
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFermentation of Illigera aromatica with Clonostachys rogersoniana producing novel cytotoxic menthane-type monoterpenoid dimers†
Jian-Wei Dong ,
Le Cai*,
Xue-Jiao Li,
Rui-Feng Mei,
Jia-Peng Wang,
Ping Luo,
Yan Shu and
Zhong-Tao Ding*
,
Le Cai*,
Xue-Jiao Li,
Rui-Feng Mei,
Jia-Peng Wang,
Ping Luo,
Yan Shu and
Zhong-Tao Ding*
Functional Molecules Analysis and Biotransformation Key Laboratory of Universities in Yunnan Province, School of Chemical Science and Technology, Yunnan University, Kunming 650091, P. R. China. E-mail: ztding@ynu.edu.cn; caile@ynu.edu.cn
First published on 9th August 2017
Abstract
Illigera aromatica, a medicinal liana, was fermented with Clonostachys rogersoniana. Two novel menthane-type monoterpenoid dimers, dimericilligerates E (1) and F (2) were isolated from the fermented material. Their structures were identified by 1D/2D NMR, electric circular dichroism, derivatization, and Mosher's method. A novel elimination reaction that formed a benzene ring from a menthane moiety afforded derivative 1a, which possesses more resolved NMR signals than the dimeric monoterpenoid, dimericilligerate E (1). The determination of 1a verified the estimation of 1. Compounds 1 and 2 showed potent cytotoxic activities; especially, 1 possessed significant activities against the liver hepatocellular carcinoma cell line (SMMC-7721) with an IC50 value of 3.89 ± 0.23 μM. Three novel precursors, dimericilligerates A–C (3–5), were identified by HPLC-MS and isolated from the original I. aromatica. This paper presents a novel class of monoterpenoid dimers and suggests that C. rogersoniana-fermented I. aromatica is effective to produce cytotoxic dimeric monoterpenoids from inactive original materials.
Introduction
The genus Illigera, one of four that make up the family Hernandiaceae, contains approximately thirty known species distributed in southern Africa and Asia.1 The chemical constituents of this genus include aporphine alkaloids,2–5 phenolic acids, and steroids,6 which have been reported to have various bioactivities, including antiplatelet aggregation,6 cytotoxic activity,4,5 and vasorelaxant activity.2Illigera aromatica (Hernandiaceae) is a small liana that mainly grows in the Yunnan and Guangxi provinces in China,7 and is frequently used by local people for promoting blood circulation and treating tuberculosis. It has been reported that the chemical composition of I. aromatica includes fatty acids and their esters,8 steroids,9 monoterpenoids8,10 and alkaloids.10
Herbal fermentation processing, which has been practised in China for more than 4000 years, frequently causes biotransformations and changes in bioactivity. For example, Kim et al.11 reported that pre-treating fermented Curcuma longa L. with Aspergillus oryzaeis effectively prevents CCl4-induced hepatic damage in rats. Hsu et al.12 reported that fermenting Radix astragali with Bacillus subtilis significantly stimulates the biosynthesis of type I procollagen in a dose-dependent manner in both young and old human dermal fibroblast cells. Our previous studies have shown that the fermentations of Bletilla striata13 and Asparagus filicinus14 both facilitate the biotransformations of their components and enhance their bioactivities.
The secondary metabolites of Clonostachys rogersoniana, a rhizosphere fungus, are cyclic peptides15 and polyketides.16 Previous studies have shown that C. rogersoniana fermentation could transform the aporphine alkaloids to 4R-hydroxyaporphine alkaloids, improving their water solubility and acetylcholinesterase inhibitory activity.17,18
In the present study, several fungi including Fusarium oxysporum, Fusarium avenaceum, Geomyces luteus, Alternaria compacta, Talaromyces purpurogenus, Scytalidium lignicola, Cladosporium cladosporioides, Stagonosporopsis cucurbitacearum, Penicillium vancouverense, and C. rogersoniana were used to ferment the tubers of I. aromatica. Fermentation with C. rogersoniana caused obvious transformation, prompting an investigation into the chemical constituents of C. rogersoniana-fermented I. aromatica. Two novel menthane-type monoterpenoid dimers, dimericilligerates E (1) and F (2) were isolated from the fermented material (Fig. 1). Afterwards, three precursors, dimericilligerates A–C (3–5), were identified by LC-MS and isolated from the original I. aromatica. The biotransformations from 3–5 to 1 and 2 were elucidated. The free monoterpenoid dimers (1–2) showed stronger cytotoxic activity than the original compounds (3–5). This paper presents an effective approach to the production of cytotoxic dimeric monoterpenoids from inactive original materials.
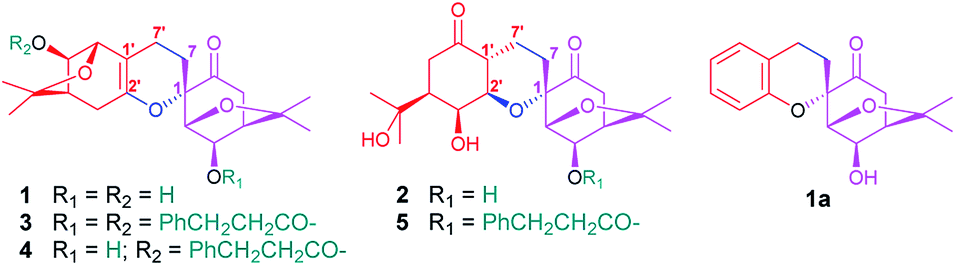 | ||
| Fig. 1 Structures of compounds 1–5 and 1a. | ||
Results and discussion
Identification of compounds isolated from fermented I. aromatica
After I. aromatica was fermented with C. rogersoniana, two compounds (1 and 2) were isolated by silica gel and Sephadex LH-20 column chromatography (CC).Compound 1, isolated as a white amorphous powder, was determined to have the molecular formula C21H32O5 based on analysis of its HRESIMS m/z 409.2235 [M + COOH]− (calcd for C22H33O7 409.2232), 13C NMR and DEPT spectra, which indicated seven degrees of unsaturation. The 1H and 13C NMR data (Table 1) showed four methyl carbons, one carbonyl, and seven oxygenated alkyl groups. Comparing its NMR data with the known isolated menthane-type monoterpenoid, cis-4-hydroxy-5-(1-hydroxy-1-methylethyl)-2-methyl-2-cyclohexen-1-one,19,20 1 possesses four angular methyl groups, which is double the number found in the known monoterpenoid, and two high-field methylenes that appear instead of the C-7 methyl group in the menthane-type monoterpenoid, suggesting 1 is a dimer of monoterpenoids, composed of the two menthane-type monoterpenoid components I and II. In component I (shown in purple, Fig. 1), an oxygenated methine at δC 77.3 (d) was assigned at C-3 based on the HMBC cross peaks of H-3/C-2, C-4, and C-8 (Fig. 2); a carbonyl at δC 206.3 (s) was assigned based on the HMBC networks of H-2/C-6, H-5/C-6, and H-7/C-6; an oxygenated quaternary carbon at δC 79.6 (s) located at C-1 was identified by the HMBC cross-peak of H-2/C-1; and a C(2)–O–C(8)–C(4) bridge was established by the HMBC cross-peaks of H-2/C-8, and CH3-9,10/C-8 and C-4, indicating a bicyclo[3,2,1]menthane-type monoterpenoid as shown in Fig. 1 (component I). Similar to the aforementioned analysis, the component II (shown in red, Fig. 1) was determined to be a bicyclo[3,2,1]menthane-type monoterpenoid based on the HMBC data in which a double bond between C-1′ and C-6′ was identified based on cross-peaks of H-7′/C-1′, C-2′, and C-6′, and H-2′/C-1′; two peaks representing oxygenated carbons at δC 146.4 (s) and 78.5 (d) were assigned to C-6′ and C-3′ based on the cross-peaks of H-3′/C-2′ and C-8′, as well as a C(4′)–C(8′)–O–C(2′) bridge that was identified based on the cross-peak of H-2′/C-8′. The absences of the two methyl carbon signals from C-7 and C-7′ in menthane-type monoterpenoid fragments, and the 1H–1H COSY correlation of H-7/H-7′ confirmed the connection between components I and II via a C(7)–C(7′) bond. Combined with the molecular formula C38H44O8, a linkage of C(1)–O–C(6′) was determined to be a part of a six-membered ring. Hence, the planar structure of 1 involves an unprecedented carbon skeleton of two menthane-type monoterpenoids.
| No. | 1 | 1a | 2 | |||
|---|---|---|---|---|---|---|
| δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | |
| 1 | 79.6 C | 80.0 C | 79.3 C | |||
| 2 | 84.6 CH | 4.11 s | 84.2 CH | 4.16 s | 89.3 CH | 3.73 s |
| 3 | 77.3 CH | 4.87 s | 77.4 CH | 5.00 s | 76.2 CH | 4.74 s |
| 4 | 47.4 CH | 2.31 s | 47.2 CH | 2.37 d (3.2) | 47.5 CH | 2.22 br s |
| 5 | 42.6 CH2 | 2.61 d (3.6), 2.59 d (3.6) | 42.6 CH2 | 2.71 d (3.2) | 42.8 CH2 | 2.50 s |
| 6 | 206.3 C | 206.5 C | 208.8 C | |||
| 7 | 23.6 CH2 | 2.09 m, 1.73 ddd (13.2, 11.4, 4.8) | 23.1 CH2 | 2.26 ddd (10.4, 6.0, 4.4), 2.03 ddd (10.4, 6.0, 4.4) | 25.5 CH2 | 1.93 m, 1.43 m |
| 8 | 84.4 C | 84.6 C | 84.3 C | |||
| 9 | 26.9 CH3 | 1.18 s | 27.0 CH3 | 1.22 s | 26.7 CH3 | 1.13 s |
| 10 | 29.7 CH3 | 1.53 s | 29.7 CH3 | 1.56 s | 29.6 CH3 | 1.47 s |
| 1′ | 108.2 C | 122.3 C | 42.1 CH | 2.73 m | ||
| 2′ | 82.3 CH | 3.91 s | 152.7 C | 75.8 CH | 3.90 s | |
| 3′ | 78.5 CH | 4.24 s | 116.8 CH | 6.74 d (8.0) | 68.8 CH | 4.19 s |
| 4′ | 48.6 CH | 2.23 s | 127.5 CH | 6.86 dd (8.0, 6.8) | 44.9 CH | 1.96 d (2.4) |
| 5′ | 32.9 CH2 | 2.36 d (2.4), 2.15 d (2.4) | 121.1 CH | 7.03 dd (8.0, 6.8) | 36.8 CH2 | 2.69 br d (13.2), 2.34 dd (13.2, 2.4) |
| 6′ | 146.4 C | 129.6 CH | 7.06 dd (8.0) | 211.7 C | ||
| 7′ | 20.2 CH2 | 2.17 m, 2.13 m | 20.8 CH2 | 2.92 ddd (10.4, 7.6, 4.4), 2.75 ddd (10.4, 7.6, 4.4) | 17.4 CH2 | 2.19 m, 2.03 m |
| 8′ | 82.1 C | 73.3 C | ||||
| 9′ | 27.2 CH3 | 1.30 s | 28.9 CH3 | 1.38 s | ||
| 10′ | 31.1 CH3 | 1.41 s | 28.4 CH3 | 1.19 s | ||
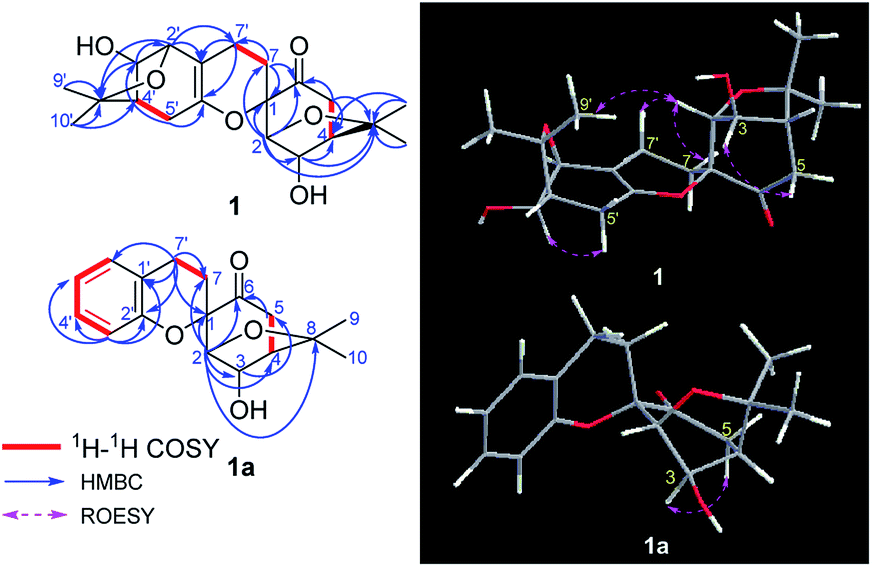 | ||
| Fig. 2 Key 1H–1H COSY, HMBC, and ROESY correlations of 1 and 1a. | ||
In the above planar structure, H-2/H-4 and H-2′/H-4′ are at the bridgeheads of bicyclo[3,2,1] ring systems and are splayed out from the bicycle in opposite directions, indicate α-orientations for H-2/H-4 and H-2′/H-4′, respectively. In the ROESY spectrum, the cross-peaks of H-3/H-5 and H-3′/H-5′, allowed the assignment of α-orientations for H-3 and H-3′, respectively. Furthermore, presence of the H-2/H-7β [δH 1.73 (1H, ddd, J = 13.2, 11.4, 4.8 Hz)] cross-peak and the absence of the H-3/H-7 cross-peak suggested a β-orientation for C-7 bound to C-1. The relative configuration of components I and II was assigned as shown in Fig. 2 by the ROESY cross-peak of H-2/H-9′. Hence, the relative configuration of 1 was determined to be 1S*,2R*,3R*,4R*,2′S*,3′R*,4′R*.
To assign the absolute configuration of 1, a further derivatization was carried out for obtaining a mono-hydroxyl derivative. 1 was treated with methanesulfonyl chloride (MsCl), to promote the novel elimination reaction yielding 1a (Scheme 1).
 | ||
| Scheme 1 Derivative reaction of dimericilligerate E (1). | ||
Compound 1a, a white powder, has a molecular formula of C38H44O8 based on analysis of its HRESIMS m/z 311.1253 (calcd for C17H20O4Na 311.1254). Its 1H and 13C NMR data showed the presence of a benzene ring, two methyl groups, one carbonyl, and four oxygenated alkyl groups. Analysis of coupling constants in the 1H NMR data in the low-field region (Table 1) showed an ortho-disubstituted benzene ring. The presence of this structural feature is supported by the 1H–1H COSY correlations of H-3′/H-4′, H-4′/H-5′, and H-5′/H-6′, and the HMBC cross-peaks of H-3′/C-4′, C-5′, C-2′ and C-1′ (Fig. 2). Comparing the rest of the NMR data of 1a with that of 1, they shared similar signals from C-1–C-7, indicating that 1a possessed the same menthane-type monoterpenoid as component I (purple), which is supported by the HMBC correlations shown in Fig. 2. The 1H–1H COSY cross-peak of H-7/H-7′ and the HMBC cross-peaks of H-7′/C-1′, C-6′, and C-2′ suggested the two components are linked by a bond at C(7)–C(7′). Combing that information with the molecular formula, leads to the conclusion that C-2′ and C-1 are linked by C(1)–O–C(2′). With that, the planar structure of 1a was elucidated. Based on comparison of structures of 1 and 1a, 1a is formed from 1 by an elimination of component II, and therefore, the relative configuration of 1a is same as component I of 1; this is supported by the ROESY cross-peak of H-3/H-5. This is a novel reaction for the construction of a benzene ring from a menthane unit.
In the present study, the NMR spectrum of 1a was better resolved than the spectrum of 1; having more separation between high-field and low-field peaks in the spectrum of 1a also helped to confirm the structure of 1. Besides that, this elimination could produce the mono-hydroxyl compound 1a, which indicates that Mosher's method is an option for determination the absolute configuration.
The absolute configuration of 1a was assigned using Mosher's method; the treatment of 1a with (R)- and (S)-(α)-methoxy(trifluoromethyl)phenylacetic acid (MTPA-OH) yielded (R)- and (S)-MTPA esters, respectively. The values of the 1H NMR ΔδS–R for the mono-MTPA esters (shown in Fig. 3) revealed an R absolute configuration at C-3 of 1a, implying the absolute configuration of 1 is 1S,2R,3R,4R,2′S,3′R,4′R. Therefore, the structure of 1 was solved as shown and named dimericilligerate E.
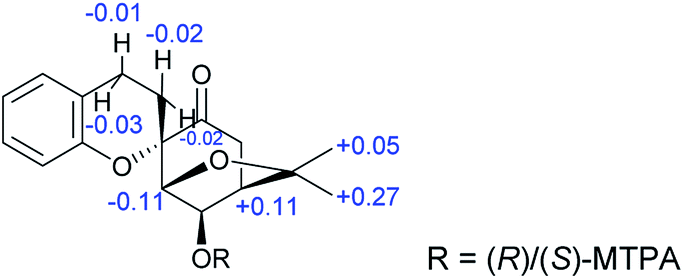 | ||
| Fig. 3 ΔδS–R of 1H-NMR for S- and R-MTPA esters of compound 1a. | ||
Compound 2, isolated as a white amorphous powder, possessed a molecular formula of C20H30O7, based on HRESIMS m/z 381.1917 [M − H]− (calcd for C20H29O7, 381.1919), 13C NMR and DEPT spectra. The 1H NMR spectrum showed four methyl groups; the 13C NMR and DEPT spectra showed two carbonyls and seven oxygenated alkyl carbons located downfield. Comparing the NMR data of 2 with that of 1 reveals that 2 has a similar dimeric monoterpenoid structure. Two partial structural components, I (in purple) and II (in red), were then constructed based on analysis of NMR data. Component I of 2 was found to be same component I of 1 through detailed comparison of the NMR data and the HMBC cross-peaks of H-2/C-1, C-3, C-4, C-6, and C-8, H-3/C-4 and C-8, and H-5/C-6, as well as the cross-peak of H-4/H-5 in 1H–1H COSY spectrum (Fig. 4). Analysing the remaining NMR data, a six-membered menthane-type monoterpenoid was identified based on the HMBC cross-peaks of CH3-9′,10′/C-8′ and C-4′, and H-7′/C-1′ and C-2′. The HMBC correlations of H-3′/C-6, H-4′/C-6′, and H-2′/C-6′ allowed C-6′ to be assigned as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and the HMBC cross-peaks of H-3′/C-2′ and H-2′/C-4′ indicated that two of the oxygenated methines were located at C-3′ and C-2′. The linkage of components I and II via the bonds of C(7)–C(7′) and C(2′)–O–C(1), forming a stable six-membered tetrahydropyran ring, was determined based on the key 1H–1H COSY interaction of H-7/H-7′ and the HMBC correlation of H-2′/C-1. Thus, the planar structure of 2 was identified as an ester of phenylpropionic acid and novel dimeric monoterpenoids composed of a bicyclo[3,2,1] group and a six-membered menthane-type monoterpenoid.
O, and the HMBC cross-peaks of H-3′/C-2′ and H-2′/C-4′ indicated that two of the oxygenated methines were located at C-3′ and C-2′. The linkage of components I and II via the bonds of C(7)–C(7′) and C(2′)–O–C(1), forming a stable six-membered tetrahydropyran ring, was determined based on the key 1H–1H COSY interaction of H-7/H-7′ and the HMBC correlation of H-2′/C-1. Thus, the planar structure of 2 was identified as an ester of phenylpropionic acid and novel dimeric monoterpenoids composed of a bicyclo[3,2,1] group and a six-membered menthane-type monoterpenoid.
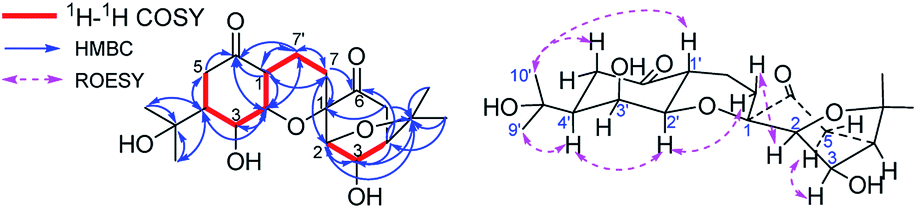 | ||
| Fig. 4 Key 1H–1H COSY, HMBC, and ROESY correlations of 2. | ||
The relative configuration of 2 was assigned based on a ROESY experiment, which showed correlations of H-3/H-5 and H-2/H-7β [δH 1.43 (1H, m)] and lacked a correlation of H-3/H-7, indicating a β,α,α,α-orientation of CH2-7, H-2, H-3, and H-4, respectively. Meanwhile, H-1′β, H-3′α, and H-4′α configurations were assigned based on the existence of cross-peaks of H-1′/H-2 and CH3-9′ and the absence of a cross-peak of H-1′/H-3′; and an H-2′α configuration was deduced by the cross-peaks of H-2′/H-7α [δH 1.93 (1H, m)], and H-4′. Therefore, the relative configuration of 2 was determined to be 1S*,2R*,3R*,4R*,1′R*,2′R*,3′S*,4′S*.
Two possible configurations, (1S,2R,3R,4R,1′R,2′R,3′S,4′S) and (1R,2S,3,4S,1′S,2′S,3′R,4′R), were optimized using the density functional theory (DFT) method at the B3LYP/6-31G(d,p) level.21 Subsequently, the ECD was calculated using time-dependent density functional theory (TDDFT) at the B3LYP/6-31G(d,p) level with the PCM solvent model22 and simulated by SpecDic 1.64.23,24 The calculated ECD curve of the (1S,2R,3R,4R,1′R,2′R,3′S,4′S)-isomer (Fig. 5A) resembled the experimental ECD curve, indicating that the absolute configuration of 2 is (1S,2R,3R,4R,1′R,2′R,3′S,4′S). The structure of 2 was thus established as depicted and named dimericilligerate F.
 | ||
| Fig. 5 Experimental and calculated ECD spectra of 1–5. | ||
Analysis of non-fermented and fermented I. aromatica
To find the precursors of dimericilligerates E (1) and F (2), HPLC-ESIMS was used to identify possibilities from the different extracted fractions of the original I. aromatica. The peaks corresponding to 1 and 2 were not detected in any fraction; and two major peaks at m/z 627.4 [M − H]− and 513.2 [M − H]− were observed in the petroleum ether extract (Fig. S52†). Therefore, 1 and 2 might be produced from the components of 627.4 [M − H]− and 513.2 [M − H]−. Inspired by that, three compounds, 3–5, were isolated from the petroleum ether extract and identified by 1D/2D NMR and ECD spectroscopy.Identification of precursors
Compound 3, isolated as a colourless gummy oil, was found to have a molecular formula of C38H44O8 based on analysis of its HRESIMS m/z 651.2920 [M + Na]+ (calcd for C38H44O8Na 651.2928), 13C NMR and DEPT spectra, which indicated seventeen degrees of unsaturation. The 1H and 13C NMR data (Table 2) showed four methyl groups, two benzene rings, three carbonyls (one ketone and two esters), and seven oxygenated alkyl groups. Comparing the NMR data with phenylpropionic acid25,26 and monoterpenoid phenylpropionic acid esters10 previously isolated from I. aromatica indicated that 3 contains two phenylpropionic esters. Comparison of the rest of the NMR data of 3 with that of 1, suggests that the rest of the structure of 3 is same as 1, which is supported by the 1H–1H COSY and HMBC correlations shown in Fig. 6. The cross-peaks of H-3/C-9′′ and H-3′/C-9′′′ revealed two phenylpropionic units attached to C-3 and C-3′, respectively. Hence, the planar structure of 3 was established as shown.| No. | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|
| δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | |
| 1 | 79.7 C | 79.6 C | 78.3 C | |||
| 2 | 81.0 CH | 4.10 s | 84.7 CH | 4.11 d (1.2) | 81.6 CH | 4.16 s |
| 3 | 78.5 CH | 5.69 s | 77.4 CH | 4.88 s | 82.1 CH | 5.67 s |
| 4 | 45.5 CH | 2.28 s | 47.5 CH | 2.33 s | 46.2 CH | 2.39 d (7.2) |
| 5 | 42.5 CH2 | 2.63 br s | 42.7 CH2 | 2.62 d (3.0), 2.59 d (3.0) | 44.8 CH2 | 2.79 d (2.4), 2.59 dd (7.2, 2.4) |
| 6 | 205.0 C | 205.9 C | 210.9 C | |||
| 7 | 19.8 CH2 | 2.05 m, 2.03 m | 23.5 CH2 | 2.11 ddd (13.8, 11.4, 5.4), 1.73 ddd (13.8, 11.4, 5.4) | 19.8 CH2 | 1.90 m, 1.81 m |
| 8 | 84.1 C | 84.3 C | 84.5 C | |||
| 9 | 26.4 CH3 | 1.09 (3H, s) | 27.0 CH3 | 1.19 s | 29.7 CH3 | 1.33 s |
| 10 | 29.1 CH3 | 1.25 (3H, s) | 29.8 CH3 | 1.53 s | 26.7 CH3 | 1.14 s |
| 1′ | 108.4 C | 108.7 C | 56.1 CH | |||
| 2′ | 79.3 CH | 3.94 s | 79.5 CH | 4.03 s | 84.1 CH | 6.74 d (6.0) |
| 3′ | 80.1 CH | 5.02 s | 80.3 CH | 5.11 s | 77.6 CH | 4.70 dd (6.0, 3.2) |
| 4′ | 46.4 CH | 2.18 s | 46.6 CH | 2.29 d (15.0) | 47.2 CH | 1.93 dd (10.0, 3.2) |
| 5′ | 32.7 CH2 | 2.41 m, 2.17 m | 32.9 CH2 | 2.46 dd (15.0, 3.6), 2.22 d (3.6) | 41.8 CH2 | 2.84 dd (10.0, 4.0), 2.53 d (4.0) |
| 6′ | 146.3 C | 146.0 C | 210.9 C | |||
| 7′ | 22.6 CH2 | 2.04 m, 1.70 ddd (8.0, 5.6, 2.4) | 20.2 CH2 | 2.19 ddd (13.8, 11.4, 5.4), 2.14 ddd (13.8, 11.4, 5.4) | 29.7 CH2 | 2.33 m, 2.22 m |
| 8′ | 82.2 C | 82.2 C | 83.9 C | |||
| 9′ | 26.7 CH3 | 1.12 s | 26.9 CH3 | 1.30 s | 29.7 CH3 | 1.44 s |
| 10′ | 30.2 CH3 | 1.19 s | 30.4 CH3 | 1.28 s | 27.1 CH3 | 1.24 s |
| 1′′ | 140.0 C | 140.5 C | 140.2 C | |||
| 2′′, 6′′ | 128.3 CH | 7.13 d (overlapped, olp) | 128.5 CH | 7.20 d (olp) | 128.4 CH | 7.23 d (olp) |
| 3′′, 5′′ | 128.5 CH | 7.19 dd (7.2, 6.0) | 128.7 CH | 7.28 dd (7.2, 3.6) | 128.0 CH | 7.28 dd (8.2, 2.4) |
| 4′′ | 126.3 CH | 7.12 dd (olp) | 126.5 CH | 7.19 d (olp) | 126.6 CH | 7.21 dd (olp) |
| 7′′ | 30.8 CH2 | 2.87 t (7.2) | 31.0 CH2 | 2.95 t (8.1) | 30.9 CH2 | 2.99 t (8.8) |
| 8′′ | 36.0 CH2 | 2.59 t (7.2) | 36.3 CH2 | 2.66 t (8.1) | 36.2 CH2 | 2.73 t (8.8) |
| 9′′ | 171.9 C | 172.5 C | 172.4 C | |||
| 1′′′ | 140.3 C | |||||
| 2′′′, 6′′′ | 128.3 CH | 7.13 d (olp) | ||||
| 3′′′, 5′′′ | 128.6 CH | 7.19 dd (7.2, 6.0) | ||||
| 4′′′ | 126.3 CH | 7.12 dd (olp) | ||||
| 7′′′ | 30.8 CH2 | 2.87 t (7.2) | ||||
| 8′′′ | 36.2 CH2 | 2.59 t (7.2) | ||||
| 9′′′ | 172.4 C | |||||
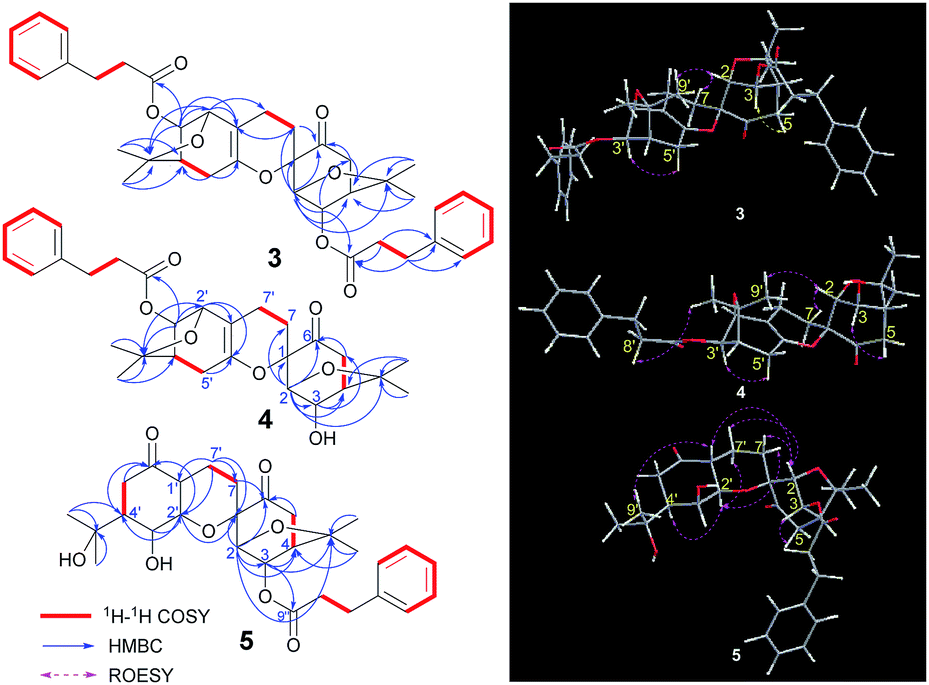 | ||
| Fig. 6 Key 1H–1H COSY, HMBC, and ROESY correlations of 3–5. | ||
The relative configurations were assigned by a ROESY experiment. The ROESY cross-peaks of H-3/H-5 and H-3′/H-5′, allowed the assignment of an α-orientations for H-3 and H-3′, respectively. Furthermore, the presence of a H-2/H-7β [δH 2.03 (1H, m)] cross-peak and the absence of a H-3/H-7 cross-peak suggested a β-orientation of C-7 linked at C-1. Combined with the ROESY cross-peak of H-2/H-9′, the relative configuration of 3 was determined to be 1S*,2R*,3R*,4R*,2′S*,3′R*,4′R*, which is same as 1.
Compound 4, isolated as a colourless gummy oil, has a molecular formula of C29H36O7 based on the HRESIMS m/z 519.2355 [M + Na]+ (calcd for C29H36O7Na 519.2353), 13C NMR and DEPT spectra. From the 1H and 13C NMR data, four methyl groups, one benzene ring, two carbonyls (one ketone and one ester), and seven oxygenated alkyl groups were identified. Comparison of the NMR data of 4 with that of dimeric menthane-type monoterpenoid 3 (Table 2) revealed that 4 possessed a same skeleton as 3, which was also supported by the 2D NMR analysis shown in Fig. 6. The main difference is that only one phenylpropionic substituent is found in 4, while 3 has two. Based on the cross-peak of H-3′/C-9′′ found in the HMBC spectrum, the phenylpropionic unit was attached at C-3′. The relative configuration of 4 was determined to be 1S*,2R*,3R*,4R*,2′S*,3′R*,4′R* (which is the same as 1 and 3) based on its ROESY cross-peaks of H-3/H-5, H-3′/H-5′, and H-2/H-7β [δH 2.14 (1H, ddd, J = 13.8, 11.4, 5.4 Hz)].
Because 3 and 4 shared skeletal features with 1, they possessed similar ECD spectra (Fig. 5B), indicating the absolute configurations of both 3 and 4 are 1S,2R,3R,4R,2′S,3′R,4′R. The structure of 3 and 4 were therefore identified and named dimericilligerates A and B, respectively.
Compound 5, obtained as a colourless gummy oil, has a molecular formula C29H38O8 based on its HRESIMS m/z 559.2552 [M + COOH]− (calcd for C30H39O10 559.2549), 13C NMR and DEPT spectra, which showed eleven degrees of unsaturation. The 1H NMR spectrum showed four methyl groups and five aromatic protons; the 13C NMR and DEPT spectra showed three carbonyls (two ketones and one ester), and seven oxygenated alkyl carbons located downfield. Comparing the NMR data of 5 with that of 3 and 4 implied that 5 consisted of one phenylpropionic substituent and one similar dimeric monoterpenoid. The dimeric monoterpenoid was confirmed to be 2 by comparing their NMR data and analysing their respective 1H–1H COSY and HMBC correlations (Fig. 6). The relative configuration of 5 was assigned as 1S*,2R*,3R*,4R*,1′R*,2′R*,3′S*,4′S* because of its ROESY cross-peaks of H-3/H-5; H-2/H-7β [δH 1.56 (1H, dd, J = 13.8, 7.2 Hz)]; H-1′/H-2 and CH3-9′; H-1′/H-3′ and H-2′α; and H-2′/H-7α [δH 2.03 (1H, dd, J = 13.8, 7.2 Hz)] and H-4′, which is the same as 2.
Subsequently, the theoretical ECD spectra were calculated using TDDFT at the B3LYP/6-31G(d,p) level. The results showed that the theoretical ECD curve of the (1S,2R,3R,4R,1′R,2′R,3′S,4′S) isomer (Fig. 5C) resembled the experimental ECD curve, indicating an absolute configuration of (1S,2R,3R,4R,1′R,2′R,3′S,4′S) for 5. The structure of 5 was thus established as depicted, and 5 was named dimericilligerate C.
Bioactivity
Compounds 1–5 were tested for cytotoxic activity27,28 and an inhibitory effect on NO production in LPS-stimulated RAW 264.7 cells.29,30 As shown in Table 3, compound 2 showed moderate inhibitory effects on NO production. Both 1 and 2 possessed cytotoxic activities against five cell lines. In particular, 1 possessed potent cytotoxic activity against a liver hepatocellular carcinoma cell line (SMMC-7721) with an IC50 value of 3.89 ± 0.23 μM, which was more potent than positive control (cis-dichlorodiammineplatinum). However, the phenylpropionic esters did not show any activity.| Sample | NO production (IC50/μM) | Cytotoxic activity (IC50/μM) | ||||
|---|---|---|---|---|---|---|
| HL-60 | A-549 | SMMC-7721 | MCF-7 | SW480 | ||
| a Positive control for inhibitory effect on NO production in RAW 264.7 at the concentration of 50 μM.b DDP (cis-dichlorodiammineplatinum) and taxol used as the positive control for cytotoxic activity. | ||||||
| 1 | >25 | 18.42 ± 0.35 | 27.40 ± 1.11 | 3.89 ± 0.23 | 17.29 ± 2.98 | 19.41 ± 0.76 |
| 2 | 23.61 ± 2.77 | 19.83 ± 1.54 | 27.47 ± 0.98 | 14.21 ± 0.87 | 16.74 ± 2.98 | 16.63 ± 0.83 |
| 3 | >25 | >40 | >40 | >40 | >40 | >40 |
| 4 | >25 | >40 | >40 | >40 | >40 | >40 |
| 5 | >25 | >40 | >40 | >40 | >40 | >40 |
| MG132a | 0.24 ± 0.02 | — | — | — | — | — |
| DDPb | — | 1.72 ± 0.22 | 6.82 ± 0.30 | 12.19 ± 0.85 | 17.40 ± 0.93 | 16.63 ± 2.90 |
| Taxolb | — | <0.008 | <0.008 | <0.008 | <0.008 | <0.008 |
Verification the biotransformation
To confirm the biotransformation of monoterpenoid dimer phenylpropionic esters (3–5) into free monoterpenoid dimers (1 and 2), the main compound 3 was selected as the substrate for fermentation. After 30 days of solid-state fermentation, 3 was almost completely transformed to 1, indicating that C. rogersoniana-fermentation of I. aromatica was effective at producing cytotoxic free monoterpenoid dimers.Conclusions
In the present study, five monoterpenoid dimer derivatives (1–5) were isolated from fermented and non-fermented C. rogersoniana. Compounds 1 and 2 isolated from fermented I. aromatica showed stronger cytotoxic activity than those isolated from the original material. This paper presents a novel class of monoterpenoid dimers and suggests that C. rogersoniana-fermented I. aromatica is effective at the production of cytotoxic dimeric monoterpenoids from inactive original materials.Experimental
General procedures
Melting points were determined on an XRC-1 melting point apparatus (Optical Instrument Factory of Sichuan University, Chengdu, China) and are uncorrected. Optical rotations were measured with a Jasco P-1020 digital polarimeter (Jasco, Tokyo, Japan). A Shimadzu UV-Vis 2550 spectrometer (Shimadzu, Tokyo, Japan) was used for the acquisition of UV-Vis spectra. Electronic circular dichroism spectra were recorded using a Chirascan circular dichroism spectrometer (Applied Photophysics Ltd., UK). NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer or Bruker Avance III 600 MHz spectrometer (Bruker, Karlsruhe, Germany) using TMS as an internal reference. HRESIMS and ESIMS analyses were carried out with an Agilent G3250AA MSD TOF-MS or Agilent 6540 Q-TOF-MS (Agilent, Santa Clara, CA, USA). Silica gel (200–300 mesh, Qingdao Marine Chemical Group Co., Qingdao, China) was used for column chromatography (CC). Fractions were monitored by TLC and visualized by spraying with a 10% H2SO4–EtOH solution.Plant material
The tubers of I. aromatica were collected from Wenshan, Yunnan, China, in December 2015 and were identified by assistant professor Shu-Da Yang from School of Pharmacy, Kunming Medical University, Kunming, China. A voucher specimen (2015-ia-01) has been deposited in School of Chemical Science and Technology, Yunnan University, Kunming, China.Microorganism and fermentation processing of I. aromatica
C. rogersoniana 828H2 was obtained from the Yunnan Institute of Microbiology, Yunnan Province, China. The nucleotide sequence data have been deposited in GenBank (accession number KT625993).PDA (1 L water, 200 g potato, 20 g dextrose, and 15 g agar) slant culture media were inoculated with the above fungus and incubated in a constant temperature incubator at 28 °C for 5 days. Afterwards, the mature fungus was cultivated in PDB (1 L water, 200 g potato, 20 g dextrose) media for 5 days at 28 °C as seed culture mediums.
The dried and powdered tubers (2.0 kg) were added to 350 mL glass spawn bottles (40 g per bottle) as the SSF culture media. After being treated with 8 mL water and sterilized at 121 °C for 30 min, the seed culture media were added and incubated at 28 °C for 30 days.
Extraction and isolation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give fractions PE1–3. PE2 (5.0 g) was chromatographed over Sephadex LH-20 gel (chloroform–methanol, 1:1) to yield PE2–1, PE2–2, and PE2–3. PE2–2 (220 mg) was further separated by repeated silica gel CC (petroleum ether–acetone) to afford 4 (3.5 mg) and 5 (1.0 mg). Further purification of PE3 (100 mg) was accomplished by repeated silica gel (petroleum ether–acetone) and Sephadex LH-20 (chloroform–methanol, 1:1) CC to yield 3 (50 mg).
:1) to give fractions PE1–3. PE2 (5.0 g) was chromatographed over Sephadex LH-20 gel (chloroform–methanol, 1:1) to yield PE2–1, PE2–2, and PE2–3. PE2–2 (220 mg) was further separated by repeated silica gel CC (petroleum ether–acetone) to afford 4 (3.5 mg) and 5 (1.0 mg). Further purification of PE3 (100 mg) was accomplished by repeated silica gel (petroleum ether–acetone) and Sephadex LH-20 (chloroform–methanol, 1:1) CC to yield 3 (50 mg).
Dimericilligerate E (1). White amorphous powder; mp 109–110 °C; [α]25D −104.2 (c = 0.19, MeOH); UV (CH3OH) λmax (log
ε) 212 (3.88), 262 (sh, 2.86), 310 (1.76); FT-IR: νKBr max (cm−1) 3432 (O–H), 2972, 2933, 1711, 1634, 1369, 1095, 1049; 1H and 13C NMR data, see Table 1; HRESIMS m/z 409.2235 [M + COOH]− (calcd for C22H33O7 409.2232).
Dimericilligerate F (2). White amorphous powder; mp 115–118 °C; [α]25D −79.1 (c = 0.1, MeOH); UV (CH3OH) λmax (log
ε) 198 (3.81), 267 (3.01), 319 (2.68); 1H and 13C NMR data, see Table 1; HR-ESI-MS m/z 381.1917 [M − H]− (calcd for C20H29O7 381.1919).
Dimericilligerate A (3). Colourless gummy oil; [α]20D −29.2 (c = 0.17, CH3CN); UV (CH3CN) λmax (log
ε) 210 (4.07), 280 (3.09), 328 (2.06); 1H and 13C NMR data, see Table 2; HRESIMS m/z 651.2920 [M + Na]+ (calcd for C38H44O8Na 651.2928).
Dimericilligerate B (4). Colourless gummy oil; [α]20D −57.5 (c = 0.12, CH3CN); UV (CH3CN) λmax (log
ε) 210 (4.17), 281 (3.24), 329 (2.45); 1H and 13C NMR data, see Table 2; HRESIMS m/z 519.2355 [M + Na]+ (calcd for C29H36O7Na 519.2353).
Dimericilligerate C (5). Colourless gummy oil; [α]20D −9.0 (c = 0.10, CH3CN); UV (CH3CN) λmax (log
ε) 210 (4.13), 274 (3.39), 325 (2.44); 1H and 13C NMR data, see Table 2; HRESIMS m/z 559.2552 [M + COOH]− (calcd for C30H39O10 559.2549).
Preparation of compounds 1a
Triethylamine (11 μL) was added to a stirred solution of 1 (10 mg, 0.027 mmol) in CH2Cl2 (2 mL) at 0 °C. After 15 min, 20 μL of methanesulfonyl chloride (MsCl) was added and the mixture stirred at room temperature for 18 h, after which, the reaction was quenched by the addition of 5 μL of water. The mixture was concentrated under vacuum and dried over anhydrous Na2SO4. The extract was further separated by silica gel CC (petroleum ether–acetone, 8:1 → 5:1) to yield 1a (6.0 mg, 77%).
White amorphous powder; mp 99–102 °C; [α]20D −92.2 (c = 0.18, CH3CN); UV (CH3CN) λmax (logε) 198 (4.43), 219 (3.99), 274 (3.22) nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 311.1253 [M + Na]+ (calcd for C17H20O4Na 311.1254).
Preparation of MTPA esters of 1a
To a stirred solution of 1a (2.0 mg, 0.007 mmol) in CH2Cl2 (2 mL), either (S)-(−)-MTPA-OH (5 μL, 0.029 mmol) or (R)-(+)-MTPA-OH (5 μL, 0.029 mmol) was added. Then, 10.0 mg of N,N′-diisopropylcarbodiimide and 10.0 mg of 4-dimethylaminopyridine were added. The reaction mixture was stirred at room temperature for 30 min and concentrated under vacuum at 45 °C. The residues were purified by silica gel CC (petroleum ether–acetone) to afford (S)-MTPA ester (2.5 mg, 70.9%) or (R)-MTPA ester (2.0 mg, 56.7%), respectively.LC-MS analysis
The dried and powdered tubers (2.0 g) were extracted with petroleum ether using an ultrasonic cleaner three times (30 min each). Afterwards, the solvent was removed under reduced pressure at 40 °C to afford the crude extract. Then, the crude extract was dissolved in acetone and filtrated through a 0.45 μm filter for LC-MS analysis. An LC-MS system equipped with an Agilent 1100 Series HPLC system, an Agilent G3250AA MSD TOF-MS, and Agilent Zorbax SB-C18 column (250 × 4.6 mm, 5 μm i.d.; Agilent, Santa Clara, CA, USA) were used to identify the isolated compounds. A gradient elution system consisting of solvents A (water) and B (acetonitrile) was used for the analysis, and the gradient program was as follows: 0–5 min, 10% B; 5–20 min, 10–60% B; and 20–30 min, 60–100% B. The flow rate was 0.8 mL min−1, the column temperature was set to 25 °C, and the injection volume was 10 μL. The ion source of the MS was set to ESI- to monitor quasi-molecular ions.Cytotoxic activity
Cytotoxicity was assayed by the trypan blue method27 using the human leukaemia cell line (HL-60) and by the MTT assay28 using the liver hepatocellular carcinoma cell line (SMMC-7721), lung carcinoma cell line (A-549), breast carcinoma cell line (MCF-7), and colon carcinoma cell line (SW480). The cell lines were purchased from America Type Culture Collection (ATCC; Rockville, MD, USA) and cultured in RPMI-1640 medium (Gibco, New York, NY, USA) supplemented with 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 1 mM glutamine and 10% heat-inactivated foetal bovine serum (Gibco). cis-Dichlorodiammineplatinum (DDP) and taxol (Sigma-Aldrich Co. LLC., Shanghai, China) were selected as the positive controls. Each sample was repeated in triplicate.NO production in RAW 264.7 macrophages
NO production in RAW 264.7 Macrophages was measured according to the method reported in the literature.29,30 Murine monocytic RAW 264.7 macrophages (Institute of Biochemistry and Cell Biology, SIBI, CAS, Shanghai, China) were seeded in 96-well cell culture plates (2 × 105 cells per well) and treated with serial dilutions of the compounds at a maximum concentration of 25 μM; this treatment was followed by stimulation with LPS (1 μg mL−1; Sigma-Aldrich) for 18 h. NO production in the supernatant was assessed using Griess reagents (Sigma-Aldrich). The absorbance at 570 nm was measured using a 2104 Envision multilabel plate reader (PerkinElmer Life Sciences, Inc., Boston, MA, USA). MG132 (Sigma-Aldrich) was used as a positive control. The viability of RAW 264.7 cells were simultaneously evaluated using the MTT assay to exclude the interference of the cytotoxicity of the test compounds.ECD calculation
The calculation of theoretical ECD spectra of compounds 3 and 4 were performed using the Gaussian Program by Yunnan Electronic Computing Center. The possible geometries were previously optimized using DFT method at the B3LYP/6-31G(d,p) level,21 and the excitation energies and rotational strengths were calculated using TDDFT at the B3LYP/6-31G(d,p) level in methanol with PCM model.22 Afterwards, the ECD spectra were generated using SpecDis.23,31Acknowledgements
This work was financially supported by two grants from the Natural Science Foundation of China (No. 81660719 and 81460648), a project of Yunling Scholars of Yunnan province, and a grant from New Academic Researcher Award for Doctoral Candidates of Yunnan Province.Notes and references
- Flora of China Editorial Committee, Flora Reipublicae Popularis Sinicae, Science Press, Beijing, 1982 Search PubMed.
- K.-S. Chen, Y.-C. Wu, C.-M. Teng, F.-N. Ko and T.-S. Wu, J. Nat. Prod., 1997, 60, 645–647 CrossRef CAS PubMed.
- S. A. Ross, R. D. Minard, M. Shamma, M. O. Fagbule, G. Olatunji and Z. Gbile, J. Nat. Prod., 1985, 48, 835–836 CrossRef CAS.
- J.-J. Chen, H.-C. Hung, P.-J. Sung, I.-S. Chen and W.-L. Kuo, Phytochemistry, 2011, 72, 523–532 CrossRef CAS PubMed.
- J.-J. Chen, T.-H. Lee, W.-L. Kuo, P.-J. Sung, I.-S. Chen, C.-W. Shu, M.-J. Cheng, T.-C. Wang, Y.-P. Lim and R. Xu, Chem. Nat. Compd., 2015, 51, 739–742 CrossRef CAS.
- C.-H. Huang, Y.-Y. Chan, P.-C. Kuo, Y.-F. Chen, R.-J. Chang, I.-S. Chen, S.-J. Wu and T.-S. Wu, Int. J. Mol. Sci., 2014, 15, 13424–13436 CrossRef CAS PubMed.
- S. Z. Huang, Guihaia, 1985, 5, 17–20 Search PubMed.
- S. Mo, Z. Li, Y. Ou, S. Wei, X. Si and D. Fan, Lishizhen Med. Mater. Med. Res., 2006, 17, 2512–2513 CAS.
- L. Xie, P. Li, Z. Gong and Y. Ou, Contemp. Biomed., 2011, 17, 31–32 Search PubMed.
- J. W. Dong, L. Cai, X. J. Li, J. P. Wang, R. F. Mei and Z. T. Ding, Arch. Pharmacal Res., 2016 DOI:10.1007/s12272-016-0860-3.
- Y. Kim, Y. You, H. G. Yoon, Y. H. Lee, K. Kim, J. Lee, M. S. Kim, J. C. Kim and W. Jun, Food Chem., 2014, 151, 148–153 CrossRef CAS PubMed.
- M. F. Hsu and B. H. Chiang, Process Biochem., 2009, 44, 83–90 CrossRef CAS.
- J. W. Dong, L. Cai, J. Xiong, X. H. Chen, W. Y. Wang, N. Shen, B. L. Liu and Z. T. Ding, Process Biochem., 2015, 50, 8–13 CrossRef CAS.
- Y. Li, L. Cai, J. W. Dong, Y. Xing, W. H. Duan, H. Zhou and Z. T. Ding, J. Agric. Food Chem., 2015, 63, 6596–6602 CrossRef CAS PubMed.
- K. Adachi, K. Kanoh, P. Wisespongp, M. Nishijima and Y. Shizuri, J. Antibiot., 2005, 58, 145–150 CrossRef CAS PubMed.
- T.-P. Yin, Y. Xing, L. Cai, J. Yu, P. Luo and Z.-T. Ding, J. Asian Nat. Prod. Res., 2017, 1–6, DOI:10.1080/10286020.2017.1314271.
- L. Cai, J.-W. Dong, L.-X. Zhao, H. Zhou, Y. Xing, Y. Li, Z.-J. Li, W.-H. Duan, X.-J. Li and Z.-T. Ding, Process Biochem., 2016, 51, 933–940 CrossRef CAS.
- X.-J. Li, J.-W. Dong, L. Cai, R.-F. Mei and Z.-T. Ding, J. Biosci. Bioeng. DOI:10.1016/j.jbiosc.2017.05.012.
- P. Ventura, M. Schiavi and S. Serafini, Xenobiotica, 1983, 13, 139–146 CrossRef CAS PubMed.
- K. Hashimoto, T. Katsuhara, K. Niitsu, Y. Ikeya, K. Hayashi, M. Maruno and T. Fujita, Phytochemistry, 1994, 35, 969–973 CrossRef CAS.
- J. W. Dong, L. Cai, X. J. Li, L. Peng, Y. Xing, R. F. Mei, J. P. Wang and Z. T. Ding, Fitoterapia, 2016, 109, 212–216 CrossRef CAS PubMed.
- Y. Liao, X. Liu, J. Yang, Y. Z. Lao, X. W. Yang, X. N. Li, J. J. Zhang, Z. J. Ding, H. X. Xu and G. Xu, Org. Lett., 2015, 17, 1172–1175 CAS.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- S. Lu, S. Nishimura, G. Hirai, M. Ito, T. Kawahara, M. Izumikawa, M. Sodeoka, K. Shin-ya, T. Tsuchida and H. Kakeya, Chem. Commun., 2015, 51, 8074–8077 RSC.
- T. Ohishi, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 8114–8117 CrossRef CAS PubMed.
- P. Devi, S. Wahidullah, C. Rodrigues and L. D. Souza, Mar. Drugs, 2010, 8, 1203–1212 CrossRef CAS PubMed.
- F. Wang, H. Hua, Y. Pei, D. Chen and Y. Jing, J. Nat. Prod., 2006, 69, 807–810 CrossRef CAS PubMed.
- K. B. Wang, Y. T. Di, Y. Bao, C. M. Yuan, G. Chen, D. H. Li, J. Bai, H. P. He, X. J. Hao, Y. H. Pei, Y. K. Jing, Z. L. Li and H. M. Hua, Org. Lett., 2014, 16, 4028–4031 CrossRef CAS PubMed.
- J. Yang, W. G. Wang, H. Y. Wu, X. Du, X. N. Li, Y. Li, J. X. Pu and H. D. Sun, J. Nat. Prod., 2016, 79, 132–140 CrossRef CAS PubMed.
- J. H. Yang, X. Du, F. He, H. B. Zhang, X. N. Li, J. Su, Y. Li, J. X. Pu and H. D. Sun, J. Nat. Prod., 2013, 76, 256–264 CrossRef CAS PubMed.
- P. M. Allard, M. T. Martin, M. E. T. H. Dau, P. Leyssen, F. Gueritte and M. Litaudon, Org. Lett., 2012, 14, 342–345 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D/2D NMR, HRESIMS, and LC-MS spectra. See DOI: 10.1039/c7ra06078e |
| This journal is © The Royal Society of Chemistry 2017 |