around 5 µL was suspended from the syringe needle and brought into contact with the surface of the discs. A series of images were acquired during 30 s. Contact angles were measured after the drop was stabilized. The contact angle measurements were performed for a dry surface. The average contact angle values and standard deviations were obtained by measuring the contact angle at one position of each of six discs.
Results and discussion
Application of lithium borohydride as efficient reducing agent for oligodiols preparation from selected PHA polyester was reported recently.25 However, most commonly members of PHA family are highly crystalline and their solubility in solvents suitable for the reduction process is limited. Therefore, we have tried to conduct the reduction of PHB in two phase system. PHB is the easiest available and cheapest member of PHA family but has also several disadvantages which limit its wide using, so the possibility of its modification seems to be a good objective intentional. The general scheme of proposed modification is presented on Scheme 1. | ||
| Scheme 1 The reduction reaction of PHB biopolyester with lithium borohydride. In the scheme, t means time and y ≪ x. | ||
In our previous work concerning reduction process of soluble PHA biopolyesters, THF was used as a suitable solvent. At the beginning of this work, the possibility of the use of other solvents for two-phase reduction process was investigated. Four different solvents or mixtures of solvents: tetrahydrofuran (THF) diethyl ether (DE), THF:toluene (1:1) and DE:toluene (1:1) were considered and the results of this investigation are presented in Table 1. The results indicated that the solvent which allows obtaining oligomers with lowest average molar mass in the same conditions (reaction were carried out for 4 h, using 0.8 mmol LiBH4 in 25 mL of solvent(s) was a mixture of THF and toluene in ratio (1:1)). Therefore, in next steps of our research concerning two-phase reduction process of PHB using lithium borohydride as reducing agent, this mixture was used as a solvent.
| No. | Solvent | M n [g mol−1] | M w/Mn | Yield [%] |
|---|---|---|---|---|
|
a The reactions were carried out for 4 hours with 8 mmol of LiBH4 in 25 mL of solvent(s). The number average molar mass of PHB was 228000 g mol−1 with polydispersity of 17.7.
|
||||
| 1 | THF | 4000 | 1.71 | 80.2 |
| 2 | DE | 4000 | 1.70 | 83.5 |
| 3 | THF:toluene (1:1) |
2800 | 1.49 | 74.2 |
| 4 | DE:toluene (1:1) |
3800 | 1.70 | 83.4 |
In the next step, the time necessary to conduct an efficient reaction of obtaining oligomers with low average molar mass was determined. The results of this study are presented in Table 2. The reaction was carried out in the range of 0.5 to 6 hours. The highest decrease of average molar mass of starting biopolyester is observed after 0.5 h what testifies that this process is relatively fast. During elongation of time of reaction, slow decrease of average molar mass is observed up to 4 h. Further time elongation (over 4 h) does not result in further decrease in average molar mass, therefore the 4 h were determined as sufficient time for this reduction process.
| No. | t [h] | M n [g mol−1] | M w/Mn | Yield [%] |
|---|---|---|---|---|
|
a The reactions were carried out with 8 mmol of LiBH4 in 25 mL of the THF-toluene solvent mixture. The number-average molar mass of PHB was 228000 g mol−1 with polydispersity of 17.7.
|
||||
| 1 | 0.5 | 4700 | 1.75 | 86.6 |
| 2 | 1 | 3500 | 1.59 | 80.7 |
| 3 | 2 | 3200 | 1.60 | 77.1 |
| 4 | 4 | 2800 | 1.49 | 74.2 |
| 5 | 6 | 2800 | 1.49 | 72.0 |
Finally, the influence of the amount of reducing agent (LiBH4) on the decrease of average molar mass of starting PHB biopolyester and the yield of synthesized PHB oligodiols were also investigated. The results are summarized in Table 3. Obtained results show that increasing the amount of lithium borohydride above the 0.8 mmol is purposeless because it does not lead to a further decrease of PHB oligodiols average molar mass.
| No. | Amount of LiBH4 [mmol] | M n [g mol−1] | M w/Mn | Yield [%] |
|---|---|---|---|---|
|
a The reactions were carried out in 25 mL of the THF-toluene solvent mixture, for 4 hours. The number-average molar mass of PHB was 228000 g mol−1 with polydispersity of 17.7.
|
||||
| 1 | 0.2 | 3900 | 1.70 | 76.6 |
| 2 | 0.4 | 3400 | 1.62 | 76.3 |
| 3 | 0.6 | 3400 | 1.68 | 81.8 |
| 4 | 0.8 | 2800 | 1.49 | 74.2 |
| 5 | 1.0 | 2900 | 1.51 | 73.7 |
| 6 | 1.2 | 2900 | 1.52 | 73.3 |
Simultaneously, a decrease of yield of reaction with decreasing of average molar mass is observed. The observed decrease of yield of reaction may be caused by PHB oligodiols purification process after the reaction. The very low molar mass PHB oligodiols may be washed out by water which is used during purification. Additionally, in the case of two-phase reduction at the end of the process we have two fractions of PHB in the reaction mixture, the first one is not soluble and the second one (PHB oligomers with very low molar mass) is soluble in the solvent. This situation leads to competition between reactions proceeding in solution and on phase boundary. Reaction in solution proceeded faster and produced PHB oligomers with very low average molar mass which were soluble in water and could be removed during the purification. The increase of reducing agent dose did not influence on the further decrease average molar masses of separated PHB oligodiols.
For the structural characterization of the obtained oligodiols, 1H NMR and mass spectrometry were used.
Proton nuclear magnetic resonance (1H NMR) spectroscopy
The 1H NMR spectrums of the oligomers obtained via the selective reduction of PHB biopolyester are presented in Fig. 2. The main signals (marked as 1, 2, 3) in the 1H NMR spectrum correspond to the protons of the 3-hydroxybutyrate repeating units. Additionally, in the presented spectrum signals corresponding to the protons of the end groups of obtained oligomers are observed. Thus, the signals denoted a, b, and c were assigned to the protons of the 3-hydroxybutyrate end group, whereas the signals denoted d, e, f, and g were assigned to the protons of the 1-methyl-3-hydroxypropyl end group. In the 1H NMR spectrum any additional signals besides those from oligodiols are not observed, thus this analysis confirmed selectivity of the reduction reaction of PHB biopolyester (Fig. 2).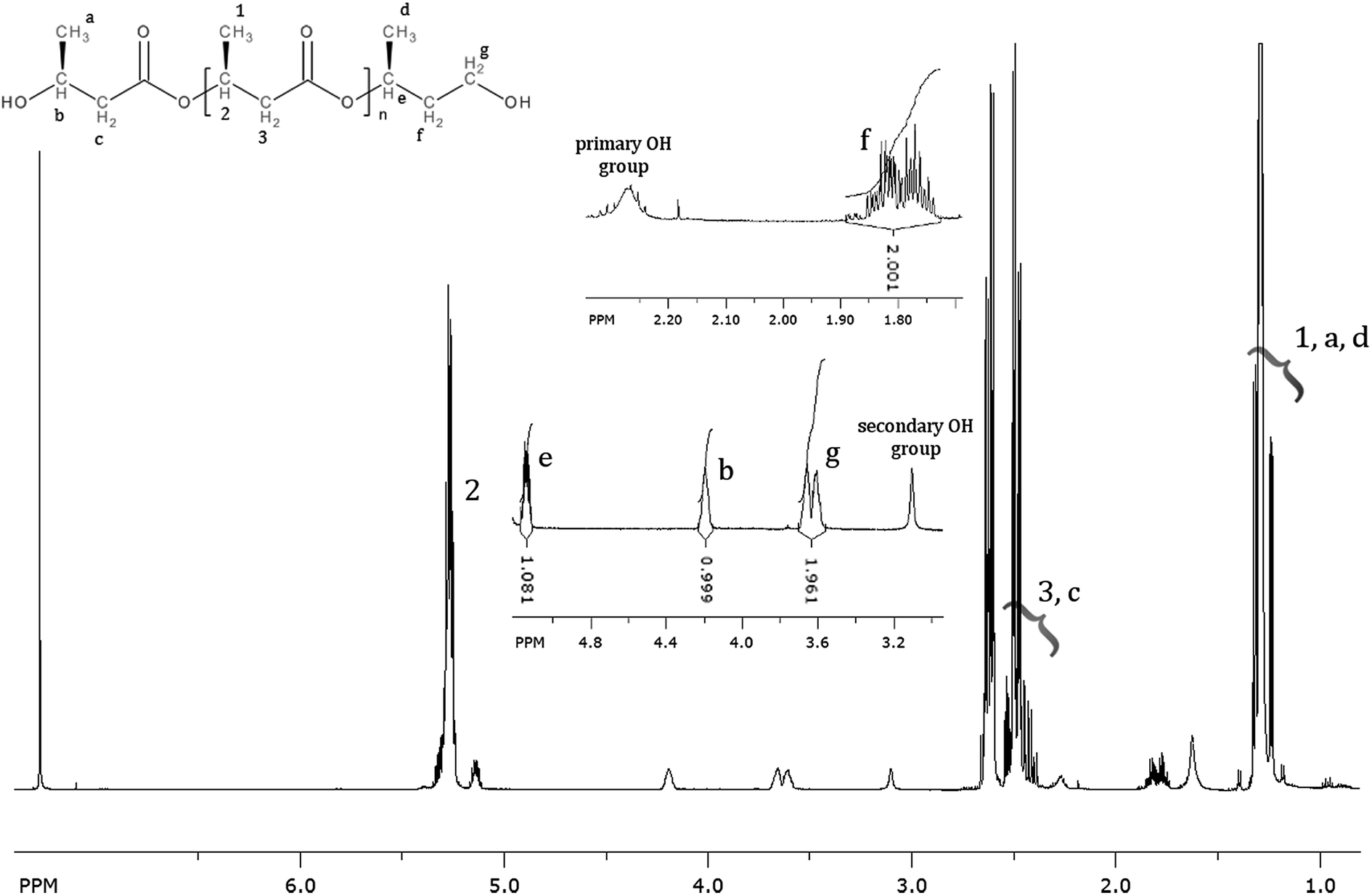 | ||
| Fig. 2 1H-NMR spectrum of PHB oligomers obtained through reduction with lithium borohydride. | ||
Electrospray ionization mass spectrometry (ESI-MS)
ESI-MS is a very useful technique for detailed analysis of synthetic polymers. This sensitive and non-averaging technique provides detailed information about individual oligomer chains as well as the chemical structure of their end groups. ESI-MS is used successfully for structural characterization PHA biopolyesters and other polyesters and copolyesters.21,25,30–32 Therefore, this technique was also used for the structural characterization of the obtained PHB oligodiols.Fig. 3 shows the ESI-MS spectrum of PHB oligomers obtained via selective reduction of high molar mass PHB biopolyester. The ions in the main series in ESI-MS spectrum occurred regularly at 86 Da what corresponds to the 3-hydroxybutyrate constitutional unit. Thus, the main series of ions present in the mass spectrum (Fig. 3) (marked as A) can be assigned to the sodium adduct of PHB oligomers. For further structural assignment of PHB oligomers, tandem mass spectrometry (ESI-MS/MS) was used. To verify the structure of individual PHB oligomers chains, ESI-MS/MS fragmentation experiments were performed for selected ions isolated from Series A.
 | ||
| Fig. 3 ESI-MS spectrum (positive ion mode) of the oligodiols obtained via the selective reduction of a PHB biopolyester. | ||
The ESI-MS/MS spectrum shown in Fig. 4, was obtained for the precursor ions at m/z 1231, which was selected from ESI-MS spectrum (Fig. 3) of PHB oligomers obtained via selective reduction of PHB biopolyester.
 | ||
| Fig. 4 ESI-MS/MS product ion spectra and fragmentation pathways of the sodium adduct at m/z 1231. | ||
The fragmentation of this ion, which occurs as a result of the statistical breakage of the ester bonds along PHB oligodiols chain, leads to the formation of two series of product ions. The series of product ions, at m/z 1127, 1041, 955, 869, 783, 697 and 611, terminated by crotonic and carboxylic end group are created: first by the expulsion of 3-hydroxybutanoic acid (104 Da) and further by the loss of crotonic acid molecules. The second series of product ions at m/z 1159, 1073, 987, 901, 815, 729 and 643 terminated by 3-hydroxybutyrate and carboxylic end group is formed in the first step by the loss of 2-butenyl alcohol (crotonyl alcohol; 72 Da) and in next steps by expulsion of crotonic acid molecules (86 Da).
Thus, the ESI-MS2 experiments performed for selected sodium adducts of PHB oligomers confirmed that the obtained PHB oligomers contained two hydroxyl end groups.
Application of NMR and ESI-MS technique for determination of the chemical structure of the obtained oligomers allows confirming that selective reduction of polyester may be carried out also in the heterogeneous system. Limited solubility of polyester in a solvent did not influence the selectivity of the reduction process using lithium borohydride as reducing agent. Moreover, in some range, the average molar mass of the resulting PHB oligodiols may be controlled by the amount of reducing agent used in the reaction. Very interesting results of studies about the heterogeneous reduction of PHB biopolyester have inspired us to apply this method for the modification of the surface chemistry of polyester materials. Surfaces bearing polar functional groups offer enhanced biological performance by improving cell attachment to the surface28,33 as well as cell growth.34 Regarding the reduction of PHB powder, which leads to obtaining oligodiols, the functionalization of the surface of polymer devices with much smaller surface area could generate hydroxyl groups on the outermost face without causing any visible destruction in the modified material. In fact, using lithium borohydride as the reducing agent generates free hydroxyl groups on the outermost face of PHB devices. Discs fabricated by extrusion of PHB were used as a model of the surface. As it is shown in Table 2, the reduction reaction of the powdered PHB proceeds in THF as well as in diethyl ether. Regarding the discs, our goal was not to obtain the oligomers with the lowest mass possible, but just to functionalize the surface. Thus, we decided to perform the borohydride reduction of PHB devices in diethyl ether instead of in THF-toluene system due to at least two facts. Firstly, diethyl ether is highly volatile, and therefore it is easier to evaporate from the surface. Secondly, diethyl ether is employed as a surface antiseptic and cleansing agent,35 so PHB devices after the functionalization could be instantly used in medical applications. To confirm that the functionalization of the surface proceeds efficiently, ATR-FTIR analyses were performed. The ATR-FTIR spectra are presented in Fig. 5.
 | ||
| Fig. 5 ATR-FTIR spectra of (a) PHB surface of blind sample and (b) chemically modified PHB surface. | ||
ATR-FTIR spectrum for extruded discs before reduction process looks like typically FTIR spectrum for PHB biopolyester. In the spectrum for disc after reduction process (presented in Fig. 4b) wide bands in the range of 3000–3700 cm are observed. This simple and fast analysis confirms presence of the hydroxyl group on the surface of discs. Generation of free hydroxyl groups on the surface of extruded discs from PHB is possible, however the longer time of reaction than in the case of powder reduction is required. The presence of free hydroxyl groups on the surface causes an increase of hydrophilicity what is very important in cell growing process. For determination of increase of hydrophilicity of the surface after the reduction process, the measurement of contact angle was performed. The pictures of a drop on the surface before and after the reduction reaction are presented in Fig. 6.
 | ||
| Fig. 6 Contact angle images of water on (a) PHB surface untreated with lithium borohydride (the blind sample; θ = 90.6°) and (b) LiBH4-treated PHB surface (θ = 58.9°). | ||
The surface untreated with the reducing agent showed noticeably higher contact angle value compared to the chemically modified surface with the average values of 90.3° ± 4.8° and 59.5° ± 1.4 respectively. The results show the increase in surface hydrophilicity. Strictly speaking, the hydrophobic PHB surface becomes hydrophilic.
Conclusions
A heterogeneous system which consists of solid PHB and a solution of lithium borohydride appears to be an efficient method for preparation of PHB oligodiols. Despite the fact that this reaction is a phase boundary reaction, it is quite selective and leads to obtaining oligomers with low polydispersity and defined end groups. The average molar masses of the obtained hydroxyl terminated oligomers can be controlled to some extent by changing the amount of reducing agent. Moreover, the results of the present investigation revealed that the developed method is useful to modify surface chemistry. Discs extruded from natural PHB and immersed to the lithium borohydride solution bear hydroxyl groups on the surface. Those hydroxyl groups appeared in the IR spectrum. Furthermore, the contact angle measurements showed that the surface becomes more hydrophilic as a result of the reduction process.The developed by us method of heterogeneous selective reduction of sparingly soluble PHAs constitute the extension of selective reduction of PHA biopolyesters soluble in standard solvents used for reduction process, recently reported by us.25 The elaborated method allows to obtain uniform PHB diols from PHB biopolyester that is produced on large scale and easily available on the market. The obtained low-molecular-weight PHB diols are useful for the further synthesis of new biodegradable polymeric materials with tailored properties.
In addition, the elaborated method can be used to modify the hydrophilicity of the surface layer of polyester devices fabricated from PHB.
Thus, this easy method seems to be useful for application in tissue engineering for creation free hydroxyl group on the surface, e.g. three-dimensional scaffold for cell growth.
Preliminary biological studies proving the usefulness of the elaborated method in the preparation of scaffolds will be the subject of our further work.
Acknowledgements
This work was supported by the National Science Centre Poland: contract No. UMO-2013/11/B/ST5/02222.References
- A. Anjum, M. Zuber, K. M. Zia, A. Noreen, M. N. Anjum and S. Tabasum, Microbial production of polyhydroxyalkanoates (PHAs) and its copolymers: A review of recent advancements, Int. J. Biol. Macromol., 2016, 89, 161–174 CrossRef CAS PubMed.
- A. Steinbüchel and H. E. Valentin, Diversity of bacterial polyhydroxyalkanoic acids, FEMS Microbiol. Lett., 1995, 128(3), 219–228 CrossRef.
- A. K. Singh and N. Mallick, SCL-LCL-PHA copolymer production by a local isolate, Pseudomonas aeruginosa MTCC 7925, Biotechnol. J., 2009, 4(5), 703–711 CrossRef CAS PubMed.
- S. F. Williams and D. P. Martin, Applications of Polyhydroxyalkanoates (PHA) in Medicine and Pharmacy, in Biopolymers Online, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed.
- S. Khanna and A. K. Srivastava, Recent advances in microbial polyhydroxyalkanoates, Process Biochem., 2005, 40(2), 607–619 CrossRef CAS.
- M. Zinn, B. Witholt and T. Egli, Occurrence, synthesis and medical application of bacterial polyhydroxyalkanoate, Adv. Drug Delivery Rev., 2001, 53(1), 5–21 CrossRef CAS PubMed.
- G. Q. Chen, A microbial polyhydroxyalkanoates (PHA) based bio- and materials industry, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- D. B. Hazer, E. Kılıçay and B. Hazer, Poly(3-hydroxyalkanoate)s: Diversification and biomedical applications: A state of the art review, Mater. Sci. Eng., C, 2012, 32(4), 637–647 CrossRef CAS.
- E. Bugnicourt, Polyhydroxyalkanoate (PHA): Review of synthesis, characteristics, processing and potential applications in packaging, eXPRESS Polym. Lett., 2014, 8(11), 791–808 CrossRef.
- A. Hoffmann, S. Kreuzberger and G. Hinrichsen, Influence of thermal degradation on tensile strength and Young's modulus of poly(hydroxybutyrate), Polym. Bull., 1994, 33(3), 355–359 CrossRef CAS.
- N. Grassie and E. J. Murray, The thermal degradation of poly(-(d)-β-hydroxybutyric acid): Part 1—Identification and quantitative analysis of products, Polym. Degrad. Stab., 1984, 6(1), 47–61 CrossRef CAS.
- D. Kai and X. J. Loh, Polyhydroxyalkanoates: Chemical Modifications Toward Biomedical Applications, ACS Sustainable Chem. Eng., 2014, 2(2), 106–119 CrossRef CAS.
- R. Nigmatullin, P. Thomas, B. Lukasiewicz, H. Puthussery and I. Roy, Polyhydroxyalkanoates, a family of natural polymers, and their applications in drug delivery, J. Chem. Technol. Biotechnol., 2015, 90(7), 1209–1221 CrossRef CAS.
- G. R. Saad, Y. J. Lee and H. Seliger, Synthesis and characterization of biodegradable poly(ester-urethanes) based on bacterial poly(R-3-hydroxybutyrate), J. Appl. Polym. Sci., 2002, 83(4), 703–718 CrossRef CAS.
- Z. Špitalský, I. Lacík, E. Lathová, I. Janigová and I. Chodák, Controlled degradation of polyhydroxybutyrate via alcoholysis with ethylene glycol or glycerol, Polym. Degrad. Stab., 2006, 91(4), 856–861 CrossRef.
- B. Hazer, Amphiphilic Poly(3-hydroxy alkanoate)s: Potential Candidates for Medical Applications, Int. J. Polym. Sci., 2010, 2010, 1–8 CrossRef.
- D. Kai and X. J. Loh, Polyhydroxyalkanoates: Chemical Modifications Toward Biomedical Applications, ACS Sustainable Chem. Eng., 2014, 2(2), 106–119 CrossRef CAS.
- D. Xue, W. Lv and H. Zhang, Telechelic diols from polyhydroxybutyrate via alcoholysis with ethylene glycol or glycerol, Proceedings of the 2015 2nd International Conference on Machinery, Materials Engineering, Chemical Engineering and Biotechnology (MMECEB 2015), Atlantis Press, 2016 Search PubMed.
- C. Lauzier, J.-F. Revol, E.-M. Debzi and R. H. Marchessault, Hydrolytic degradation of isolated poly(β-hydroxybutyrate) granules, Polymer, 1994, 35(19), 4156–4162 CrossRef CAS.
- G. Adamus, W. Sikorska and M. Kowalczuk, Sequence Distribution and Fragmentation Studies of Bacterial Copolyester Macromolecules: Characterization of PHBV Macroinitiator by Electrospray Ion-Trap Multistage Mass Spectrometry, Macromolecules, 2000, 33(16), 5797–5802 CrossRef CAS.
- E. Žagar, A. Kržan, G. Adamus and M. Kowalczuk, Sequence Distribution in Microbial Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Co-polyesters Determined by NMR and MS, Biomacromolecules, 2006, 7(7), 2210–2216 CrossRef PubMed.
- S. Nguyen, G. Yu and R. H. Marchessault, Thermal Degradation of Poly(3-hydroxyalkanoates): Preparation of Well-Defined Oligomers, Biomacromolecules, 2002, 3(1), 219–224 CrossRef CAS PubMed.
- M. Kawalec, G. Adamus, P. Kurcok, M. Kowalczuk, I. Foltran, M. L. Focarete and M. Scandola, Carboxylate-Induced Degradation of Poly(3-hydroxybutyrate)s, Biomacromolecules, 2007, 8(4), 1053–1058 CrossRef CAS PubMed.
- J. M. Bergamaschi, E. J. Pilau, F. C. Gozzo and M. I. Felisberti, Synthesis of Polyurethane from Poly(3-hydroxybutyrate) and Poly(p-dioxanone): Molar Mass Reduction via Sodium Borohydrate, Macromol. Symp., 2011, 299–300(1), 10–19 CrossRef.
- M. Kwiecień, G. Adamus and M. Kowalczuk, Selective Reduction of PHA Biopolyesters and Their Synthetic Analogues to Corresponding PHA Oligodiols Proved by Structural Studies, Biomacromolecules, 2013, 14(4), 1181–1188 CrossRef PubMed.
- S. Montoro, Master Dissertation, Universidade de São Paulo, Lorena, 2005.
- H. C. Brown, H. l. Schlesinger and A. B. Burg, Hydrides of Boron. XI. The Reaction of Diborane with Organic Compounds Containing a Carbonyl Group, J. Am. Chem. Soc., 1939, 61(3), 673–680 CrossRef CAS.
- R. Vasita, K. Shanmugam and D. S. Katti, Improved Biomaterials for Tissue Engineering Applications: Surface Modification of Polymers, Curr. Top. Med. Chem., 2008, 8(4), 341–353 CrossRef CAS PubMed.
- K. Anselme, L. Ploux and A. Ponche, Cell/Material Interfaces: Influence of Surface Chemistry and Surface Topography on Cell Adhesion, J. Adhes. Sci. Technol., 2010, 24(5), 831–852 CrossRef CAS.
- I. Kwiecień, I. Radecka, M. Kwiecień and G. Adamus, Synthesis and Structural Characterization of Bioactive PHA and γ-PGA Oligomers for Potential Applications as a Delivery System, Materials, 2016, 9(5), 307 CrossRef.
- M. Kwiecień, I. Kwiecień, I. Radecka, V. Kannappan, M. R. Morris and G. Adamus, Biocompatible terpolyesters containing polyhydroxyalkanoate and sebacic acid structural segments – synthesis and characterization, RSC Adv., 2017, 7(33), 20469–20479 RSC.
- G. Adamus, Molecular Level Structure of (R,S)-3-Hydroxybutyrate/(R,S)-3-Hydroxy-4-ethoxybutyrate Copolyesters with Dissimilar Architecture, Macromolecules, 2009, 42, 4547 CrossRef CAS.
- Y. Arima and H. Iwata, Effects of surface functional groups on protein adsorption and subsequent cell adhesion using self-assembled monolayers, J. Mater. Chem., 2007, 17, 4079–4087 RSC.
- A. Zühlke, B. Röder, H. Widdecke and J. Klein, Synthesis and application of new microcarriers for animal cell culture. Part I: Design of polystyrene based microcarriers, J. Biomater. Sci., Polym. Ed., 1993, 5(1–2), 65–78 Search PubMed.
- P. Viccellio, Emergency Toxicology, Lippincott-Raven Publishers, 1998, p. 293 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |