
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible loading of thiol-modified curcumin in an engineered protein capsid†
Q. Guo‡
a,
G. C. Thomas‡b and
K. J. Woycechowsky§
 *ab
*ab
aSchool of Pharmaceutical Science and Technology, Tianjin University, 92 Weijin Road, Nankai District, Tianjin, 300072, China. E-mail: kenneth@tju.edu.cn
bDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, UT 84112, USA
First published on 11th July 2017
Abstract
The dodecahedral capsid formed by Aquifex aeolicus lumazine synthase (AaLS) is a promising protein scaffold for bionanotechnological applications. A cysteine was installed at the inner surface of the AaLS capsid to give a variant (AaLS-IC) that can covalently capture small-molecule thiols in its hollow interior. Cargo loading utilizes a two-stage thiol–disulfide exchange process, involving the initial formation of an activated disulfide adduct between AaLS-IC and 2-nitro-5-thiobenzoate (NTB) followed by displacement of the NTB by an incoming guest molecule. Using a thiol-containing curcumin derivative (cur-SH) as a model guest, we show that about 41 guest molecules can be loaded per capsid. The sequestration of cur-SH inside the capsid increases its solubility in aqueous buffer by more than 30-fold. Further, the guest can be released upon treatment with tris(2-carboxyethyl)phosphine, which reduces the disulfide bond tethering cur-SH to the capsid. Thus, the AaLS-IC capsid can act as a container for small-molecule thiols, and guest release can be triggered by reducing agents.
1. Introduction
Protein capsids can act as nanoscale molecular containers. Such self-assembled structures form a closed shell which encases a hollow interior that can host various guest molecules.1 The capsid wall physically separates the guest molecules inside from the bulk environment.2 Nature provides a wide array of capsid structures, with diameters ranging from ∼10 nm to >100 nm and functions, including storage and transport of nucleic acids (by virus capsids),3 metal storage (by ferritin),4 and biocatalysis (by bacterial microcompartments,5 enapsulin,6 and some lumazine synthases7). For nanotechnology, protein capsids generally possess several additional desirable traits, such as well-ordered and uniform structures, biocompatibility, and ready modification or functionalization.8,9 Consequently, these scaffolds have great potential for applications such as drug delivery, materials synthesis, and the construction of nanoreactors.10–12 Nevertheless, the development of general methods for the controlled loading and release of cargo molecules into and out of protein capsids remains an important challenge.Several virus capsids have been repurposed to encapsulate non-native guests.13–16 For example, the capsids from bacteriophage MS2,17 bacteriophage Qβ,18,19 bacteriophage P22,20 polyoma virus,21 cucumber mosaic virus,22 and cowpea chlorotic mottle virus (CCMV)23–25 have been loaded with a variety of cargoes, including RNA, proteins, and small molecules. For macromolecular cargoes, like RNA and protein, capsid loading is usually coupled with capsid assembly, since these guests are typically too big to diffuse into the capsid, and encapsulation is driven by non-covalent interactions between the capsid and guest. Small molecules can often be loaded into intact capsids, since virus capsids typically possess pores that allow these cargoes access to the capsid interior. However, small-molecule encapsulation typically requires covalent conjugation of the guest molecule to the inner capsid wall to prevent the guest from simply diffusing back out of the capsid again.
The enzyme lumazine synthase from Aquifex aeolicus (AaLS) has shown much promise as a scaffold for bionanotechnology. The AaLS capsid (Fig. 1A) is built from 60 identical subunits,26 which assemble as a dodecamer-of-pentamers.27 A central tunnel, which is ∼0.9 nm wide at its narrowest point, runs through each of the pentameric building blocks, and potentially provides a means for small molecules to diffuse across the capsid shell.28 Compared with viral nanoparticles, the AaLS capsid is relatively small, with an outer diameter of 16 nm and an inner diameter of 9 nm. Previously, AaLS has been engineered to encapsulate either RNA29 or proteins bearing a high number of positively charged residues,30–34 based on charge complementarity strategies. Despite these successes with biomacromolecular encapsulation, methods for the encapsulation of small molecules by AaLS are lacking.
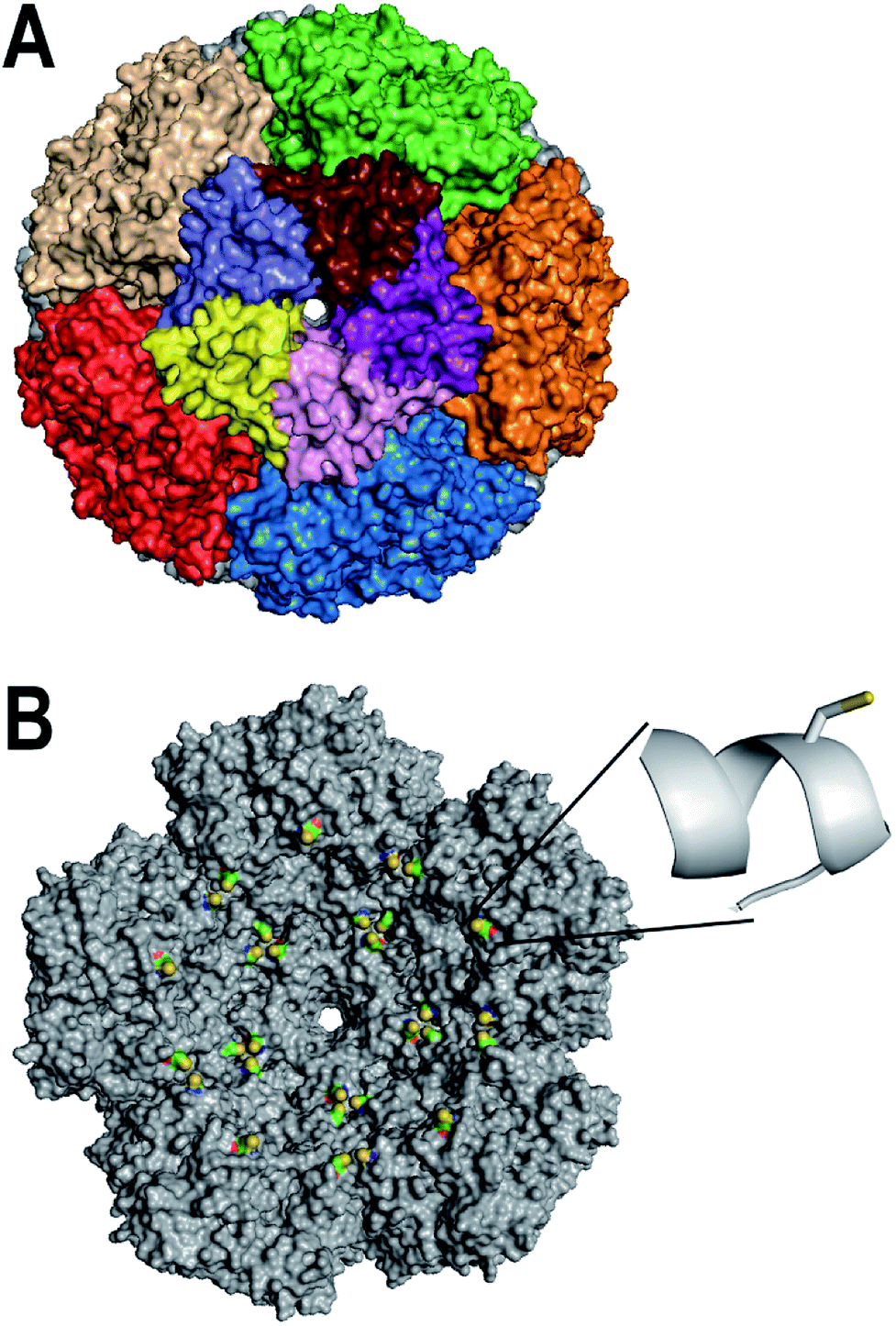 | ||
| Fig. 1 Design of AaLS-IC. (A) The surface of the AaLS capsid (pdb id: 1HQK) is shown, viewed down one of the five-fold symmetry axes. Individual subunits of the central pentameric building block are individually colored; the other pentamers are each colored uniformly. (B) The interior surface of the rear half of the AaLS capsid is shown. The E122C mutation was modeled using the program Pymol. The engineered cysteines are colored by atom type (sulfur in yellow, carbon in green, oxygen in red, and nitrogen in blue); the rest of the protein is colored gray. The inset shows a close-up of the region around amino acid 122 in which the E122C has been modeled. The side-chain of the engineered cysteine is shown as a stick representation; the protein backbone around it is shown in a ribbon representation. | ||
Here, we describe a novel AaLS-based system for the controlled encapsulation of small-molecule thiols by the intact capsid. An engineered cysteine was installed at the inner surface of AaLS (Fig. 1B) as a means to capture potential cargo molecules covalently inside the capsid. Using a two-stage thiol–disulfide exchange process, we successfully conjugated an analog of the natural product curcumin, as a model guest, to the engineered cysteine via a disulfide linkage. The encapsulated curcumin analog was subsequently released from the capsid by adding a reducing agent. Interestingly, the encapsulated curcumin analog was much more soluble in aqueous buffer than its free counterpart, which could be advantageous for delivery applications.
2. Materials and methods
2.1 Materials
Curcumin (AR grade) was purchased from Biotopped (Beijing, China) and was purified before use (see below). All other chemicals were of reagent grade or better and used without further purification, unless otherwise noted. Reverse-phase high-performance liquid chromatography (HPLC) separations were performed on an Agilent 1200 HPLC (Agilent, Santa Clara, CA, USA). Determinations of molecular mass were carried out using an Agilent 6310 ion trap mass spectrometer or an Agilent 1260 infinity 6420 triple quad LC/MS. NMR spectroscopy was performed with an 400 MHz Bruker Avance III NMR spectrometer (Bruker, Karlsruhe, Germany). UV-Vis absorbance measurements were made with either a TECAN Infinite M200 Pro plate reader (TECAN, Männedorf, Switzerland), Eppendorf BioPhotometer (Eppendorf, Hamburg, Germany), or a Hitachi U-3900 spectrophotometer (Hitachi High-Tech Science Corporation, Tokyo, Japan). Fluorescence spectroscopy experiments were performed with either the TECAN Infinite M200 plate reader or an FLS 980 fluorimeter (Edinburgh Instruments, Livingston, UK). Protein purifications were carried out using either an AKTA Pure or an AKTA Prime Plus fast protein liquid chromatography instrument (GE Healthcare Life Sciences, Pittsburgh, PA, USA).2.2 Site-directed mutagenesis
To generate a unique cysteine in the AaLS sequence whose side chain should be accessible at the inner capsid surface and be oriented towards the capsid lumen, two successive rounds of site-directed mutagenesis were carried out. In the first round, the naturally occurring cysteine at position 37 was changed to alanine. For this mutagenesis reaction, the plasmid pMG-AaLS-noHis27 was used as the template and the primers GT_C37AF and GT_C37AR (5′-GTGGAGGGTGCAATTGATGCTATAGTCCGTCATGGCGGC-3′ and 5′-GCCGCCATGACGGACTATAGCATCAATTGCACCCTCCAC-3′, respectively, mutations indicated in bold) were used to introduce the C37A mutation, yielding plasmid pMG-AaLS-C37A. In a second round of site-directed mutagenesis, glutamate at position 122 was replaced by cysteine. In this case the plasmid pMG-AaLS-C37A was used as the template and the primers GT_E122CF and GT_E122CR (5′-GTTATTACAGCTGACACCTTGTGCCAGGCTATCGAGCGCGCCGGC-3′ and 5′-GCCGGCGCGCTCGATAGCCTGGCACAAGGTGTCAGCTGTAATAAC-3′, respectively) were used to make the E122C mutation. The resulting plasmid after both rounds of mutagenesis, pMG-AaLS-IC, encodes the double variant C37A/E122C-AaLS (AaLS-IC). For both mutations, the mutagenesis PCR was carried out similarly to a previously reported procedure27 and involved 18 cycles of denaturation (95 °C for 30 s), annealing (58 °C for 30 s), and primer extension (72 °C for 6 min). The coding portions of the resulting plasmids (pMG-AaLS-C37A and pMG-AaLS-IC) were confirmed by DNA sequencing (University of Utah DNA sequencing core facility).2.3 Production and purification of AaLS-IC
AaLS-IC capsids were produced in Escherichia coli BL21 (DE3) cells from plasmid pMG-AaLS-IC and purified using a combination of ammonium sulfate treatment, anion exchange chromatography, and size-exclusion chromatography, according to a previously published procedure.27 The purity of each protein was confirmed by SDS-PAGE. The yield was determined by Bradford assay. The typical yield of pure AaLS-IC was ∼20 mg L−1 of cell culture.2.4 Synthesis of cur-SH
The curcumin used for this derivatization was purified by silica gel chromatography using CH2Cl2 as the mobile phase. Purified curcumin gave a single spot with an Rf value of 0.5 by thin-layer chromatography using 1,2-dichloroethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (20:1, v/v) as the mobile phase. Purified curcumin was derivatized using a previously reported procedure35 to generate cur–SS–COOH (Fig. 2). The crude product was re-dissolved in dichloromethane (4 mL) and purified by silica gel column chromatography using 1,2-dichloroethane:methanol:acetic acid (98:0.98:1, v/v) as the mobile phase. Individual fractions were analyzed by thin-layer chromatography using 1,2-dichloroethane:methanol:acetic acid (90:3:0.93, v/v) as the developing solvent. The Rf value of the desired singly modified curcumin product is about 0.27. The mass of the purified product was 0.16 g (28% yield). The structure of this product was confirmed by 1H and 13C NMR (400 MHz, CDCl3) spectroscopy. 1H NMR, δ [ppm]: 7.63–7.57 (m, 2H), 7.14–6.92 (m, 6H), 6.57–6.47 (m, 2H), 5.83 (s, 1H), 3.94 (s, 3H), 3.88 (s, 3H), 3.07–3.05 (m, 4H), 3.00–2.96 (t, 2H), 2.85–2.81 (t, 2H). 13C NMR, δ [ppm]: 184.64, 181.77, 169.73, 151.25, 148.07, 146.87, 141.26, 140.99, 139.35, 134.19, 127.52, 124.30, 123.16, 123.07, 121.71, 120.95, 114.92, 111.50, 109.78, 101.59, 55.96, 55.93, 43.51, 33.91, 33.05, 29.71, 22.73. IR (KBr) cm−1: 3600–3900 (broad, m), 3000–3300 (broad, m), 1754 (w), 1629 (w), 1581 (w), 1511 (s), 1384 (w), 1123 (m). MS (ESI) calculated for [C27H27O9S2]− 559.12, observed 559.1.
:methanol (20:1, v/v) as the mobile phase. Purified curcumin was derivatized using a previously reported procedure35 to generate cur–SS–COOH (Fig. 2). The crude product was re-dissolved in dichloromethane (4 mL) and purified by silica gel column chromatography using 1,2-dichloroethane:methanol:acetic acid (98:0.98:1, v/v) as the mobile phase. Individual fractions were analyzed by thin-layer chromatography using 1,2-dichloroethane:methanol:acetic acid (90:3:0.93, v/v) as the developing solvent. The Rf value of the desired singly modified curcumin product is about 0.27. The mass of the purified product was 0.16 g (28% yield). The structure of this product was confirmed by 1H and 13C NMR (400 MHz, CDCl3) spectroscopy. 1H NMR, δ [ppm]: 7.63–7.57 (m, 2H), 7.14–6.92 (m, 6H), 6.57–6.47 (m, 2H), 5.83 (s, 1H), 3.94 (s, 3H), 3.88 (s, 3H), 3.07–3.05 (m, 4H), 3.00–2.96 (t, 2H), 2.85–2.81 (t, 2H). 13C NMR, δ [ppm]: 184.64, 181.77, 169.73, 151.25, 148.07, 146.87, 141.26, 140.99, 139.35, 134.19, 127.52, 124.30, 123.16, 123.07, 121.71, 120.95, 114.92, 111.50, 109.78, 101.59, 55.96, 55.93, 43.51, 33.91, 33.05, 29.71, 22.73. IR (KBr) cm−1: 3600–3900 (broad, m), 3000–3300 (broad, m), 1754 (w), 1629 (w), 1581 (w), 1511 (s), 1384 (w), 1123 (m). MS (ESI) calculated for [C27H27O9S2]− 559.12, observed 559.1.
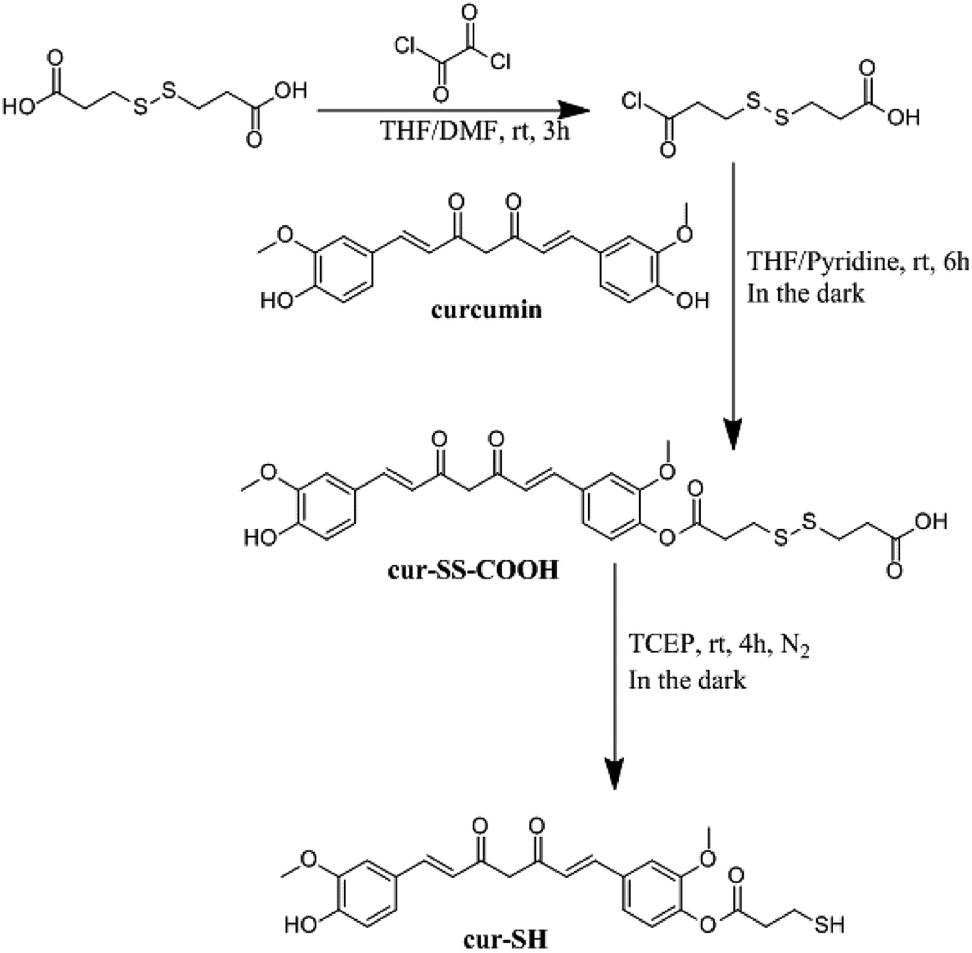 | ||
| Fig. 2 Synthetic scheme for the derivatization of curcumin to make cur-SH. | ||
Cur–SS–COOH contains a disulfide bond which was reduced to generate the thiol–curcumin derivate used for loading into the AaLS-IC capsid. This reduction was carried out by first dissolving the disulfide-containing curcumin derivative (0.1 mmol, 57.8 mg) in THF (20 mL) and then adding 183 mM aqueous tris(2-carboxyethyl)phosphine (TCEP) (3 mL). The reaction mixture was incubated at room temperature under N2 and in the dark for 4 h. The solvent was then removed by rotary evaporation and the resulting orange solid was purified using silica gel column chromatography using a solvent system consisting of 1,2-dichloroethane:methanol:acetic acid (98:0.98:1, v/v). Individual fractions were analyzed by thin-layer chromatography using 1,2-dichloroethane:methanol:acetic acid (90:3:0.93, v/v) as the developing solvent. The Rf value of cur-SH is about 0.6. The collected product has a mass of 7 mg (15% yield). Cur-SH (7 mg) was then dissolved in acetonitrile (1 mL), passed through a 0.22 μM nylon syringe filter, and further purified by reverse-phase HPLC using an Agilent 1260 infinity HPLC system equipped with a SunFire Prep C8 column (19 × 250 mm, 5 μM particle size, OBD). The HPLC purification was carried out at 40 °C under isocratic conditions using a solvent system consisting of acetonitrile:water:formic acid (50:50:0.1) and at a flow rate of 10 mL min−1. The detection wavelength was 210 nm. Cur-SH eluted as the main peak with an elution time of 7.23 min. The material corresponding to this peak was collected. The final isolated yield of cur-SH was 5 mg. The resulting orange-yellow solid was confirmed to be cur-SH by 1H and 13C NMR spectroscopy (400 MHz, CDCl3). 1H NMR, δ [ppm]: 7.54–7.51 (m, 2H), 7.06–6.87 (m, 6H), 6.49–6.41 (m, 2H), 5.76 (s, 2H), 3.87 (s, 3H), 3.80 (s, 3H), 2.88 (t, 1H), 2.82 (m, 1H), 1.74 (t, 1H). 13C NMR, δ [ppm]: 184.59, 181.76, 169.57, 151.26, 148.04, 146.84, 141.22, 140.97, 139.34, 134.24, 127.55, 124.35, 123.22, 123.07, 121.76, 120.98, 114.89, 111.45, 109.70, 101.59, 55.98, 55.92, 38.37, 19.85. IR (KBr) cm−1: 3002 (s), 2882 (s), 2811 (s), 1578 (m), 1543 (m), 1510 (m), 1476 (m), 1420 (w), 1386 (w). MS (ESI) calculated for [C24H25O7S]+ [M + H]+ 457.1, observed 457.5; calculated for [C24H23O7S]− [M − H]− 455.1, observed 455.0.
2.5 Conjugation of cur-SH to AaLS-IC
Loading of cur-SH into AaLS-IC capsids was carried out via a two-stage thiol–disulfide exchange process. AaLS-IC (3 mL of a 1 mg mL−1 solution) was slowly added dropwise with stirring into a solution (353 μL) containing 5 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB). Both reactants (AaLS-IC and DTNB) were dissolved in SEC buffer (50 mM sodium phosphate, 200 mM NaCl, pH 8.0). The final concentration of AaLS-IC was 53 μM; the final concentration of DTNB was 530 μM. The reaction was incubated at room temperature for 20 min. The solution immediately turned bright yellow, indicating disulfide bond formation between the cysteine of AaLS-IC and one half of DTNB with concomitant release of NTB (the other half of DTNB). The concentration of NTB released can be calculated by measuring the absorbance at 412 nm (ε412 nm = 14150 M−1 cm−1).36 The product mixture was dialyzed into fresh SEC buffer (1 L) overnight at 4 °C in the dark to remove the free NTB and unreacted DTNB. Cur-SH (660 μL of a 0.17 mg mL−1 solution, dissolved in ethanol and kept in the dark) was slowly added to the AaLS-IC–NTB conjugate (6 mL of a 0.5 mg mL−1 solution, dissolved in SEC buffer), which resulted in a two-fold molar excess of cur-SH over AaLS-IC–NTB. The final concentration of ethanol in the reaction was 10% (v/v). The reaction was incubated at room temperature for 20 min in the dark. Since both curcumin and NTB have strong absorbance at 412 nm, the amount of NTB released was calculated by measuring the ΔA412 nm, since the ε412 nm of cur-SH should not change significantly between the thiol and disulfide forms. Before measuring the absorbance, an aliquot of the product mixture was diluted five-fold with SEC buffer, so that the final concentration of ethanol is 2% (v/v). The ΔA412 nm was calculated by subtracting the A412 nm of 3.4 μg mL−1 unreacted cur-SH (corresponding to a 50-fold dilution of the original 0.17 mg mL−1 cur-SH stock solution into SEC buffer) from the A412 nm of the product mixture. For all subsequent experiments, the remaining crude product mixture was dialyzed into SEC buffer (2 L) overnight at 4 °C in the dark.
2.6 UV-Vis spectroscopy
Since curcumin has a distinct absorbance spectrum from the AaLS-IC protein, UV-Vis spectroscopic analysis was carried out on the AaLS-IC–cur conjugate. The absorbance of AaLS-IC–cur (8 μM) in SEC buffer with 2% ethanol (200 μL final volume) was measured from 600 nm to 300 nm in 1 nm increments using a TECAN plate reader and a flat-bottomed polystyrene 96-well plate (Jet Biofil, Guangzhou, China). SEC buffer with 2% ethanol was used as the blank. UV-Vis absorbance spectra of AaLS-IC–NTB conjugate (8 μM) and free cur-SH (5.8 μM) were obtained in the same way, as controls.2.7 Fluorescence spectroscopy
Since curcumin is fluorescent, fluorescence spectroscopy was used to further characterize the AaLS-IC–cur conjugate. The fluorescence spectrum of AaLS-IC–cur (8 μM) in SEC buffer with 2% ethanol was obtained by measuring the emission from 422 nm to 600 nm in 1 nm increments, after excitation at 412 nm. Fluorescence spectra of AaLS-IC–NTB (8 μM) and free cur-SH (5.8 μM) were also measured in the same way, as controls.2.8 Size-exclusion chromatography analysis of AaLS-IC–cur
To verify the attachment of cur-SH (a small molecule) to AaLS-IC (a large protein), the AaLS-IC–cur conjugate was subjected to analytical SEC. The A280 nm and curcumin-based fluorescence were then measured for each individual fraction. Prior to SEC analysis, the conjugate was concentrated by ultrafiltration using a VIVASPIN 20 (30000 MWCO) spin concentrator (Sartorius) to a final concentration of 2.5 mg mL−1 and then passed through a 0.22 μm syringe filter. The AaLS-IC–cur was then loaded onto an AKTA Pure system equipped with a Hiprep 16/60 sephacryl S-400HR column. The running buffer, flow rate, and temperature were the same as described above for the purification of AaLS-IC. Immediately after the sample was injected onto the column, fractions (4 mL each) were collected over a total eluent volume of 120 mL. For each fraction, the A280 nm and fluorescence emission at 525 nm (λexcitation = 412 nm) were recorded.
2.9 HPLC of AaLS-IC–cur
Analytical reverse-phase HPLC was used to check for non-covalently bound cur-SH in the AaLS-IC–cur sample. To prepare the AaLS-IC–cur for HPLC analysis, the conjugate was dialyzed into PBS buffer containing 10 mM sodium phosphate, 2 mM potassium phosphate, 150 mM NaCl, and 3 mM KCl at pH 7.4 (1 L) overnight at 4 °C in the dark, concentrated by ultrafiltration to a final protein concentration of ∼2 mg mL−1, diluted with pure water to a final protein concentration of 0.2 mg mL−1, and then filtered with a 0.22 μM nylon membrane. AaLS-IC–cur (20 μL) was then injected onto a HPLC system equipped with a Thermo Biobasic C4 column (150 mm × 4.6 mm, 5 μm particle size, 30 nm pore size) and a diode array detector. The HPLC running conditions were 25 °C, 1 mL min−1 flowrate; the detection wavelength was 410 nm. AaLS-IC–cur was eluted from the column using a gradient from 5:95:0.05 (acetonitrile:water:formic acid) to 95:5:0.05 (acetonitrile:water:formic acid) over 20 min.
2.10 Reductive release of cur-SH from AaLS-IC–cur
The ability of the reducing agent TCEP to release cur-SH from the conjugate was examined using reverse-phase HPLC. AaLS-IC–cur was prepared for the reaction by dialysis into PBS and ultrafiltration, as described above. TCEP (1.6 μL of a 125 mM stock solution in PBS) was added to five aliquots (38.4 μL each) of AaLS-IC–cur (2.3 mg mL−1 protein, 90.5 μM curcumin). The final concentration of TCEP in these aliquots was 5 mM. The aliquots were incubated at 4 °C in the dark. At certain time points following the addition of TCEP (0.5 h, 1 h, 2 h, 4 h, and 8 h), 0.05% formic acid in acetonitrile (160 μL) was added to a single aliquot to dissolve precipitated free cur-SH. The sample was then filtered through a 0.22 μm nylon membrane and then immediately injected (20 μL injection volume) onto an HPLC system equipped with a Thermo Biobasic C4 column and a diode array detector. Free cur-SH was separated from the protein and other components of the reaction mixture using a gradient from 5% acetonitrile/95% water/0.05% formic acid to 95% acetonitrile/5% water/0.05% formic acid over 20 min using a flow rate of 1 mL min−1 at 25 °C. Cur-SH was detected at 410 nm. PBS (1.6 μL) was added to seven additional aliquots of AaLS-IC–cur, which were treated and analyzed in the same fashion as the other aliquots. Five of these additional aliquots served as negative controls for cur-SH release for each of the above time points. The remaining two TCEP-free aliquots were analyzed immediately and used to represent the 0 h time point. Time courses of the reaction between TCEP and AaLS-IC–cur were performed in triplicate. Reductive release of cur-SH from AaLS-IC was also carried out at a higher concentration of TCEP (10 mM), using essentially the same procedure, but with a slightly different concentration of AaLS-IC–cur conjugate (2 mg mL−1 protein, 76.5 μM curcumin).The concentration of free cur-SH in each sample was determined by integrating the appropriate peak in the HPLC chromatogram (15.2 min) and comparing the peak area to a calibration curve. To obtain the calibration curve, different concentrations of free cur-SH (0.39 μM to 50 μM) were prepared by two-fold serial dilutions of a 50 μM cur-SH stock solution in acetonitrile. Following filtration through a 0.22 μm nylon membrane, each cur-SH standard was subjected to the same HPLC conditions as described above for the release reaction. The volume of each injection was 20 μL. The areas of the A410 nm peaks at 15.2 min were then plotted against the concentration of cur-SH and fit to a linear equation using the program OriginPro 2015 (OriginLab, Northampton, MA, USA). The data were linear over the entire concentration range (r2 = 0.9958). The free cur-SH concentration at each time point was corrected for the non-covalently bound cur-SH present in the AaLS-IC–cur conjugate by subtracting the free cur-SH concentration of the corresponding sample lacking TCEP.
2.11 Stability of cur-SH
Curcumin is known to degrade slowly in aqueous buffers and in the presence of thiols.37 To check for curcumin degradation under the experimental conditions, the loss of absorbance by cur-SH was measured over time. TCEP (160 μL of a 250 mM stock solution in water) was added to a solution containing 19 μM cur-SH dissolved in 90% SEC buffer/10% ethanol (3840 μL). The final concentration of TCEP was 10 mM and the final volume was 4 mL. After mixing, the A412 nm was measured for 2 h. As a control, another sample was prepared and analyzed in the same way, except that SEC buffer (160 μL) was added instead of TCEP.3. Results and discussion
3.1 Design & production of AaLS-IC
Disulfide bonds are useful linkages for conjugating small molecules to proteins. To enable disulfide conjugation at the interior surface of the AaLS capsid, two point mutations (C37A/E122C) were made in order to remove a naturally-occurring cysteine and to introduce a new one in the desired location. Visual inspection of the AaLS structure (Fig. 1B) suggested that the side-chain of the engineered cysteine at position 122, which is located in the middle of an α-helix, should be oriented toward the capsid lumen and thus be accessible for reaction with molecules that diffuse into the interior space. The resulting variant (AaLS-IC) was produced in Escherichia coli BL21 (DE3) cells and purified by ammonium sulfate treatment, anion exchange chromatography, and size-exclusion chromatography (SEC), yielding about 20 mg of pure protein per L of cell culture. The mutations do not noticeably affect the capsid assembly, as assessed by analytical SEC (Fig. S1†) and transmission electron microscopy (Fig. S2†).3.2 Synthesis & loading of AaLS-IC
Small molecules containing a thiol group can potentially be loaded into the AaLS-IC capsid by disulfide bond formation. Towards this end, a derivative of curcumin containing a thiol group (cur-SH) (Fig. 2) was synthesized by a three-step process. Cur-SH represents an appealing model guest because curcumin is fluorescent38 and there has been great recent interest in developing vehicles for the delivery of curcumin into cells that can overcome its low bioavailability.39–41Cur-SH can be loaded into the AaLS-IC capsid using a two-stage thiol–disulfide exchange reaction involving reduction of DTNB via the formation of an intermediate containing a mixed disulfide bond between the AaLS-IC protein and 2-nitro-5-thiobenzoic acid (NTB) (Fig. 3). Upon addition of DTNB, a bright yellow color formed instantaneously, which was accompanied by the appearance of a large new peak in the UV-Vis spectrum at 412 nm (Fig. S3†). This color change is characteristic of the specific reaction between cysteine thiols and DTNB.42 Upon removal of the unreacted DTNB and the free NTB product, the spectrum of the AaLS-IC–NTB product revealed a new shoulder at 330 nm, which can be attributed to the formation of a mixed disulfide between NTB and AaLS-IC. The yield of AaLC-IC–NTB conjugate in the first stage was 71%, as assessed by UV-Vis spectroscopy (Table S1†).
 | ||
| Fig. 3 Scheme for loading and unloading of AaLS-IC with cur-SH. (A) Cur-SH can be loaded into the intact AaLS-IC capsid by a two-stage thiol–disulfide exchange reaction at the engineered cysteine on the inner capsid surface, yielding a disulfide bonded conjugate (AaLS-IC–cur) between cur-SH and AaLS-IC. (B) The disulfide bond between cur-SH and AaLS-IC can be broken by the reducing reagent TCEP, allowing free cur-SH to be released from the capsid. | ||
The disulfide bond in this intermediate is activated for reaction with incoming thiols, as NTB is a good leaving group. Indeed, during the second stage, the addition of cur-SH to AaLS-IC–NTB led to rapid displacement of NTB, forming the desired AaLS-IC–cur conjugate with a 96% yield (Table S2†). The overall yield for these two steps indicates that about 41 cur-SH molecules were conjugated per capsid.
Following the removal of free NTB and unreacted cur-SH by dialysis, the AaLS-IC–cur conjugate was characterized by UV-Vis spectroscopy and fluorescence spectroscopy. Natural curcumin and the cur-SH derivative have similar UV-Vis absorbance and fluorescence emission spectra, although λmax is about 7–10 nm lower for cur-SH (Fig. S4†). The UV-Vis spectrum of AaLS-IC–cur showed a large absorbance peak at 410 nm (Fig. 4), similar to that of curcumin43,44 and cur-SH, but absent from the AaLS-IC–NTB intermediate. The fluorescence spectrum of AaLS-IC–cur showed a large emission peak at 525 nm (Fig. 5), which is characteristic of curcumin43,44 and cur-SH but not AaLS-IC–NTB. Together, these spectra are consistent with what would be expected for the desired AaLS-IC–cur conjugate. SEC analysis shows that the AaLS-IC–cur conjugate elutes as a single peak with the same elution volume as the unreacted AaLS-IC capsid, indicating that curcumin loading does not change the assembly state of AaLS-IC (Fig. 6). Further, during SEC the curcumin-based fluorescence co-elutes with the capsid protein, signifying that the cur-SH is attached to the protein. AaLS-IC–cur was also subjected to reverse-phase HPLC analysis. The resulting chromatogram (Fig. 7) showed a large peak with a retention time of 18 min, which is about 3 minutes later than the elution time of free cur-SH. A small peak was observed at 15.2 min, similar to free cur-SH, but this species represents only about 5% of the total curcumin in the sample, or about 2 cur-SH molecules per capsid. This free cur-SH observed in the chromatogram can be attributed to non-covalently bound cur-SH that is released from the protein under the denaturing conditions of the reverse-phase HPLC analysis.
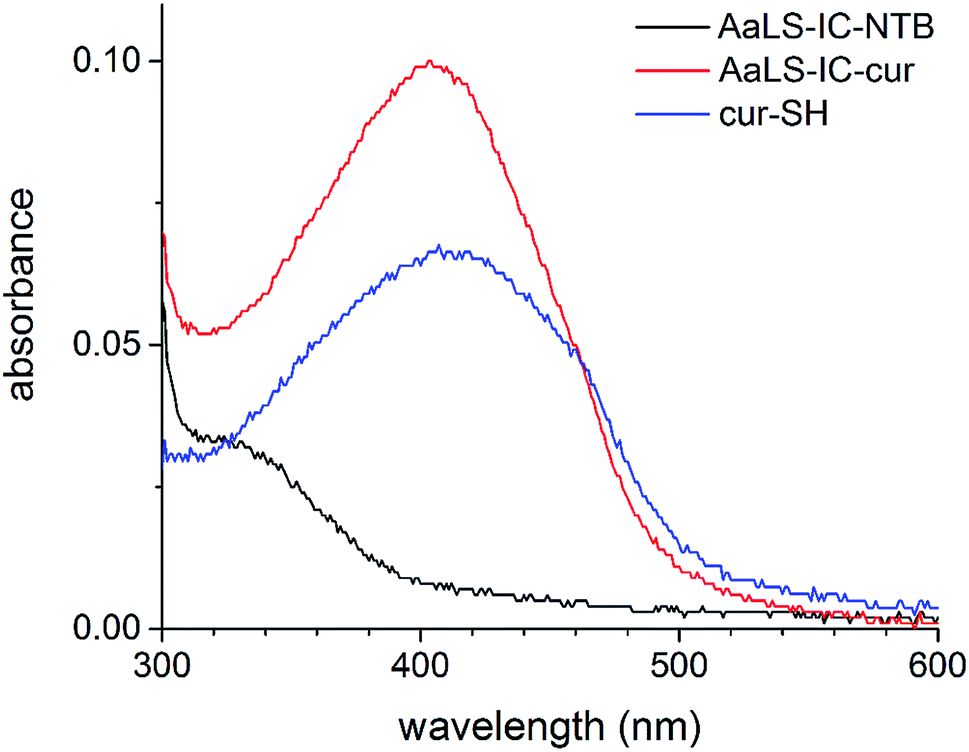 | ||
| Fig. 4 UV-Vis spectra of cur-SH, AaLS-IC–NTB and AaLS-IC–cur. The absorbance of AaLS-IC–cur (red), AaLS-IC–NTB (black), and cur-SH (blue) in SEC buffer with 2% ethanol were measured from 600 nm to 300 nm. SEC buffer with 2% ethanol was used as the blank. The concentrations of AaLS-IC–cur, AaLS-IC–NTB, and cur-SH were 8 μM, 8 μM, and 5.8 μM, respectively. | ||
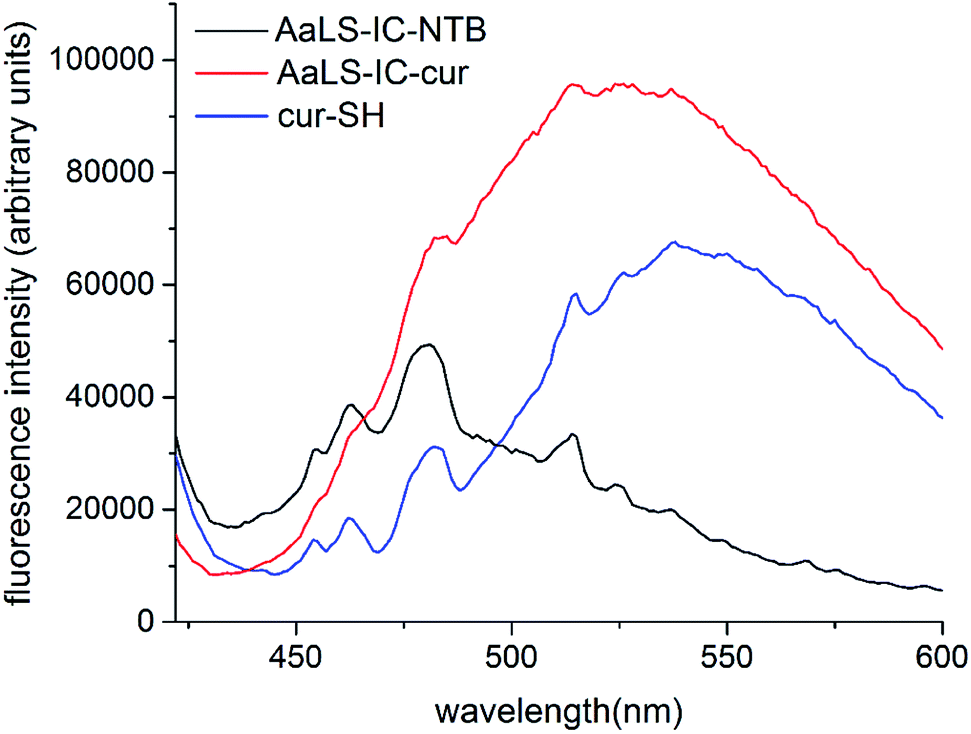 | ||
| Fig. 5 Fluorescence spectra of cur-SH, AaLS-IC–NTB and AaLS-IC–cur. The fluorescence emission of AaLS-IC–cur (red), AaLS-IC–NTB (black), and cur-SH (blue) in SEC buffer with 2% ethanol were measured from 422 nm to 600 nm, after excitation at 412 nm. SEC buffer with 2% ethanol was used as the blank. The concentrations of AaLS-IC–cur, AaLS-IC–NTB, and cur-SH were 8 μM, 8 μM, and 5.8 μM, respectively. | ||
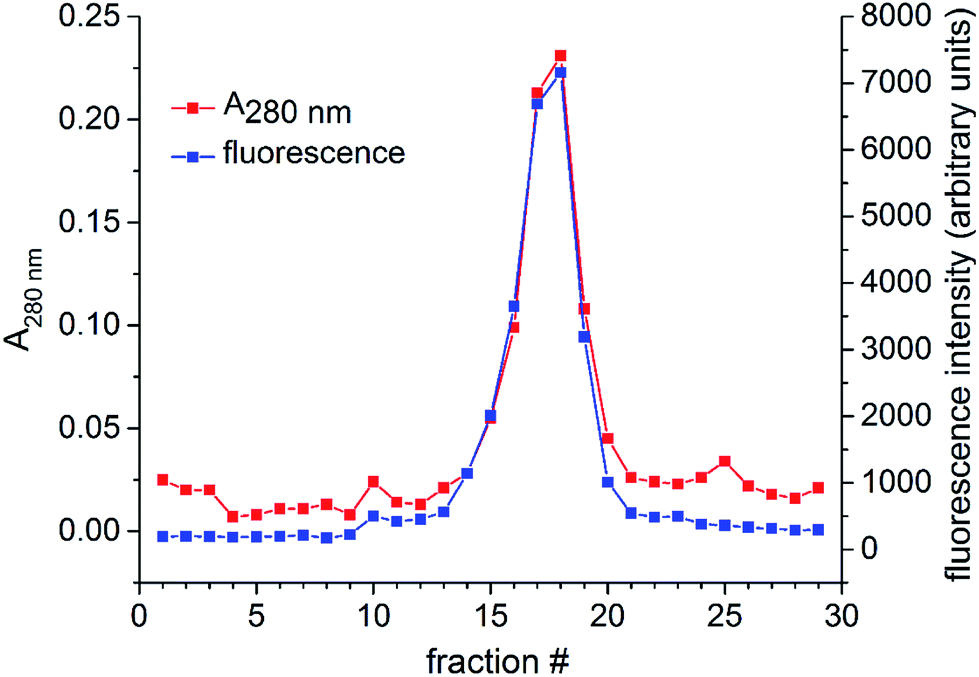 | ||
| Fig. 6 SEC analysis of AaLS-IC–cur. The AaLS-IC–cur conjugate was injected onto a Hiprep 16/60 sephacryl S-400HR column and eluted over a total volume of 120 mL. The A280 nm and fluorescence emission at 525 nm (excitation at 412 nm) were then measured for each of the individual fractions. The size of each fraction was 4 mL. | ||
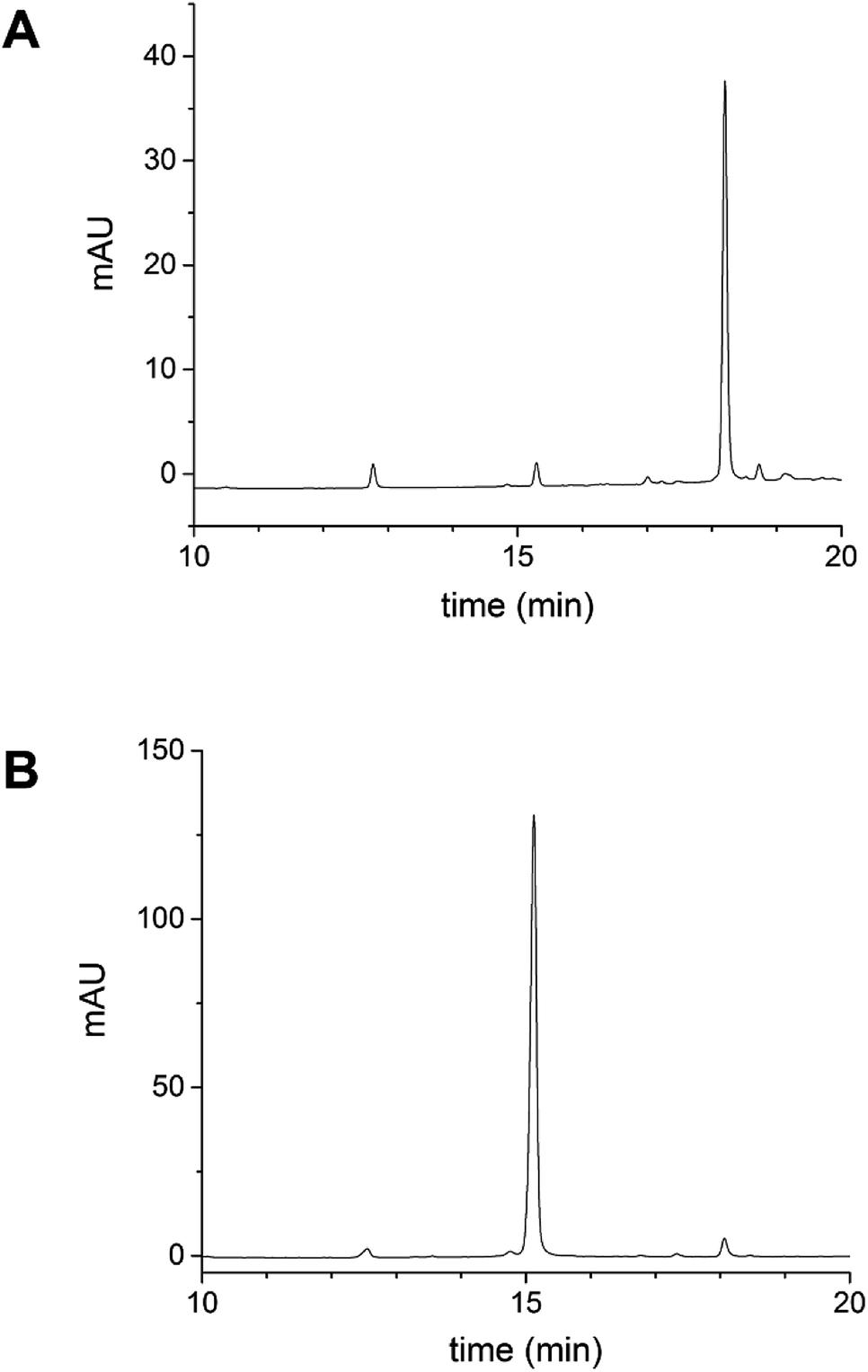 | ||
| Fig. 7 Reverse-phase HPLC analysis of cur-SH and AaLS-IC–cur. (A) AaLS-IC–cur in PBS buffer was injected onto an analytical C4 HPLC column to check for non-covalently bound cur-SH in the AaLS-IC–cur sample. Under reverse-phase HPLC conditions, AaLS-IC protein should be denatured, allowing the separation of any noncovalent bound curcumin from the protein. (B) Pure cur-SH was subjected to the same HPLC analysis as a reference. The detection wavelength was 410 nm. | ||
3.3 Solubility of AaLS-IC–cur
Curcumin is notoriously insoluble in aqueous solution. However, the sequestration of curcumin inside the AaLS capsid should prevent the encapsulated curcumin from aggregating and precipitating. To test whether the encapsulation of curcumin increases its solubility in PBS buffer containing 0.8% ethanol, samples of both AaLS-IC–cur and free cur-SH, each containing a total amount of curcumin that would correspond to a concentration of 30 μM (if fully soluble), were analyzed by subjecting them to centrifugation (to pellet any insoluble material) and measuring the absorbance values of the supernatants at 412 nm (to provide a relative estimate of the curcumin remaining in solution). Indeed, the A412 of AaLS-IC–cur, which did not produce a visible pellet, was double that of free cur-SH, which gave a yellow pellet. The AaLS-IC–cur sample was then further concentrated by ultrafiltration and the analysis was repeated. For total curcumin concentrations as high as 430 μM, precipitation was still not evident and the A412 value increased proportionately (Table 1). Thus, the curcumin in AaLS-IC–cur is >30-fold more soluble in buffer than free cur-SH. At the highest concentration of AaLS-IC–cur tested, the total protein concentration is ∼10 mg mL−1, and the solubility limit had still not been reached.| Sample | Theoreticala [curcumin] (μM) | Pellet observed?b | A412 nmc |
|---|---|---|---|
| a The theoretical [curcumin] represents the amount of cur-SH (either free or conjugated to AaLS-IC) expected in solution if the curcumin is fully soluble. The solvent is PBS buffer with 0.8% ethanol.b The samples were subjected to centrifugation in 1.5 mL microcentrifuge tubes at 13000 rpm for 5 min at room temperature in a Cence H1650 benchtop centrifuge. The bottoms of the tubes were then examined to check for insoluble material.c After centrifugation, the supernatants were filtered through a 0.22 μm membrane. The reported A412 nm values correspond to the filtered supernatants.d The pellet was yellow.e After correction for a 2-fold dilution. |
|||
| Cur-SH | 30 | Yesd | 0.156 |
| AaLS-IC–cur | 30 | No | 0.330 |
| AaLS-IC–cur | 86 | No | 1.383 |
| AaLS-IC–cur | 430 | No | 5.556e |
The poor solubility of curcumin in aqueous media contributes to its low bioavailability.45 The aqueous solubility of natural curcumin has been reported to be 30 nM.38 After dialysis into pure water, AaLS-IC–cur also remained soluble up to a total curcumin concentration of 430 μM (Table S3†). Thus the solubility of cur-SH encapsulated by AaLS-IC is >14000-fold higher than that of the free natural product in water. The increased solubility of encapsulated cur-SH might also improve the bioavailability of this potential drug.
3.4 Release of cur-SH
Reduction of the disulfide bond connecting cur-SH to the AaLS-IC protein should allow release of the cur-SH cargo from the capsid. This disulfide bond should be sensitive to small-molecule reducing agents. To test this hypothesis, AaLS-IC–cur was treated with TCEP. Analysis of this reaction at different time points by reverse-phase HPLC showed the rapid accumulation of free cur-SH over the first two hours (Fig. 8A). Doubling the concentration of TCEP increased the apparent rate of cur-SH release over the first hour (Fig. 8B). In contrast, no free cur-SH was apparent after 8 h in the negative control containing no TCEP (Fig. 8C). Together, these results demonstrate that cur-SH can be released from AaLS-IC capsid quickly, and that cur-SH release can be triggered by a reducing agent in a concentration-dependent manner.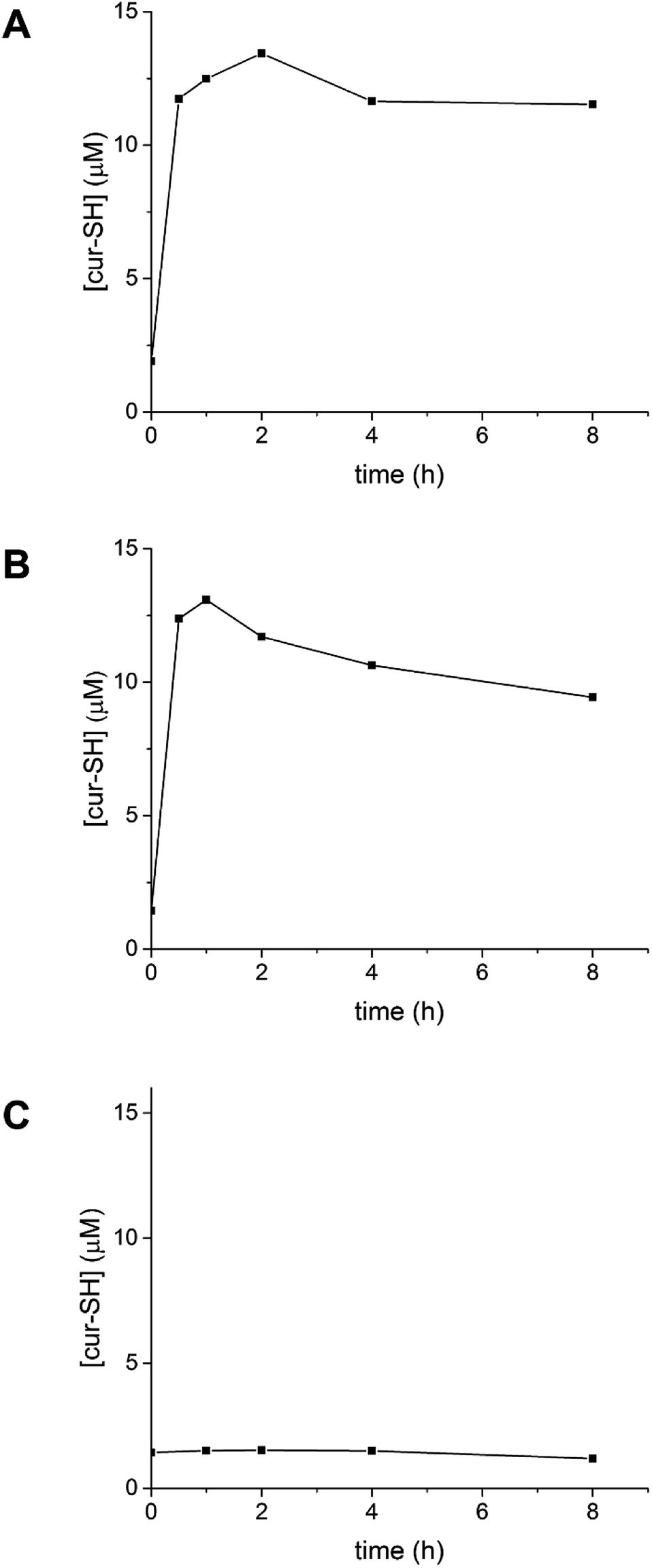 | ||
| Fig. 8 Time course of cur-SH release from AaLS-IC–cur. AaLS-IC–cur in PBS buffer was incubated with either 5 mM TCEP (A), 10 mM TCEP (B), or 0 mM TCEP (C) at 4 °C in the dark. The amount of free cur-SH was measured over time using analytical HPLC. | ||
Following the addition of 5 mM TCEP to AaLS-IC–cur, the amount of free cur-SH reaches a maximum within 2 h, after which the concentration slowly declines. A similar pattern is also observed for 10 mM TCEP, although in this case the maximum concentration of free cur-SH occurs after only 1 h. The maximum free cur-SH concentrations correspond to 66% and 80% of the covalently bound cur-SH in AaLS-IC–cur, for 5 mM TCEP and 10 mM TCEP, respectively. These yields indicate that the majority of the covalently bound cur-SH can be released from the capsid by TCEP.
Several other small-molecule encapsulation systems have previously been developed by engineering cysteine residues on the inner surfaces of virus capsids. For example, an engineered cysteine on the interior surface of the 180 subunit CCMV capsid was conjugated to a maleimide-derivatized spin label, which was used to probe structural changes associated with capsid swelling.46 Similarly, the bacteriophage MS2 capsid was labeled with a maleimide containing derivative of taxol with a yield of 65%.47 Using the same conjugation chemistry, this MS2 variant was also quantitatively loaded with porphyrin48 and an imaging agent.49 Likewise, engineered variants of the bacteriophage P22 capsid have been generated at various positions along the inner capsid surface that have been loaded with guests (including an imaging agent, a radical polymerization initiator, and a biotin derivative) bearing either maleimide or iodoacetamide functional groups with yields ranging from 50–100%.20,50,51 The loading system used for AaLS-IC with cur-SH gives a yield in this range. The reaction between cysteine and maleimide or iodoacetamide produces a thioether bond, and thiol alkylation is generally irreversible. In contrast, disulfide bonds can be readily reduced under physiological conditions. The controlled release of the cargo molecules triggered by reduction extends the functional capability of this encapsulation system, which could be useful for applications such as drug delivery.
Given its broad array of pharmacological activities, there has been much interest in developing curcumin nanocarriers for drug delivery applications. Many different curcumin encapsulation systems have been reported, mostly based on non-covalent hydrophobic partitioning of curcumin into the non-polar core of polymersomes, micelles, liposomes, solid lipid nanoparticles, or silica nanoparticles.40,41 Recently, covalent conjugation strategies for attaching curcumin analogs to polymer-based micelles have been devised to provide triggered release of curcumin. Using a hydrazone linkage to methoxy-poly(ethylene glycol)–poly(lactic acid) (mPEG–PLA) copolymers, curcumin analog loadings of 5–15% were reported in the resulting micelles.52 Upon lowering the pH, the hydrazone was broken, releasing a maximum of 40–60% of the conjugated curcumin over >10 h. Similarly, an acetal conjugation between a curcumin analog and mPEG–PLA gave a loading yield of 7.1% (w/w) in the resulting micelles and acidic release of ∼50% of the bound curcumin over >30 h.53 Similar to this study, a disulfide bond was also used to conjugate a curcumin analog to mPEG–PLA.35 The loading yield in the micelles was 8.2% (w/w), and the bound curcumin could be released by reducing agents. In this case, the addition of 10 mM DTT triggered about 50% of the curcumin to be released over >30 h. In comparison, the cur-SH content of AaLS-IC–cur is 2% (w/w), however this apparently lower loading yield is mostly due to the higher molecular weight of the AaLS-IC polypeptide compared to the mPEG–PLA copolymer. For both the protein capsid and the polymeric micelle, each subunit of the nanoparticle assembly has one site for guest attachment and the majority of the subunits are conjugated to the guest after loading. The addition of TCEP to AaLS-IC–cur gives a somewhat higher maximum yield of released cur-SH, with much faster guest release, than the analogous mPEG–PLA micelles. The more rapid guest release kinetics in the AaLS-IC–cur system probably suggest that the disulfide bond connecting the cur-SH to AaLS-IC is relatively exposed compared to the disulfide bond in the analogous mPEG–PLA micelles, in which the curcumin analog and its conjugation site on the polymer are likely buried in the non-polar core of the particle, making them less accessible to reducing agents.
The decrease in free cur-SH at later time points could be caused by degradation of cur-SH. Indeed, previous studies have shown that curcumin is unstable in aqueous buffer37 and in the presence of free thiol groups.54 The mechanism of curcumin degradation is complex, and several pathways have been identified, many of which involve either nucleophilic attack at one of the carbonyl groups or a Michael-type addition at one of the adjacent α,β double bonds.55 Both the thiol group of cur-SH and the phosphine of TCEP could conceivably promote curcumin degradation after the release of cur-SH from the AaLS-IC capsid. To examine whether TCEP causes degradation of cur-SH, the absorbance of free cur-SH at 412 nm was measured over time in the presence and absence of 10 mM TCEP. In the absence of TCEP, the absorbance of cur-SH decreases by about 15% over 2 h at pH 8.0 (90% SEC buffer/10% ethanol) and room temperature (Fig. 9). Cur-SH lost absorbance much more rapidly in the presence of 10 mM TCEP, with only about 30% of its original absorbance remaining after 2 h. Although the cur-SH release experiments described above were carried out at lower pH and temperature conditions, at which the reaction of cur-SH with TCEP is likely somewhat slower, the decreased yield of free cur-SH observed at later time points (Fig. 8B) can be attributed to this type of degradation, given that the HPLC detection of cur-SH was also done at 410 nm.
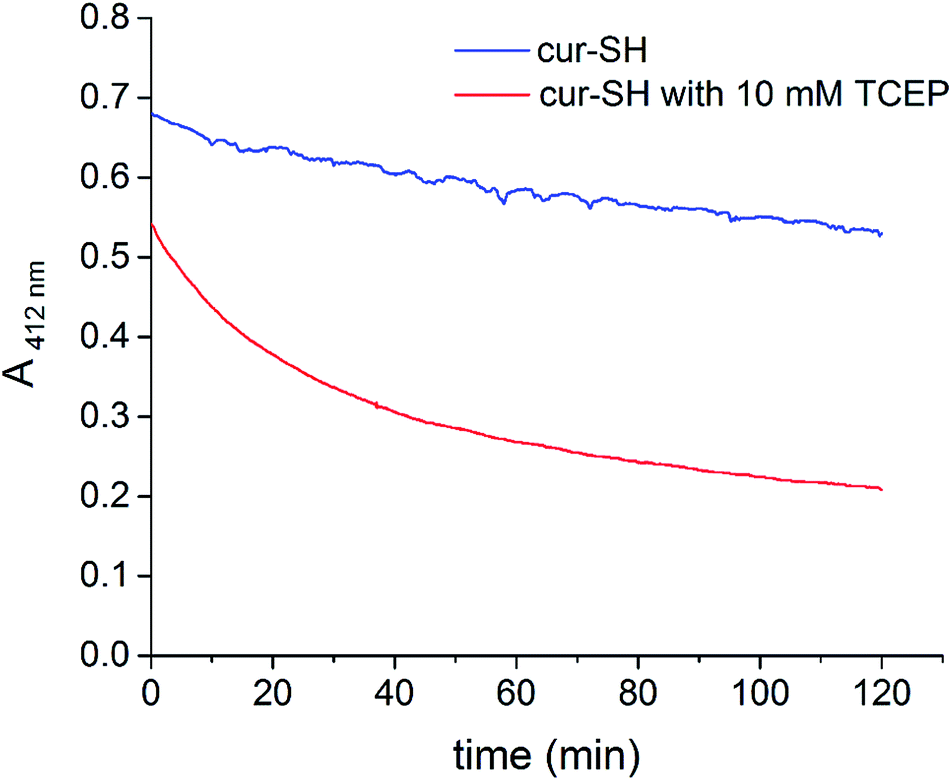 | ||
| Fig. 9 Time course of cur-SH degradation. The absorbance at 412 nm of cur-SH in SEC buffer with 10% ethanol was measured over 2 h in the presence (red) and absence (blue) of TCEP. | ||
The addition of TCEP to AaLS-IC–cur causes free cur-SH to accumulate rapidly (Fig. 8), suggesting that reduction of the disulfide bond in the AaLS-IC–cur conjugate is faster than cur-SH degradation. Over time, however, the accumulated free cur-SH degrades. Although initially this degradation is compensated for by the continuing reductive release of cur-SH from the capsid, eventually the supply of AaLS-IC–cur conjugates becomes depleted, the cur-SH degradation rate overtakes the cur-SH release rate, and the concentration of free cur-SH declines. Thus, the maximum observed yields for cur-SH release (66% and 80% for 5 mM TCEP and 10 mM TCEP, respectively) represent lower limits for the reduction of the disulfide bond in AaLS-IC–cur. For other potential guests that are not susceptible to degradation by TCEP, the yield of guest release would likely be even higher.
4. Conclusions
To encapsulate small molecules, a reactive cysteine was installed at the interior surface the AaLS capsid. A novel thiol–disulfide exchange process was carried out at this interior cysteine which lead to the capture of the 41 cur-SH molecules per capsid. Cur-SH can be released from the AaLS-IC capsid by treatment with the reducing agent TCEP. This reductive release mechanism may be a useful feature for applications such as drug delivery, as the reducing environment of the cytosol could potentially trigger drug release in the cell. In addition, encapsulated cur-SH demonstrated a marked improvement in aqueous solubility, suggesting that its encapsulation within AaLS-IC might improve its bioavailability for drug delivery applications. This system may be generally useful for encapsulation of small molecules containing a thiol group and could also be easily extended to the capture of cargo molecules that possess electrophilic groups.Acknowledgements
We thank Prof. Bob Borris for technical assistance and advice regarding the HPLC experiments. Prof. Nat Finney graciously provided access to the fluorimeter used to obtain the emission spectra. We are grateful to Profs. Yanjun Zhao and Zheng Wang for helpful discussions and technical assistance with the synthesis of cur-SH. Funding for this research was provided by Tianjin University and the University of Utah.References
- M. Uchida, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- T. W. Giessen and P. A. Silver, J. Mol. Biol., 2016, 428, 916–927 CrossRef CAS PubMed.
- J. A. Speir and J. E. Johnson, Curr. Opin. Struct. Biol., 2012, 22, 65–71 CrossRef CAS PubMed.
- X. Liu and E. C. Theil, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- T. A. Bobik, B. P. Lehman and T. O. Yeates, Mol. Microbiol., 2015, 98, 193–207 CrossRef CAS PubMed.
- M. Sutter, D. Boehringer, S. Gutmann, S. Gunther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban and N. Ban, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CAS.
- R. Ladenstein, M. Fischer and A. Bacher, FEBS J., 2013, 280, 2537–2563 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–759 CrossRef CAS PubMed.
- R. M. Putri, J. J. Cornelissen and M. S. T. Koay, ChemPhysChem, 2015, 16, 911–918 CrossRef CAS PubMed.
- L. Schoonen and J. C. M. van Hest, Adv. Mater., 2016, 28, 1109–1128 CrossRef CAS PubMed.
- Z. P. Sahandi, M. Karimi, F. Mehidizadeh, A. Ghasemi, S. Bahrami, H. Zare, M. Moghoofei, A. Hekmatmanesh and M. R. Hamblin, Nanoscale, 2017, 9, 1356–1392 RSC.
- Y. Zhang, M. S. Ardejani and B. P. Orner, Chem.–Asian J., 2016, 11, 2814–2828 CrossRef CAS PubMed.
- R. L. Garcea and L. Gissmann, Curr. Opin. Biotechnol., 2004, 15, 513–517 CrossRef CAS PubMed.
- E. Strable and M. G. Finn, Curr. Top. Microbiol. Immunol., 2009, 327, 1–21 CAS.
- Z. Wu, K. Chen, I. Yildiz, A. Dirksen, R. Fischer, P. E. Dawson and N. F. Steinmetz, Nanoscale, 2012, 435, 67–76 Search PubMed.
- Y. Ma, R. J. Nolte and J. J. Cornelissen, Adv. Drug Delivery Rev., 2012, 64, 811–825 CrossRef CAS PubMed.
- E. W. Kovacs, J. M. Hooker, D. W. Romanini, P. G. Holder, K. E. Berry and M. B. Francis, Bioconjugate Chem., 2007, 18, 1140–1147 CrossRef CAS PubMed.
- D. Banerjee, A. P. Liu, N. R. Voss, S. L. Schmid and M. G. Finn, ChemBioChem, 2010, 11, 1273–1279 CrossRef CAS PubMed.
- J. D. Fiedler, S. D. Brown, J. L. Lau and M. G. Finn, Angew. Chem., Int. Ed., 2010, 49, 9648–9651 CrossRef CAS PubMed.
- M. Uchida, D. S. Morris, S. Kang, C. C. Jolley, J. Lucon, L. O. Liepold, B. LaFrance, P. E. J. Prevelige and T. Douglas, Langmuir, 2012, 28, 1998–2006 CrossRef CAS PubMed.
- A. Abbing, U. K. Blaschke, S. Grein, M. Kretschmar, C. M. B. Stark, M. J. W. Thies, J. Walter, M. Weigand, D. C. Woith, J. Hess and C. O. A. Reiser, J. Biol. Chem., 2004, 279, 27410–27421 CrossRef CAS PubMed.
- X. Lu, J. R. Thompson and K. L. Perry, J. Gen. Virol., 2012, 93, 1120–1126 CrossRef CAS PubMed.
- M. Kwak, I. J. Minten, D. M. Anaya, A. J. Musser, M. Brasch, R. J. Nolte, K. Müllen, J. J. Cornelissen and A. Herrmann, J. Am. Chem. Soc., 2010, 132, 7834–7835 CrossRef CAS PubMed.
- I. J. Minten, L. J. Hendriks, R. J. Nolte and J. J. Cornelissen, J. Am. Chem. Soc., 2009, 131, 17771–17773 CrossRef CAS PubMed.
- P. A. Suci, Z. Varpness, E. Gillitzer, T. Douglas and M. Young, Langmuir, 2007, 23, 12280–12286 CrossRef CAS PubMed.
- X. Zhang, W. Meining, M. Fischer, A. Bacher and R. Ladenstein, J. Mol. Biol., 2001, 306, 1099–1114 CrossRef CAS PubMed.
- H.-N. Chen and K. J. Woycechowsky, Biochemistry, 2012, 51, 4704–4712 CrossRef CAS PubMed.
- R. Ladenstein, K. Ritsert, R. Huber, G. Richter and A. Bacher, Eur. J. Biochem., 1994, 223, 1007–1017 CrossRef CAS PubMed.
- S. Lilavivat, D. Sardar, S. Jana, G. C. Thomas and K. J. Woycechowsky, J. Am. Chem. Soc., 2012, 134, 13152–13155 CrossRef CAS PubMed.
- Y. Azuma, R. Zschoche, M. Tinzl and D. Hilvert, Angew. Chem., Int. Ed., 2016, 55, 1531–1534 CrossRef CAS PubMed.
- R. Frey, S. Mantri, M. Rocca and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 10072–10075 CrossRef CAS PubMed.
- F. P. Seebeck, K. J. Woycechowsky, W. Zhuang, J. P. Rabe and D. Hilvert, J. Am. Chem. Soc., 2006, 128, 4516–4517 CrossRef CAS PubMed.
- B. Wörsdörfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589–592 CrossRef PubMed.
- B. Wörsdörfer, Z. Pianowski and D. Hilvert, J. Am. Chem. Soc., 2012, 134, 909–911 CrossRef PubMed.
- Y. Cao, M. Gao, C. Chen, A. Fan, J. Zhang, D. Kong, Z. Wang, D. Peer and Y. Zhao, Nanotechnology, 2015, 26, 115101 CrossRef PubMed.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Methods Enzymol., 1983, 91, 49–60 CAS.
- O. Naksuriya, M. J. van Steenbergen, J. S. Torano, S. Okonogi and W. E. Hennink, AAPS J., 2015, 18, 777–787 CrossRef PubMed.
- H. H. Tønnesen, M. Másson and T. Loftsson, Int. J. Pharm., 2002, 244, 127–135 CrossRef.
- B. B. Aggarwal and K. B. Harikumar, Int. J. Biochem. Cell Biol., 2009, 41, 40–59 CrossRef CAS PubMed.
- S. Shome, A. D. Talukdar, M. D. Choudhury, M. K. Bhattacharya and H. Upadhyaya, J. Pharm. Pharmacol., 2016, 68, 1481–1500 CrossRef CAS PubMed.
- M. Mehanny, R. M. Hathout, A. S. Geneidi and S. Mansour, J. Controlled Release, 2016, 225, 1–30 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- S. H. Kim, S. Y. Gwon, S. M. Burkinshaw and Y. A. Son, Spectrochim. Acta, Part A, 2010, 76, 384–387 CrossRef PubMed.
- S. Mondal, S. Ghosh and S. P. Moulik, J. Photochem. Photobiol., B, 2016, 158, 212–218 CrossRef CAS PubMed.
- Z. Stanic, Plant Foods Hum. Nutr., 2017, 72, 1–12 CrossRef CAS PubMed.
- R. J. Usselman, E. D. Walter, D. Willits, T. Douglas, M. Young and D. J. Singel, J. Am. Chem. Soc., 2011, 133, 4156–4159 CrossRef CAS PubMed.
- W. Wu, S. C. Hsiao, Z. M. Carrico and M. B. Francis, Angew. Chem., Int. Ed., 2009, 48, 9493–9497 CrossRef CAS PubMed.
- N. Stephanopoulos, G. J. Tong, S. C. Hsiao and M. B. Francis, ACS Nano, 2010, 4, 6014–6020 CrossRef CAS PubMed.
- P. D. Garimella, A. Datta, D. W. Romanini, K. N. Raymond and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 14704–14709 CrossRef CAS PubMed.
- J. Lucon, S. Qazi, M. Uchida, G. J. Bedwell, B. LaFrance, P. E. Prevelige and T. Douglas, Nat. Chem., 2012, 4, 781–788 CrossRef CAS PubMed.
- S. Kang, M. Uchida, A. O'Neil, R. Li, P. E. Prevelige and T. Douglas, Biomacromolecules, 2010, 11, 2804–2809 CrossRef CAS PubMed.
- M. Gao, C. Chen, A. Fan, J. Zhang, D. Kong, Z. Wang and Y. Zhao, Nanotechnology, 2015, 26, 275101 CrossRef PubMed.
- M. Li, M. Gao, Y. Fu, C. Chen, X. Meng, A. Fan, D. Kong, Z. Wang and Y. Zhao, Colloids Surf., B, 2016, 140, 11–18 CrossRef CAS PubMed.
- S. Amslinger, N. Al-Rifai, K. Winter, K. Wörmann, R. Scholz, P. Baumeister and M. Wild, Org. Biomol. Chem., 2013, 11, 549–554 CAS.
- M. Heger, R. F. van Golen, M. Broekgaarden and M. C. Michel, Pharmacol. Rev., 2013, 66, 222–307 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05890j |
| ‡ These authors contributed equally to this work. |
| § Present address: School of pharmaceutical Science and Technology, Tianjin University, 92 Weijin Road, Nankai District, Tianjin, 300072, China. |
| This journal is © The Royal Society of Chemistry 2017 |