
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyoxometalate-MgF2 hybrids as heterogeneous solid acid catalysts for efficient biodiesel production†
Kai-Li Yang,
Shan Huang,
Hu Pan,
Heng Zhang,
Xiao-Fang Liu and
Song Yang *
*
State Key Laboratory Breeding Base of Green Pesticide & Agricultural Bioengineering, Key Laboratory of Green Pesticide & Agricultural Bioengineering, Ministry of Education, State-Local Joint Laboratory for Comprehensive Utilization of Biomass, Center for Research & Development of Fine Chemicals, Guizhou University, Guiyang 550025, China. E-mail: jhzx.msm@gmail.com; Fax: +86 851 88292170; Tel: +86 851 88292171
First published on 3rd July 2017
Abstract
A series of highly active and stable Keggin heteropolyacid catalysts were prepared through mixing of 12-tungstophosphoric acid (TPA) with magnesium fluoride (MgF2). Among those acidic catalysts, the Mg20F39TPA-1.0 hybrid with moderate acidity (0.96 mmol g−1) and good dispersion of active sites presented pronounced catalytic performance in esterification of oleic acid (up to 95% oleic acid conversion). Particularly, Mg20F39TPA-1.0 was also efficient for the transesterification of Jatropha oil with a high acid value, giving biodiesel in 93% yield. Moreover, the catalyst showed good durability and reusability for at least five consecutive cycles.
1. Introduction
In the wake of the consumption of fossil fuels and the concomitant aggravation of environmental concerns, there is currently an urgent need to search for renewable and clean energy sources.1–3 Due to the merits of biodegradability, non-sulfur, renewability, and high lubricity, biodiesel is considered to be one of promising alternative fuels.4–6 Biodiesel is composed of long chain fatty acid monoesters, which are generally derived from renewable oils or fats by (trans)esterification with short carbon-chain alcohols.7–9 In the process of producing biodiesel, cheaper oils (e.g., Jatropha oil) have been elected as raw materials to cut the production cost.10,11 However, the use of an alkaline catalyst may lead to saponification, difficult separation of products, and high catalyst consumption.12–15 In addition, abundant free fatty acids (FFAs) can be generated as important byproducts during food processing.16 Therefore, it is a beneficial route to use acidic catalysts to esterify FFAs and (trans)esterify Jatropha oil for biodiesel production.Traditionally, homogeneous acid catalysts (e.g., H2SO4, HCl or H3PO4) are applied for transesterification and esterification.17 However, these types of catalytic systems not only need extra neutralization, are energy-intensive and require complicated purification steps, but they are also difficult to recover and separate, which may increase the cost of biodiesel production.18,19
Therefore, the design and use of solid acid catalysts for either transesterification or esterification have been widely developed to overcome these drawbacks. For example, an ionic liquid functionalized polymer (e.g., poly([VSIM][HSO4])) exhibited good catalytic activity in oleic acid esterification, giving biodiesel in a conversion of 92%.20 Sulfated zirconia possessing Brönsted and Lewis acid sites afforded 88% biodiesel yield in esterification of stearic acid, while the loss of the Brönsted acid sites directly resulted in significant deactivation of the catalyst.21 Sulfonic acid-functionalized solid acids (e.g., sulphonated carbon and SO3H-SBA-15) were also explored to be used for producing biodiesel from plant oils or fatty acids, giving 82–92% yield of biodiesel at high temperatures (e.g., 200 °C).22,23 However, wide application of those materials in biodiesel production was restricted possibly due to their low thermal stability, need of costly raw materials, and rigorous and complicated preparation procedures.
Heteropolyacids (HPAs) have stronger Brönsted acidy than most of mineral acids.24–27 However, low surface areas (<10 m2 g−1) of HPAs greatly reduce the dispersion and accessibility of acid sites to substrates during reactions, and they are difficult to recover in polar solvents because of high miscibility.28 To address these issues, HPAs were usually immobilized onto porous materials (e.g., HPMo/A20 and TPA/ZrO2),29,30 while the problem of leaching remained heavily in the production of biodiesel. Instead, solvothermal method seems to be a valid solution to overcome this drawback, with active sites dispersed more evenly.31,32 The solidification with cations is another way to overcome the solubility of HPAs. However, a comparison of immobilization and solidification methods showed that the latter possessed higher density of acidic sites and better catalytic performance.33,34
It was reported that MgF2 possessed high thermal stability, mesoporous structure, Brönsted and Lewis acid sites.35 Besides, it also contains moderate week base sites (0.65 mmol g−1). Therefore, MgF2 can be a promising support to solidify TPA, without destructing the structure of TPA. Herein, a series of polyoxometalate-MgF2 (Mg20F39TPA-x) were synthesized from mixing of TPA with MgF2. For the sake of evaluating the catalytic performance of Mg20F39TPA-x, the catalysts were used for producing biodiesel from both oleic acid and Jatropha oil with a high acid value of 19.35 mg KOH/g via esterification and transesterification, respectively.
2. Experimental
2.1 Materials
Mg (99.9%) and HF (49 wt% in water, 99.99998%) were bought from Aladdin Industrial Inc. (Shanghai). Methanol (>99.5%) was supplied by Guanghua scientific Ltd. (Guangdong). H3PW12O40·nH2O (AR) and Oleic acid (AR) were purchased from Kemiou Chemical Reagent, Co., Ltd. (Tianjin). Jatropha oil (≥98%, Table 1) procured from Luodian County, Guizhou Province, China.| Fatty acid | Palmitic acid (C16:0) | Palmitoleic acid (C16:1) | Stearic acid (C18:0) | Oleic acid (C18:1) | Linoleic acid (C18:2) | Linolenic acid (C18:3) |
|---|---|---|---|---|---|---|
| Content (wt%) | 13.23 | 0.85 | 5.40 | 41.62 | 36.99 | 0.22 |
2.2 Catalyst preparation
2.3 Catalyst characterization
Fourier transform-infrared (FT-IR) spectra of samples were recorded on a Nicolet 360 FT-IR spectrometer by using a KBr disc method. Energy dispersive X-ray spectroscopy (EDS; S-4800, Hitachi) under 5 kV primary electron voltages was employed to measure the elemental components. X-ray diffraction (XRD) patterns in the 2θ angle from 5 to 80 °C were obtained on D8 ADVANCE X-ray diffractometer (Cu Kα radiation, λ = 0.154 nm). The surface properties were determined by nitrogen adsorption–desorption over Micromeritics ASAP 2020 M analyzer at −196 °C. The surface areas, pore-volumes and pore-size distributions were calculated by Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) method, respectively. Transmission electron microscopy (TEM) observations were achieved on a HT7700 system operated at 200 kV. Thermogravimetric (TG) test was conducted on a METTLER TGA/DSC1 apparatus in nitrogen atmosphere with heating rate of 10 °C min−1. According to the curves of ammonia/carbon dioxide temperature-programmed desorption (NH3/CO2-TPD), the content of acid sites were measured and calculated on AutoChem 2920 chemisorption analyzer. Approximately 0.1 g catalyst was pretreated under He flow (30 mL min−1) at 300 °C for 2 h, then saturated with ammonia by exposing to 10% NH3–He or 10% CO2–He at 50 °C. The physically adsorbed ammonia was flushed with He. The desorbed ammonia was recorded under a temperature rate of 10 °C min−1 until heated up to 500–700 °C. Elemental analyzer (Vario EL III) was used to measure the carbon contents of the catalysts.2.4 Catalytic test
The conversion of oleic acid was determined via a titration method by employing 0.1 M standard potassium hydroxide ethanol solution as titrant and phenolphthalein as indicator. It was calculated depending on the below equation:
| Oleic acid conversion (%) = (1 – AV0/AV1) × 100% |
According to the weights and GC peak areas of crude biodiesel and heptadecanoic acid methyl ester, biodiesel yield was calculated by following equation:
| Yield (wt%) = {[(AC16:0/fC16:0 + AC16:1/fC16:1 + AC18:0/fC18:0 + AC18:1/fC18:1 + AC18:2/fC18:2 + AC18:3/fC18:3 + ACothers)/AC17:0] × weight of C17:0}/(weight of crude biodiesel) × 100% |
3. Results and discussion
3.1 Catalysts characterization
In order to demonstrate that the TPA was not in a crystalline state but molecularly dispersed on the surface of magnesium fluoride material, XRD patterns of Mg20F39TPA-1.0, bare TPA and MgF2 were conducted (Fig. 1). TPA (curve c) exhibited a series of sharp characteristic diffraction peaks of Keggin ion at 10.23°, 17.78°, 20.43°, 25.09°, 29.42° and 37.66°, respectively.37 The catalysts all (curve a, curve b) displayed the typical reflections of MgF2 at 27.14°, 40.41°, 53.21° and 67.62°.35 And the resultant Mg20F39TPA-1.0 catalyst (curve a) also showed the characteristic diffraction peaks of Keggin ion at 8.86°, 17.98°, 20.29° and 28.51°. Even though characteristic peaks of Mg20F39TPA-1.0 are very similar to Keggin ion of TPA and MgF2, the slight shift were happened, especially at 10.23°, indicating that the metal salt was formed for the exchange of protons with metal ions.38 Besides, the peaks widths of Mg20F39TPA-1.0 broadened and the intensity of peaks decreased, demonstrating that the chemical interactions was occurred between TPA and Mg20F39, and the TPA was semidispersed on catalyst.39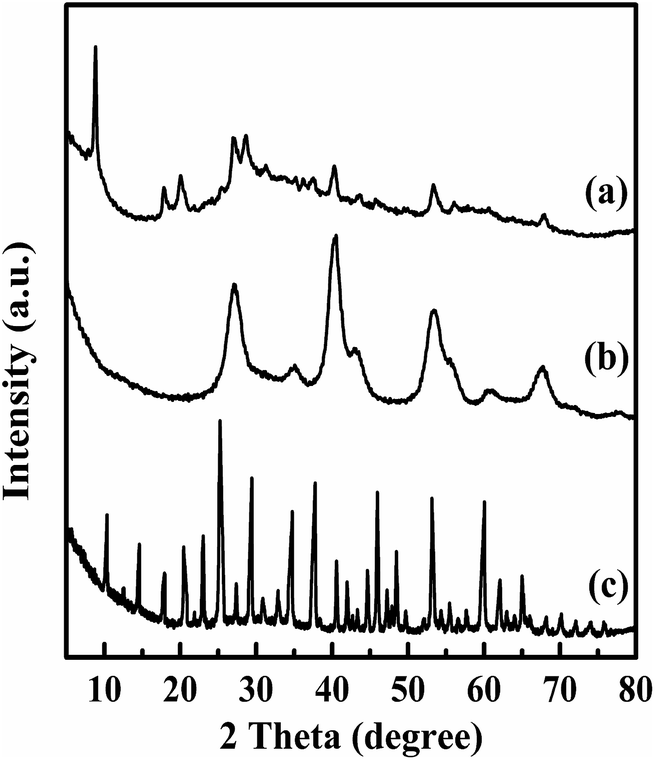 | ||
| Fig. 1 XRD patterns of (a) Mg20F39TPA-1.0, (b) MgF2, and (c) TPA. | ||
EDS analysis results were obtained by EDS measurement: Mg, 12.65, W, 9.08 (Mg20F39TPA-1.2); Mg, 12.41 and W, 7.63 (Mg20F39TPA-1.0); Mg, 13.08, W, 6.47 (Mg20F39TPA-0.8); Mg, 15.68 and W, 4.91 (Mg20F39TPA-0.6). The W/Mg atomic ratios of Mg20F39TPA-x were described in Table 2, and the results were consistent with the theoretical values, indicating that there was no sole TPA in Mg20F39TPA-x. From the EDS spectra of Mg20F39TPA-x catalysts in Fig. S1(b–e),† extra characteristic peaks of P and W elements were appeared, which were not presented on the spectrum of MgF2 (Fig. S1(a)†), suggesting that the Mg20F39TPA-x hybrids were formed.40 These results were in good agreement with those of XRD patterns.
| Catalyst | W/Mg nominal ratio | W/Mg average value from EDS |
|---|---|---|
| a Mg20F39TPA-x denoted as Mg20F39H2.6P1.2W14.4O48, Mg20F39H2.0PW12O40, Mg20F39H1.4P0.8W9.6O32, and Mg20F39H0.8P0.6W9.6O32. | ||
| Mg20F39TPA-1.2 | 0.72 | 0.72 |
| Mg20F39TPA-1.0 | 0.60 | 0.61 |
| Mg20F39TPA-0.8 | 0.48 | 0.49 |
| Mg20F39TPA-0.6 | 0.36 | 0.31 |
FT-IR spectra of Mg20F39TPA-1.0 and TPA were exhibited in Fig. 2. Four featured peaks of TPA Keggin ion could be observed at 1080, 980, 893 and 800 cm−1, which might be attributed to P–Oa, W = Od, W–Ob–W and W–Oc–W, respectively.32 The FT-IR spectra of other Mg20F39TPA-x catalysts (x = 1.2, 0.8, and 0.6) were provided in Fig. S2.† Keggin structure of all Mg20F39TPA-x catalysts was found to remain unchanged, despite slight weakening in some peak strengths, which could be ascribed to the decrease of the TPA ratio in Mg20F39TPA-x.41
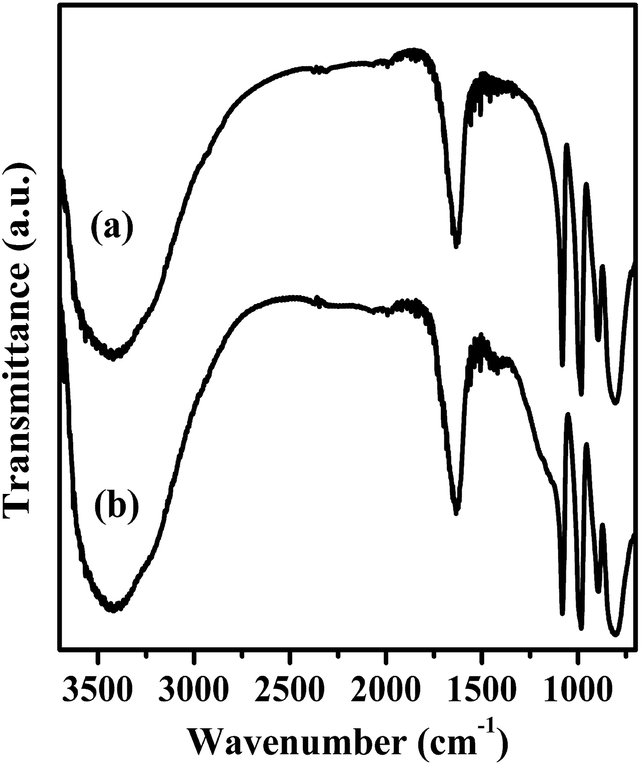 | ||
| Fig. 2 FTIR spectra of (a) TPA and (b) Mg20F39TPA-1.0. | ||
The acidity of Mg20F39TPA-x was determined by NH3-TPD, and the results were listed in Table 3. As expected, the increase of proton number in Mg20F39TPA-x, obtained by the increase of the TPA contents led to the rise of acidity of the solid. As shown in Fig. S3,† TPA was characterized by weak and strong acidic sites, and MgF2 showed weak and moderate acidic sites as evidenced from TPD determination. Correspondingly, NH3-TPD curves of Mg20F39TPA-x displayed the weak, moderate and strong acidic sites located at 100–300 °C, 300–500 °C and 500–700 °C. In this respect, it could be explained that the moderate acidic sites were mainly from the Mg20F39 moiety.42 In Fig. S5,† the pore size and surface area of Mg20F39TPA-1.0 was found to be 5.4 nm and 25.4 m2 g−1, respectively. Nitrogen adsorption–desorption experiment displayed that the mesoporous structure was formed in Mg20F39TPA-1.0, which is beneficial to mass transfer during the transesterification and esterification. Although the surface area of Mg20F39TPA-1.0 was small, the narrow pore size distribution was observed, which was mainly centered at 4–6 nm. It was indicated that the pores were distributed in the surface of Mg20F39TPA-1.0 are uniform. As shown in Fig. S4,† the week basicity (0.65 mmol g−1) of MgF2 could be measured. As expected, it is advantageous to form polyoxometalate-MgF2 hybrids without destroying the structure of TPA due to the weak basicity of MgF2. This fact verified the results of XRD and FT-IR.
| Catalysts | Acidity (mmol g−1) | Molar ratio | |||||
|---|---|---|---|---|---|---|---|
| Weak (100–300 °C) | Moderate (300–500 °C) | Strong (500–700 °C) | Total | W/Ta | M/Ta | S/Ta | |
| a W = density of week acid sites, M = density of moderate acid sites, S = density of strong acid sites, and T = density of total acid sites. | |||||||
| Mg20F39TPA-1.2 | 0.35 | 0.23 | 0.43 | 1.01 | 0.35 | 0.23 | 0.43 |
| Mg20F39TPA-1.0 | 0.30 | 0.26 | 0.40 | 0.96 | 0.31 | 0.27 | 0.42 |
| Mg20F39TPA-0.8 | 0.50 | 0.13 | 0.30 | 0.93 | 0.54 | 0.14 | 0.32 |
| Mg20F39TPA-0.6 | 0.49 | 0.08 | 0.06 | 0.63 | 0.78 | 0.13 | 0.09 |
The dispersion of Mg29F30TPA-1.0 particles was analyzed by TEM (Fig. 3), and a large number of worm-like mesoporous structure could be observed. These nanoparticles in small size of 2–7 nm were clearly observed. This result is in good agreement with nitrogen adsorption–desorption analysis of Mg29F30TPA-1.0 that shows uniform mesoporous structure with average pore size of 5.3 nm.
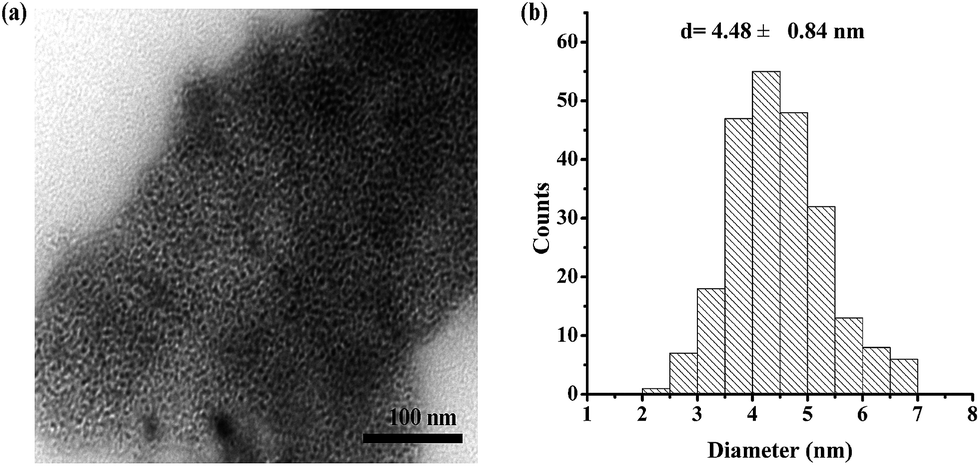 | ||
| Fig. 3 (a) TEM image, (b) particle size distribution of Mg29F30TPA-1.0. | ||
Fig. S6† exhibits the thermal stability of Mg20F39TPA-1.0 catalyst studied by TG analyses. It can be seen that Mg20F39TPA-1.0 has an excellent thermal stability with less weight loss (about 5 wt%) at 100–350 °C, which is possibly attributed to desorption of water molecules inside the catalyst pores. Thus, it has great potential to be used at high reaction temperatures.
3.2 The comparison of catalytic activities
The catalytic activities of different catalyst were tested for oleic acid esterification. In the reaction, the relatively low methanol/oleic acid molar ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was employed at 80 °C with 5 wt% catalyst dosage for 4 h, and the results were displayed in Fig. 4. The conversion of oleic acid reached to 10% without any catalyst under the above reaction conditions, indicating that the catalytic activity was attributed to the weak acidity of oleic acid, wherein it might act as both catalyst and reactant.43 Sole MgF2 presented conversion of 15%. The slight increase of conversion during esterification could be resulted from the existence of Brönsted and Lewis acid sites in MgF2. Although the conversion over TPA was 94% in oleic acid conversion, the catalyst was soluble in this reaction system, which made it difficult to reuse. Gratifyingly, excellent conversions were also gained when the esterification was performed in the presence of Mg20F39TPA-x catalysts. From Fig. 4, the sequence of catalytic activities for these TPA salt catalysts was Mg20F39TPA-1.2 ∼ Mg20F39TPA-1.0 > Mg20F39TPA-0.8 > Mg20F39TPA-0.6, according to the oleic acid conversion. It is obviously to see that the total acidity of the catalysts directly influenced their catalytic activity, which is in accordance with the proposal that Brönsted acid take main effect in esterification.44 In addition, the conversion of oleic acid was increased significantly from 74% to 95% with the protons of TPA rose from 1.4 to 2.0. It was worth noticing that the total acidity of Mg20F39TPA-x increased slightly (0.93–0.96 mmol g−1), however, the density of strong acid sites was raised obviously from 0.30 to 0.40 mmol g−1. Further increasing of protons of TPA from 2.0 to 2.4 resulting slightly enhanced strong acid sites (0.40 to 0.43 mmol g−1) with a conversion of 95%, which is identical to that of over Mg20F39TPA-1.0. On the other hand, the acidity of 30 wt% TPA/MgF2 prepared by impregnation method was demonstrated to be 0.41 mmol g−1 (Fig. S3†), which was far less than that of Mg20F39TPA-x (0.63–1.01 mmol g−1). As a result, only 42% conversion was obtained over 30 wt% TPA/MgF2, which is much lower than that of over Mg20F39TPA-x. Thus, the solvothermal solidified Mg20F39TPA-x gives higher density of acidic sites and provides better catalytic performance than that of the immobilized TPA/MgF2.
:1 was employed at 80 °C with 5 wt% catalyst dosage for 4 h, and the results were displayed in Fig. 4. The conversion of oleic acid reached to 10% without any catalyst under the above reaction conditions, indicating that the catalytic activity was attributed to the weak acidity of oleic acid, wherein it might act as both catalyst and reactant.43 Sole MgF2 presented conversion of 15%. The slight increase of conversion during esterification could be resulted from the existence of Brönsted and Lewis acid sites in MgF2. Although the conversion over TPA was 94% in oleic acid conversion, the catalyst was soluble in this reaction system, which made it difficult to reuse. Gratifyingly, excellent conversions were also gained when the esterification was performed in the presence of Mg20F39TPA-x catalysts. From Fig. 4, the sequence of catalytic activities for these TPA salt catalysts was Mg20F39TPA-1.2 ∼ Mg20F39TPA-1.0 > Mg20F39TPA-0.8 > Mg20F39TPA-0.6, according to the oleic acid conversion. It is obviously to see that the total acidity of the catalysts directly influenced their catalytic activity, which is in accordance with the proposal that Brönsted acid take main effect in esterification.44 In addition, the conversion of oleic acid was increased significantly from 74% to 95% with the protons of TPA rose from 1.4 to 2.0. It was worth noticing that the total acidity of Mg20F39TPA-x increased slightly (0.93–0.96 mmol g−1), however, the density of strong acid sites was raised obviously from 0.30 to 0.40 mmol g−1. Further increasing of protons of TPA from 2.0 to 2.4 resulting slightly enhanced strong acid sites (0.40 to 0.43 mmol g−1) with a conversion of 95%, which is identical to that of over Mg20F39TPA-1.0. On the other hand, the acidity of 30 wt% TPA/MgF2 prepared by impregnation method was demonstrated to be 0.41 mmol g−1 (Fig. S3†), which was far less than that of Mg20F39TPA-x (0.63–1.01 mmol g−1). As a result, only 42% conversion was obtained over 30 wt% TPA/MgF2, which is much lower than that of over Mg20F39TPA-x. Thus, the solvothermal solidified Mg20F39TPA-x gives higher density of acidic sites and provides better catalytic performance than that of the immobilized TPA/MgF2.
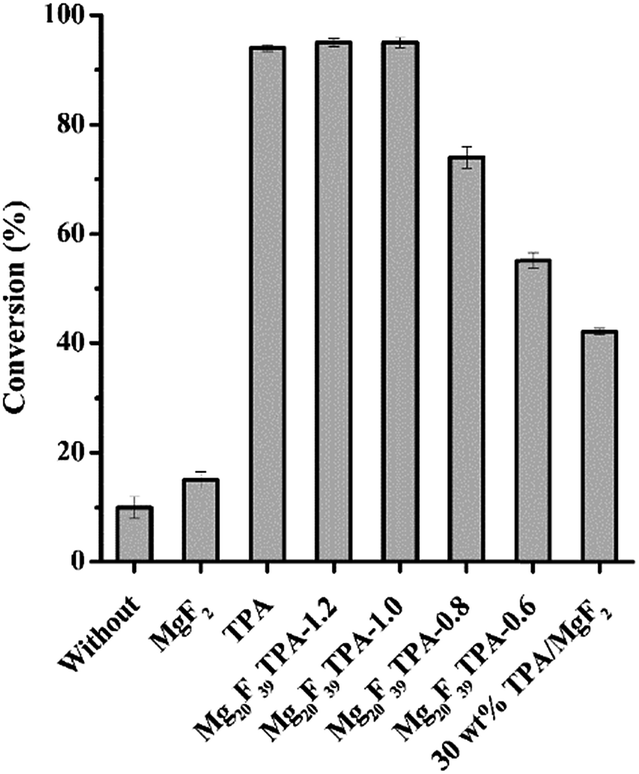 | ||
| Fig. 4 Catalytic performance of different catalysts in the conversion of oleic acid to methyl oleate. Reaction conditions: methanol/oleic acid molar ratio = 9/1, catalyst dosage = 5 wt%, reaction time = 4 h, and reaction temperature = 80 °C. | ||
3.3 Esterification of oleic acid with methanol
Obviously, the most important advantage of single factor is adaptable, and the plans of experiment can be easily updated based on the result of one test.45 In order to get the optimal reaction conditions, single factor design was thus applied to evaluate the effect of methanol/oleic acid molar ratio, catalyst dosage, reaction time and temperature in the esterification of oleic acid, as illustrated in Fig. 5.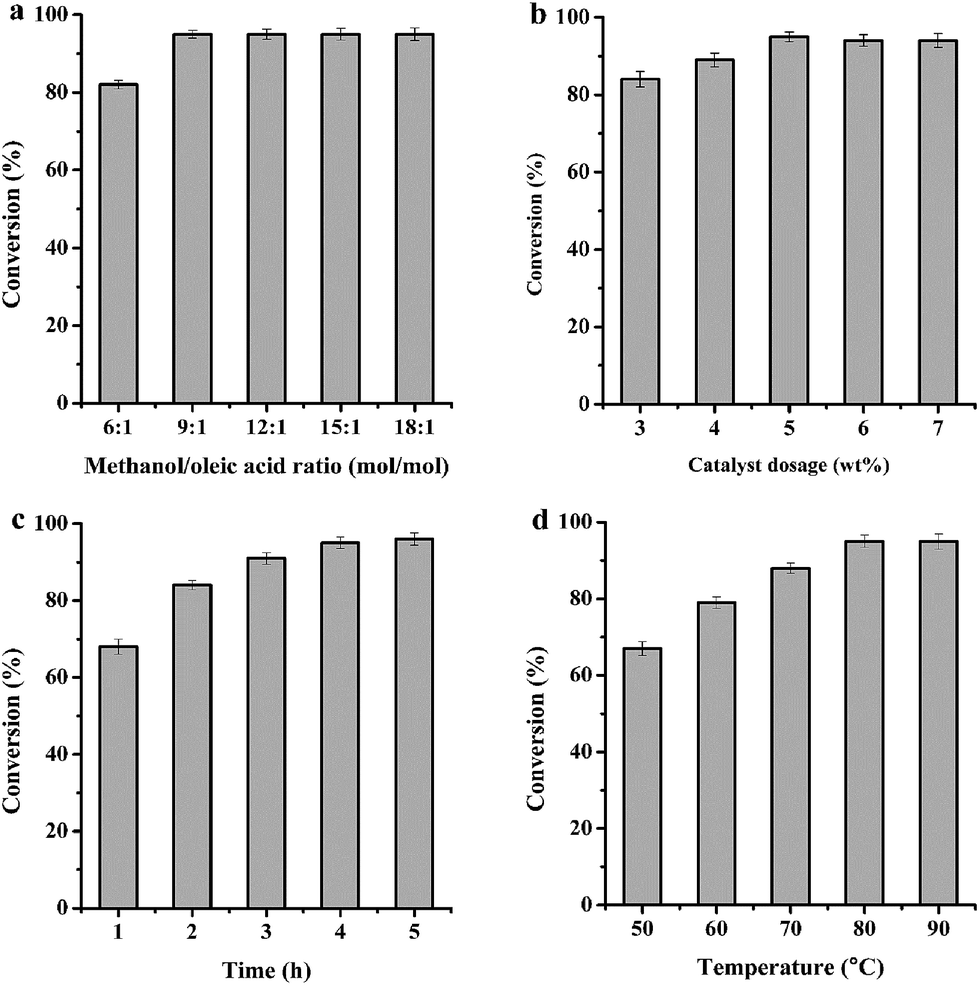 | ||
| Fig. 5 Conversion of oleic acid over Mg20F39TPA-1.0 based on (a) methanol/oleic acid molar ratio = 6/1–18/1, (b) catalyst dosage = 3–7 wt% (catalyst/oleic acid mass ratio), (c) reaction time = 1–5 h, (d) reaction temperature = 50–90 °C. The optimum reaction conditions: methanol/oleic acid molar ratio = 9/1, catalyst dosage = 5 wt%, reaction time = 4 h, reaction temperature = 80 °C. | ||
3.4 Leaching experiments
To verify the heterogeneity of catalyst, leaching experiment was carried out. As shown in Fig. S7,† a conversion of 68% was obtained after 1 h at 80 °C. Then Mg20F39TPA-1.0 catalyst was filtered and removed from mixtures. The filtrate was continued to react for further 2–5 h without catalyst under the same conditions and the oleic acid conversion remained stable. This result revealed that Mg20F39TPA-1.0 was heterogeneous in this reaction system.3.5 Recyclability of the catalysts
To assess the recyclability feature of Mg20F39TPA-1.0 in esterification of oleic acid, the solid catalyst was separated by centrifugation after each recycling test, washed several times with n-hexane, dried at 80 °C, and used for the next cycle. To estimate the catalytic ability of Mg20F39TPA-1.0 more propitious, about 50% of oleic acid conversion was set for comparison.49 As shown in Fig. S8,† The conversion decreased slightly in five consecutive cycles. As demonstrated in Table S1,† residual carbon of 3.70 wt% on used Mg20F39TPA-1.0 could be detected by elemental analysis. It could be speculated that the active sites of the catalyst partially covered by organic matter led to the decrease in the conversion.50,51 Furthermore, FT-IR and XRD was carried out to fully understand the reasons of catalyst deactivation. Fig. 6 displayed that FT-IR spectra (1200–700 cm−1) of fresh and recycled Mg20F39TPA-1.0 catalysts were similar, indicating that Keggin structure remained intact after reused. However, the small peaks were appeared at 2926 cm−1 and 2526 cm−1, which were belonged to CH3 asymmetrical stretching vibrations and CH2 symmetrical stretching vibrations of methyl oleate, respectively.52 As expected, the results of FT-IR and elemental analysis were consistent. The XRD patterns (Fig. 7) of recycled Mg20F39TPA-1.0 catalyst (after five recycles) did allow all characteristic diffraction peaks of Mg20F39TPA-1.0 to be appeared, although the intensity of diffraction were weak. These results illustrated that Mg20F39TPA-1.0 was stable and the decrease of reflection could be due to the adsorption of organic matter.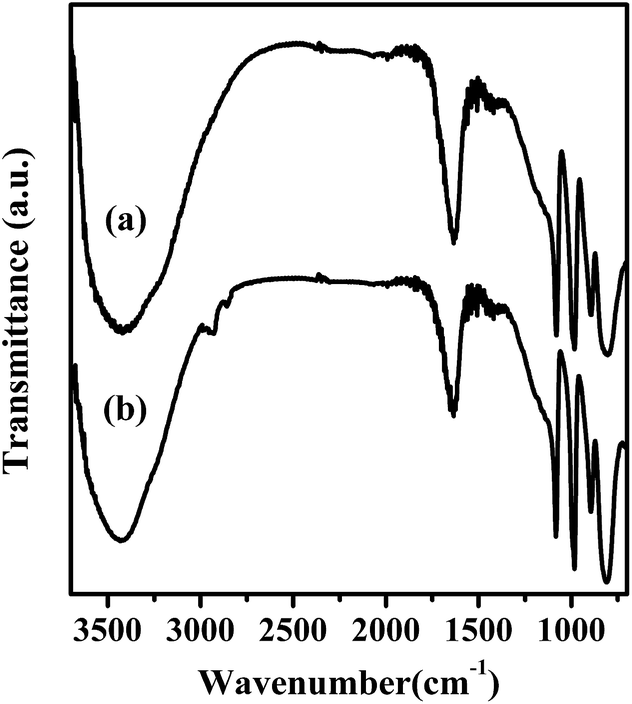 | ||
| Fig. 6 FT-IR spectra of (a) fresh and (b) reused 5th Mg20F39TPA-1.0. | ||
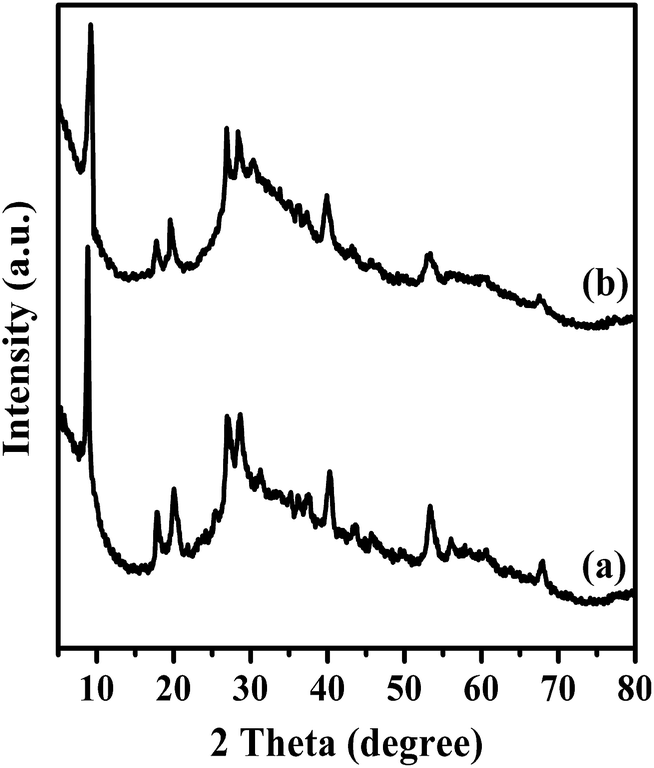 | ||
| Fig. 7 XRD patterns of (a) fresh and (b) reused 5th Mg20F39TPA-1.0. | ||
As shown in Table 4, [BMIM][HSO4] ionic liquid and sulfonated cation exchange resin (SCER) exhibited good catalytic performance, and oleic acid conversion of 81% and 93% was attained by using the remarkably high catalyst amount of 98 wt% and 47 wt%, respectively (entries 1, 2). Although the phosphorylated carbon (POMC) solid acid catalyst has well-ordered mesoporous structure and large surface area (530 m2 g−1), the low conversion of oleic acid (74%) was obtained (entry 3). In addition, long time and high temperature were typically required to get impressive conversion of oleic acid. For example, 100% conversion could be achieved while prolonging the reaction time to 12 h for HClSO3–ZrO2 (entry 4). WO3/USY displayed moderate catalytic performance of 74% conversion at 200 °C (entry 5). Even though mesoporous polymeric solid acid (MPD–SO3H–IL) possessed high acidity (2.6 mmol g−1) and large surface area (281 m2 g−1), good catalytic activity (up to 98% conversion) was achieved when the large molar ratio of methanol and oleic acid (30:1) was required (entry 6). Notably, Mg20F39TPA-1.0 showed compatible catalytic activity to other heteropoly acid catalysts, but higher temperature (100–120 °C), longer reaction time (7.5 h) and larger molar ratio of methanol and oleic acid (20:1) were needed for HPW/H-Y and SWIL/SiO2 (entries 7–9). These results clearly demonstrated that the catalytic activity of Mg20F39TPA-1.0 presented in this work was more superior to those of previously reported solid acidic catalysts. These results clearly demonstrated that the catalytic activity of Mg20F39TPA-1.0 presented in this work was more superior to those of previously reported solid acidic catalysts.
| No. | Catalyst | Reaction conditions | Conversion (%) | Ref. |
|---|---|---|---|---|
| 1 | [BMIM][HSO4] | T = 87 °C, CA = 98 wt%, M/O = 9:1, t = 5 h |
81 | 53 |
| 2 | SCER | T = 82 °C, CA = 47 wt%, M/O = 9:1, t = 8 h |
93 | 54 |
| 3 | POMC | T = 80 °C, CA = 5 wt%, M/O = 15:1, t = 5 h |
73 | 55 |
| 4 | HClSO3–ZrO2 | T = 100 °C, CA = 3 wt%, M/O = 8:1, t = 12 h |
100 | 56 |
| 5 | WO3/USY | T = 200 °C, CA = 10 wt%, M/O = 6:1, t = 2 h |
74 | 57 |
| 6 | MPD–SO3H–IL | T = 100 °C, CA = 10 wt%, M/O = 30:1, t = 4 h |
98 | 14 |
| 7 | TPA/H–Y | T = 120 °C, CA = 13 wt%, M/O = 20:1, t = 7.5 h |
98 | 58 |
| 8 | SWIL/SiO2 | T = 100 °C, CA = 5 wt%, M/O = 30:1, t = 4 h |
97 | 59 |
| 9 | Mg20F39TPA-1.0 | T = 80 °C, CA = 5 wt%, M/O = 9:1, t = 4 h |
95 ± 0.6% | This work |
3.6 Simultaneous esterification and transesterification of Jatropha oil
Simultaneous esterification and transesterification is highly desired for producing biodiesel from high acid value inedible oil. Thus, Jatropha oil (19.35 mg KOH/g) was selected as feedstock to produce biodiesel over Mg20F39TPA-1.0. The high biodiesel yield 93% was achieved under moderate reaction conditions (methanol/oil molar ratio of 30/1, catalyst dosage of 8 wt%, reaction time of 12 h, reaction temperature of 130 °C), as can be seen in Table S2.† For the simultaneous esterification and transesterification of Jatropha oil, some more sufficient comparison results have been listed in Table 5. It was clear to see that high temperature (160–220 °C) was required to get high biodiesel yield, except for producing biodiesel over Mg20F39TPA-1.0. Especially, higher temperature and longer time were necessary over TPA/Nb2O5 and TPA/Al2O3 to achieve comparable catalytic activity to Mg20F39TPA-1.0 (entries 6, 7). These results indicated that Mg20F39TPA-1.0 was an excellent catalyst in the simultaneous esterification and transesterification.| No. | Catalyst | Feedstock | Reaction conditions | Yield (%) | Ref. |
|---|---|---|---|---|---|
| 1 | E-260-20-SO3H | Jatropha oil | T = 220 °C, CA = 5 wt%, M/O = 12:1, t = 5 h |
93 | 60 |
| 2 | Zr–PMOs | Palm oil | T = 209 °C, CA = 13 wt%, M/O = 49:1, t = 6 h |
85 | 61 |
| 3 | FS-B-L-IL | K. integrifoliola oil | T = 160 °C, CA = 10 wt%, M/O = 40:1, t = 10 h |
93 | 62 |
| 4 | Fe2O3–MnO–SO42−/ZrO2 | Waste cooking oil | T = 180 °C, CA = 3 wt%, M/O = 20:1, t = 4 h |
95 | 63 |
| 5 | [BMIm][TS]–ZnCl2 | Jatropha oil | T = 200 °C, CA = 3 wt%, M/O = 12:1, t = 5 h |
88 | 64 |
| 6 | TPA/Nb2O5 | Cooking oil | T = 200 °C, CA = 3 wt%, M/O = 18:1, t = 20 h |
92 | 65 |
| 7 | TPA/Al2O3 | Canola oil | T = 200 °C, CA = 3 wt%, M/O = 6:1, t = 10 h |
65 | 66 |
| 8 | Mg20F39TPA-1.0 | Jatropha oil | T = 130 °C, CA = 8 wt%, M/O = 30:1, t = 12 h |
93 ± 0.7 | This work |
As mentioned above, the total acidity of the catalysts played an important role in biodiesel production. Thus, the catalytic performance of the catalysts bearing the same acid density was compared, and the results are shown in Table S3.† The conversion of oleic acid (79 ± 0.9%) and yield of biodiesel (45 ± 1.2%) over Nafion NR50 were higher than those of Amberlyst 15, but relatively higher catalyst amount was needed due to its lower acid density. It was worthy to note that the highest conversion and yield could be obtained over Mg20F39TPA-1.0, which was 95 ± 0.6% and 93 ± 0.7% respectively, possibly due to the stronger acidity resulted from the moiety of heteropoly acids compared with the conventional solid acids.67
4. Conclusions
In summary, a range of Mg20F39TPA-x hybrids catalysts were prepared by solvothermal method and their catalytic activities were investigated for esterification of oleic acid with methanol. Mg20F39TPA-1.0 was found to exhibit excellent catalytic performance with the conversion of 95% of oleic acid under the optimum esterification reaction conditions (methanol/oleic acid molar ratio of 9:1, 5 wt% catalyst dosage at 80 °C for 4 h). Moreover, Mg20F39TPA-1.0 also shows satisfied catalytic activity for the simultaneous esterification and transesterification reaction of high acid value oil, during which 93% yield of biodiesel was achieved at mild conditions. It was found that solvothermal solidified Mg20F39TPA-x gives higher density of acidic sites and provides better catalytic performance than that of the immobilized TPA/MgF2. Further leaching experiment confirms that Mg20F39TPA-1.0 was heterogeneous in this reaction system. And the catalyst still keeps high catalytic activity after five consecutive cycles. Therefore, the Mg20F39TPA-1.0 catalyst described herein shows great potential for biodiesel production.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21576059, 21666008), the Key Technologies R&D Program (2014BAD23B01), and the Innovation Fund for Graduate Students of Guizhou University (No. 2016075).References
- C. Z. Liu, F. Wang, A. R. Stiles and C. Guo, Appl. Energy, 2012, 92, 406 CrossRef CAS
.
- W. Liu, P. Yin, X. G. Liu, S. H. Zhang and R. J. Qu, J. Ind. Eng. Chem., 2015, 21, 893 CrossRef CAS
- Y. Chang, C. Lee and C. Bae, RSC Adv., 2014, 4, 47448 RSC
- R. Luque, J. C. Lovett, B. Datta, J. Clancy, J. M. Campelo and A. A. Romero, Energy Environ. Sci., 2010, 3, 1706 CAS
- S. Sankaranarayanan, C. A. Antonyraj and S. Kannan, Bioresour. Technol., 2012, 109, 57 CrossRef CAS PubMed
- I. K. Hong, H. Jeon, H. Kim and S. B. Lee, J. Ind. Eng. Chem., 2016, 42, 107 CrossRef CAS
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285 RSC
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887 RSC
- L. J. Konwar, J. Boro and D. Deka, Renewable Sustainable Energy Rev., 2014, 29, 546 CrossRef CAS
- D. Meloni, D. Perra, R. Monaci, M. G. Cutrufello, E. Rombi and I. Ferino, Appl. Catal., B, 2016, 184, 163 CrossRef CAS
- Q. Zhou, H. Zhang, F. Chang, H. Li, H. Pan, W. Xue, D. Y. Hu and S. Yang, J. Ind. Eng. Chem., 2015, 31, 385 CrossRef CAS
- X. Deng, Z. Fang and Y. H. Liu, Energy Convers. Manage., 2010, 51, 2802 CrossRef CAS
- S. Jansri, S. B. Ratanawilai, M. L. Allen and G. Prateepchaikul, Fuel Process. Technol., 2011, 92, 1543 CrossRef CAS
- H. Pan, H. Li, X. F. Liu, H. Zhang, K. L. Yang, S. Huang and S. Yang, Fuel Process. Technol., 2016, 150, 50 CrossRef CAS
- H. Amani, Z. Ahmad, M. Asif and B. H. Hameed, J. Ind. Eng. Chem., 2014, 20, 4437 CrossRef CAS
- D. A. G. Aranda, R. T. P. Santos, N. C. O. Tapanes, A. L. D. Ramos and O. A. C. Antunes, Catal. Lett., 2008, 122, 20 CrossRef CAS
- K. Y. Nandiwale and V. V. Bokade, Ind. Eng. Chem. Res., 2014, 53, 18690 CrossRef CAS
- R. M. N. Kalla, M. R. Kim, Y. N. Kim and L. Kim, New J. Chem., 2016, 40, 687 RSC
- D. Y. Zhang, M. H. Duan, X. H. Yao and Y. J. Fu, Fuel, 2016, 172, 293 CrossRef CAS
- Z. W. Wu, C. Chen, Q. R. Guo, B. X. Li, Y. G. Que, L. Wang, H. Wan and G. F. Guan, Fuel, 2016, 184, 128 CrossRef CAS
- K. Saravanan, B. Tyagi and H. C. Bajaj, Appl. Catal., B, 2016, 192, 161 CrossRef CAS
- S. Y. Chen, S. Lao-ubol, T. Mochizuki, Y. Abe, M. Toba and Y. Yoshimura, Bioresour. Technol., 2014, 157, 346 CrossRef CAS PubMed
- K. Malins, J. Brinks, V. Kampars and I. Malina, Appl. Catal., A, 2016, 519, 99 CrossRef CAS
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS PubMed
- T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113 CAS
- T. Okuhara, T. Nishimura and M. Misono, Stud. Surf. Sci. Catal., 1996, 101, 581 CrossRef CAS
- M. Misono, Chem. Commun., 2001, 13, 1141 RSC
- J. Li, X. H. Wang, W. M. Zhu and F. H. Cao, ChemSusChem, 2009, 2, 177 CrossRef CAS PubMed
- J. A. Monge, G. Trautwein and J. P. M. Lozar, Appl. Catal., A, 2013, 468, 432 CrossRef
- C. F. Oliveira, L. M. Dezaneti, F. A. C. Garcia, J. L. Macedo, J. A. Dias, S. C. L. Dias and K. S. P. Alvim, Appl. Catal., A, 2010, 372, 153 CrossRef CAS
- J. W. Sun, P. F. Yan, G. H. An, J. Q. Sha, G. M. Li and G. Y. Yang, Sci. Rep., 2016, 6, 25595 CrossRef CAS PubMed
- Z. Bian, S. Zhang, X. Zhu, Y. Li, H. Liu and J. Hu, RSC Adv., 2015, 5, 31502 RSC
- J. Li, D. F. Li, J. Y. Xie, Y. Q. Liu, Z. J. Guo, Q. Wang, Y. Lyu, Y. Zhou and J. Wang, J. Catal., 2016, 339, 123 CrossRef CAS
- S. H. Zhu, X. Q. Gao, F. Dong, Y. L. Zhu, H. Y. Zheng and Y. W. Li, J. Catal., 2013, 306, 155 CrossRef CAS
- S. Wuttke, S. M. Coman, G. Scholz, H. Kirmse, A. Vimont, M. Daturi, S. L. M. Schroeder and E. Kemnitz, Chem.–Eur. J., 2008, 14, 11488 CrossRef CAS PubMed
- F. Zhang, Z. Fang and Y. T. Wang, Appl. Energy, 2015, 155, 637 CrossRef CAS
- K. Jagadeeswaraiah, C. R. Kumar, P. S. S. Prasad and N. Lingaiah, Catal. Sci. Technol., 2014, 4, 2969 CAS
- M. X. Cheng, T. Shi, H. Y. Guan, S. T. Wang, X. H. Wang and Z. J. Jiang, Appl. Catal., B, 2011, 107, 104 CrossRef CAS
- L. Y. Meng, S. R. Zhai, Z. C. Sun, F. Zhang, Z. Y. Xiao and Q. D. An, Microporous Mesoporous Mater., 2015, 204, 123 CrossRef CAS
- Z. Zillillah, T. A. Ngu and Z. Li, Green Chem., 2014, 16, 1202 RSC
- F. Richard, S. Célérier, M. Vilette, J. D. Comparot and V. Montouillout, Appl. Catal., B, 2014, 152–153, 241 CrossRef CAS
- I. A. Telleria, Y. Guo, F. Hemmann, P. L. Arias and E. Kemnitz, Catal. Sci. Technol., 2014, 4, 1357 Search PubMed
- X. Mo, D. E. López, K. Suwannakarn, Y. Liu, E. Lotero, J. G. Goodwin and C. Lu, J. Catal., 2008, 254, 332 CrossRef CAS
- X. X. Han, H. Du, C. T. Hung, L. L. Liu, P. H. Wu, D. H. Ren, S. J. Huang and S. B. Liu, Green Chem., 2015, 17, 499 RSC
- N. Sudarsanam and D. Frey, Qual. Reliab. Eng. Int., 2011, 27, 947 CrossRef
- Y. Zhang, W. T. Wong and K. F. Yung, Appl. Energy, 2014, 116, 191 CrossRef CAS
- X. X. Han, K. K. Chen, W. Yan, C. T. Hung, L. L. Liu, P. H. Wu, K. C. Lin and S. B. Liu, Fuel, 2016, 165, 115 CrossRef CAS
- F. Zhang, Z. Fang and Y. T. Wang, Appl. Energy, 2015, 155, 637 CrossRef CAS
- H. Li, Z. Fang, J. Luo and S. Yang, Appl. Catal., B, 2017, 200, 182 CrossRef CAS
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2016, 18, 726 RSC
- J. He, H. Li, Y. X. Liu, W. F. Zhao, T. T. Yang, W. Xue and S. Yang, J. Ind. Eng. Chem., 2016, 43, 133 CrossRef CAS
- J. Erkelens and T. J. Liefkens, J. Catal., 1966, 5, 542 CrossRef CAS
- A. H. M. Fauzi and N. Amin, Energy Convers. Manage., 2013, 76, 818 CrossRef
- Y. Jiang, J. Lu, K. Sun, L. Ma and J. Ding, Energy Convers. Manage., 2013, 76, 980 CrossRef CAS
- L. Wang, X. Dong, H. Jiang, G. Li and M. Zhang, Catal. Commun., 2014, 56, 164 CrossRef CAS
- Y. Zhang, W. Wong and K. Yung, Appl. Energy, 2014, 116, 191 CrossRef CAS
- A. Costa, P. Braga, J. de Macedo, J. Dias and S. Dias, Microporous Mesoporous Mater., 2014, 147, 142 CrossRef
- C. Baroi and A. K. Dalai, Appl. Catal., A, 2014, 485, 99 CrossRef CAS
- B. Zhen, H. Li, Q. Jiao, Y. Li, Q. Wu and Y. Zhang, Ind. Eng. Chem. Res., 2012, 51, 10374 CrossRef CAS
- M. Huang, J. Luo, Z. Fang and H. Li, Appl. Catal., B, 2016, 190, 103 CrossRef CAS
- R. Sánchez-Vázquez, C. Pirez, J. Iglesias, K. Wilson, A. F. Lee and J. A. Melero, ChemCatChem, 2013, 5, 994 CrossRef
- H. Zhang, H. Li, H. Pan, X. Liu, K. Yang, S. Huang and S. Yang, Energy Convers. Manage., 2017, 138, 45 CrossRef CAS
- F. Alhassan, U. Rashid and Y. Taufiq-Yap, Fuel, 2015, 142, 38 CrossRef CAS
- F. Guo, Z. Fang, X. Tian, Y. Long and L. Jiang, Bioresour. Technol., 2013, 140, 447 CrossRef PubMed
- V. Brahmkhatri and A. Patel, Fuel, 2012, 102, 72 CrossRef CAS
- K. Narasimharao, D. R. Brown, A. F. Lee, A. D. Newman, P. F. Siril, S. J. Tavener and K. Wilson, J. Catal., 2007, 248, 226 CrossRef CAS
- N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06080g |
| This journal is © The Royal Society of Chemistry 2017 |