DOI:
10.1039/C7RA05856J
(Paper)
RSC Adv., 2017,
7, 33402-33407
Structural, mechanical and electronic properties of Nb2C: first-principles calculations†
Received
25th May 2017
, Accepted 16th June 2017
First published on 3rd July 2017
Abstract
Nb–C compounds are potential candidates to achieve high hardness and refractory nature. We performed a crystal structure search for the Nb–C system using an ab initio evolutionary algorithm implemented in the USPEX code. By comparing the formation enthalpy, a P–x phase diagram was calculated, and an orthorhombic Pnnm structure of Nb2C was predicted and denoted as Nb2C-I, which was both mechanically and dynamically stable. In this Pnnm phase, there are four-sided rings continuously along the c-axis, which probably contributes to the relatively high incompressibility of Nb2C-I along the crystallographic c-axis. Moreover, the hardness and Young's modulus were calculated to be 28.5 GPa and 448.9 GPa, respectively; hence, the Nb2C-I is considered as a potential ultra-stiff and hard material.
1 Introduction
In the past few decades, superhard materials with excellent mechanical properties have drawn extensive attention from the researchers and have been well-studied in terms of both theoretical and experimental aspects.1 Traditional superhard materials, such as diamond,2,3 γ-B,4 c-BN,5 and c-BC2N,6,7 are mainly composed of light elements. Diamond, the earliest discovered and hardest (70–150 GPa) superhard material, shows promising mechanical properties and is hence among the most widely used superhard materials. However, diamond shows its own practical defects. At a temperature greater than 800 °C, diamond tends to oxidize. Moreover, when the diamond tool is used in the grinding or cutting of ferrous materials, the tool easily reacts with iron-based materials. The c-BN, another typical superhard material, is difficult to synthesise, which limits its practical application. Therefore, development of novel superhard multifunctional materials is an urgent requirement.
During the past decade, scientists were focused on the design of new materials in transition-metal (TM) light-element (LE) systems. One of the advantages of transition metals is their large bulk modulus. For example, the bulk modulus of Os is about 395–462 GPa,8 which is close to that of diamond (446 GPa). However, the hardness of metal is always low, which is normally one-thirtieth that of diamond. This is mainly due to the huge difference of chemical bonds in the compound structures. Classical superhard materials usually hold three-dimensional networks composed of covalent bonds with sp3 hybridization, which is the basis of superhard properties. Therefore, the type of TM-LE bonding is the key factor for improving the hardness. Due to the behavior of high valence electron density and large interatomic distance, the transition metal can be intercalated with light elements, which may lead to the formation of covalent bonding between light elements and transition metals. Thus, the relatively high directional covalent bonds and valence electron density are considered to favor the formation of new superhard materials. It is believed that the carbides of the group-IV, -V, and -VI transition metals show an unusual combination of physical and chemical properties.9–14 The transition-metal carbide (TMC) compounds combine the physical properties of three different kinds of materials: transition metals, covalent solids and ionic crystals,15 making TMC a family of industrially relevant compounds with outstanding physical properties.
The niobium carbides are an emblematical TMC system, which exhibits meaningful physicochemical properties.10,16 Moreover, niobium carbides are significant candidates for improving the mechanical properties of niobium alloys.10,16–19 Niobium and carbide could form weak metallic or semiconductors compounds with different compositions and space symmetries. In the Nb–C binary phase diagram, niobium carbides have four experimentally known compositions: NbC, Nb6C5, Nb4C3, and Nb2C.20–22 Stoichiometric NbC is in the rocksalt structure,23 in which Nb atoms form the cubic lattices, and C atoms occupy the octahedral interstitial positions. Nb4C3 was reported to adopt a R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, and Nb6C5 is in the P31 space group.24,25 Among all the niobium carbides, Nb2C is the least understood carbide;26 it exists in three polymorphic forms: γ-Nb2C, β-Nb2C, and α-Nb2C,27–31 as reported in previous studies. The γ- and β-Nb2C are hexagonal phases. The γ-Nb2C is in L'3-type structure at temperatures above 2770 K. The β-Nb2C has ζ-Fe2N structure at relatively low temperatures. The α-Nb2C is an orthorhombic low temperature phase. As Hugosson reported,20 some controversy exists in the determination of the structure of the orthorhombic phase. In his study, both structures showed energies of formation higher than those of the hexagonal phase. However, other studies showed that the formation energy of α-Nb2C was the lowest, which indicated that the formation of α-Nb2C was feasible.26,32 To date, controversy still remains regarding the orthorhombic phase structure of Nb2C.
m space group, and Nb6C5 is in the P31 space group.24,25 Among all the niobium carbides, Nb2C is the least understood carbide;26 it exists in three polymorphic forms: γ-Nb2C, β-Nb2C, and α-Nb2C,27–31 as reported in previous studies. The γ- and β-Nb2C are hexagonal phases. The γ-Nb2C is in L'3-type structure at temperatures above 2770 K. The β-Nb2C has ζ-Fe2N structure at relatively low temperatures. The α-Nb2C is an orthorhombic low temperature phase. As Hugosson reported,20 some controversy exists in the determination of the structure of the orthorhombic phase. In his study, both structures showed energies of formation higher than those of the hexagonal phase. However, other studies showed that the formation energy of α-Nb2C was the lowest, which indicated that the formation of α-Nb2C was feasible.26,32 To date, controversy still remains regarding the orthorhombic phase structure of Nb2C.
In this article, we performed a crystal structure search for the Nb–C system using an ab initio evolutionary algorithm implemented in the USPEX code. Full P–x phase diagrams of the Nb–C system at 0 K were calculated. We report a new phase for Nb2C at high pressures. The predicted new phase of Nb2C belongs to the orthorhombic Pnnm space group. Moreover, this phase is dynamically and mechanically stable at ambient and high pressures. Through a detailed electronic structure analysis, we found that niobium and carbon formed strong bonds with sp3 hybridization style, which further knitted into three-dimensional networks. Collectively, we proved that Nb2C can be a potential candidate for an ultra-stiff and hard material.
2 Methods
To search the stable and low-enthalpy structures of the Nb–C system, the evolutionary algorithm USPEX33–35 coupled with an ab initio evolutionary algorithm (EA) was employed. The calculation was designed to find the most stable structure for the given stoichiometries under given external conditions as implemented in the USPEX code. In this study, evolutionary crystal structure prediction calculations were performed at 0, 10, 30, and 50 GPa for the NbxCy system (x = 1–8, y = 1–8) at zero temperature. We performed ab initio calculations with the local density approximation (LDA), as implemented in the Vienna ab initio simulation package (VASP) code,36,37 which is based on density functional theory. The electron–ion interaction was described via the projector augmented wave (PAW) scheme.38 The orbitals 2s and 2p for C and 4p, 4d, and 5s for Nb were treated as valence states. During structural relaxation, a tested energy cutoff of 600 eV was used for the plane wave basis sets, and Monkhorst–Pack39 k-point meshes with a resolution of 0.03 Å−1 in the reciprocal space were used for all structures to minimize error from the k-point meshes. The atomic positions, lattice parameters, and cell volume were fully relaxed until the force on each atom was less than 1 meV Å−1. Phonons were calculated using the supercell method implemented in the PHONOPY package.40,41 The strain–stress method was used to calculate the elastic constants. The shear modulus, bulk modulus, Young's modulus, and Pugh's indicator were derived from the Voigt–Reuss–Hill approximation.42–44 The structure diagram of Nb2C was drawn using VESTA.45
3 Results and discussion
In this study, we uncovered a stable structure of Nb2C, denoted as Nb2C-I. At first, we calculated the formation enthalpy of NbxC1−x using a fractional representation of NbxC1−x with respect to its decomposition into Nb and C as
| ΔH(NbxC1−x) = H(NbxC1−x) − [x × H(Nb) + (1 – x) × H(C)] |
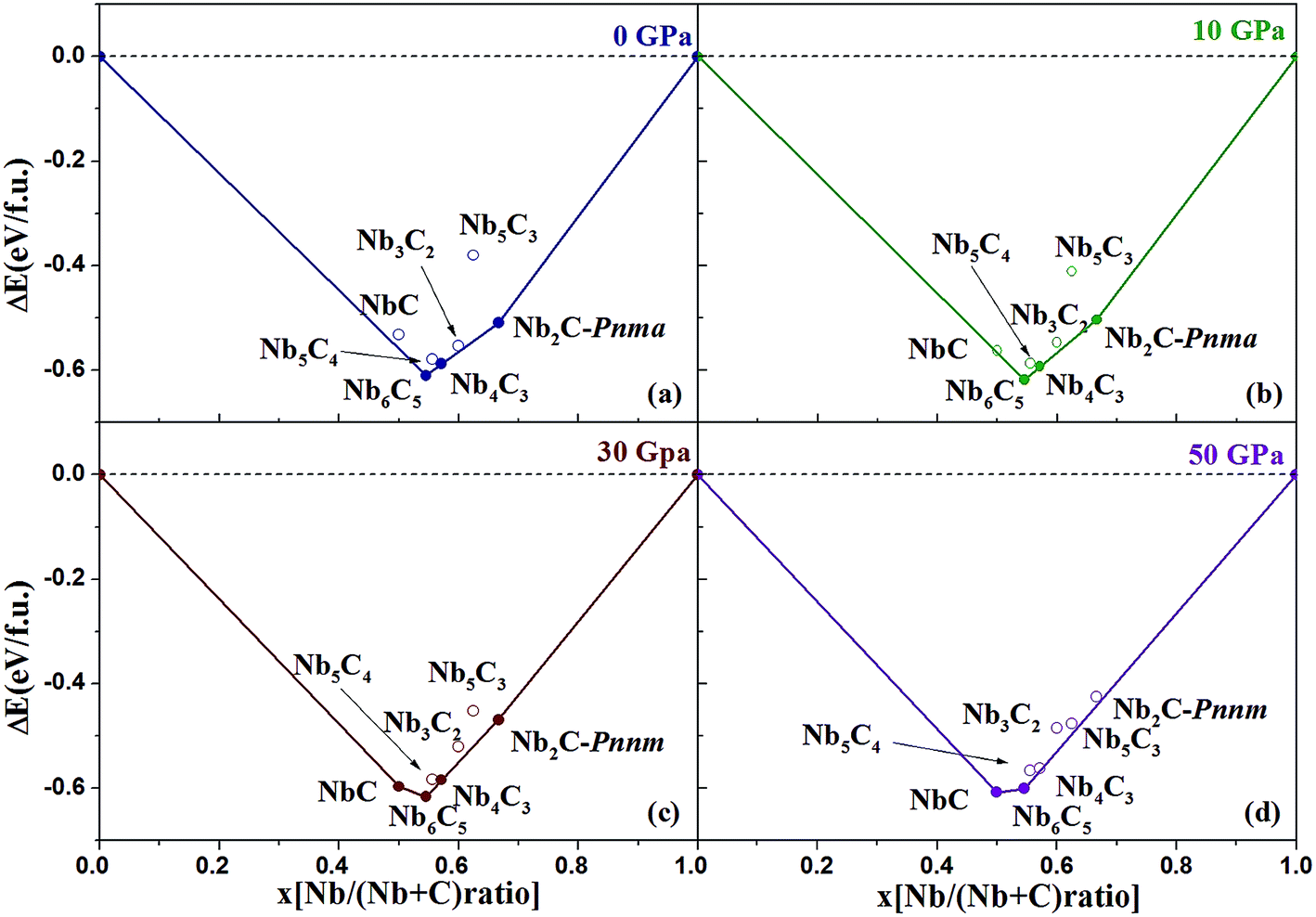
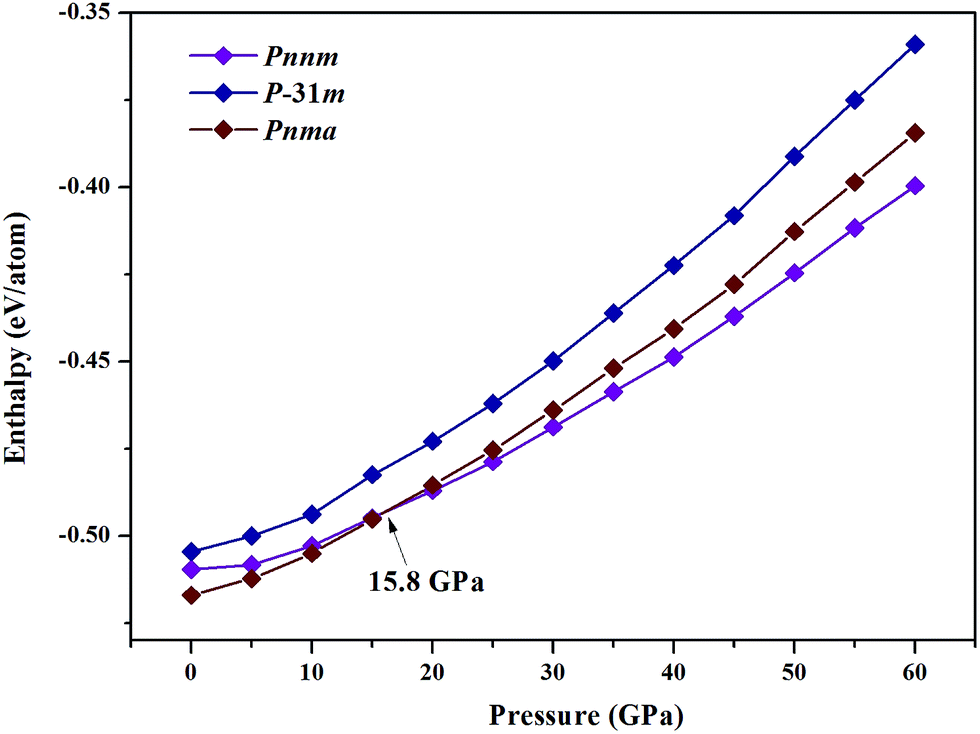
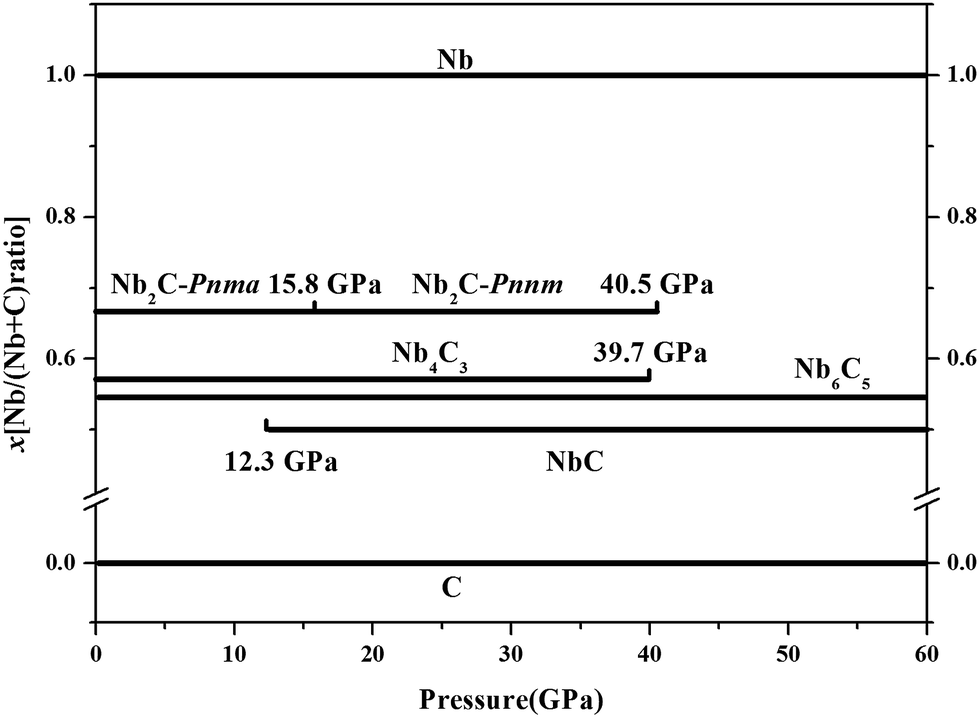
where x is the concentration of Nb. The formation enthalpies from 0 to 50 GPa were evaluated as the difference in the enthalpy of the predicted Nb–C system with respect to Nb metal and C in their most stable forms (bcc phase for Nb and graphite for C), as depicted in Fig. 1. The formation enthalpy of NbC (Fm3m) in our calculation is −0.531 eV, similar to the previous results20 (−43 mRy = −0.585 eV). Structures lying on the convex hull are either thermodynamically stable or metastable and can be synthesized in principle. According to Fig. 1(a), at 0 GPa, the Nb2C-Pnma and Nb6C5 phases are thermodynamically stable, and Nb4C3 is also located on the convex hull, which is similar to the previous results.32 With an increase in the pressure, the stoichiometric NbC phase becomes thermodynamically stable, and Nb2C-Pnnm is more stable than Nb2C-Pnma at 30 GPa. We calculated the formation enthalpies of three candidates of Nb2C. As Fig. 2 shows, the enthalpy curves of Nb2C structures are presented. It can be clearly seen that the Nb2C-Pnma is the most stable structure at 0 GPa, which is consistent with the Wu's studies.22 When the pressure is greater than 15.8 GPa, the formation enthalpies of the proposed Nb2C-I are lowest negative values, which indicates that Nb2C-I is thermodynamically stable under pressure. From Fig. 1(d), it can be observed that at 50 GPa, the stable phases are only NbC and Nb6C5. We calculated the detailed enthalpy differences as a function of pressure. As shown in the ESI Fig. S1(a),† the enthalpy difference of NbC is calculated against decomposition into Nb6C5 and C; thus, we concluded that the NbC phase becomes stable at 12.3 GPa. The enthalpy differences of Nb4C3 and Nb2C-Pnnm are calculated against decomposition into the constituent carbides. As shown in Fig. S1(b) and (c),† the structures of Nb4C3 and Nb2C-Pnnm become completely unstable at 39.7 GPa and 40.5 GPa, respectively. With these detailed calculations shown in Fig. S1,† a pressure–constituent (P–x) phase diagram of niobium carbide was constructed, which is plotted in Fig. 3. With the increase in C concentration, the Nb–C system first undergoes an orthorhombic to monoclinic transition and then transforms back to the cubic phase. Moreover, some phases of niobium carbides appear and decompose with the increase of pressure. However, the structure with high carbon concentration is not stable, even if the pressure is increased to 50 GPa.
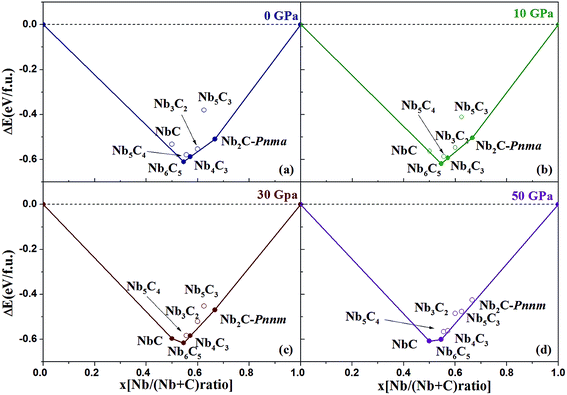 |
| | Fig. 1 Convex hull diagram for the Nb–C compounds. The formation enthalpies (ΔH, with respect to Nb and C of their most stable phases) of NbxC1−x. The abscissa x is the fraction of Nb in the structures. Circles on the solid lines represent stable ground-state compounds. | |
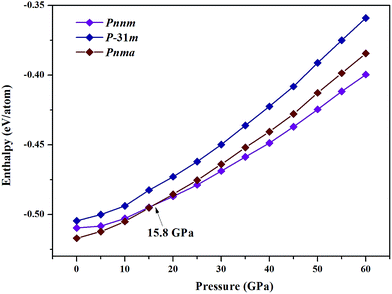 |
| | Fig. 2 Enthalpy–pressure diagrams. Calculated enthalpies as the function of pressure. | |
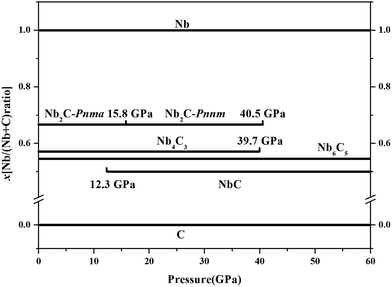 |
| | Fig. 3 Pressure–composition phase diagrams. | |
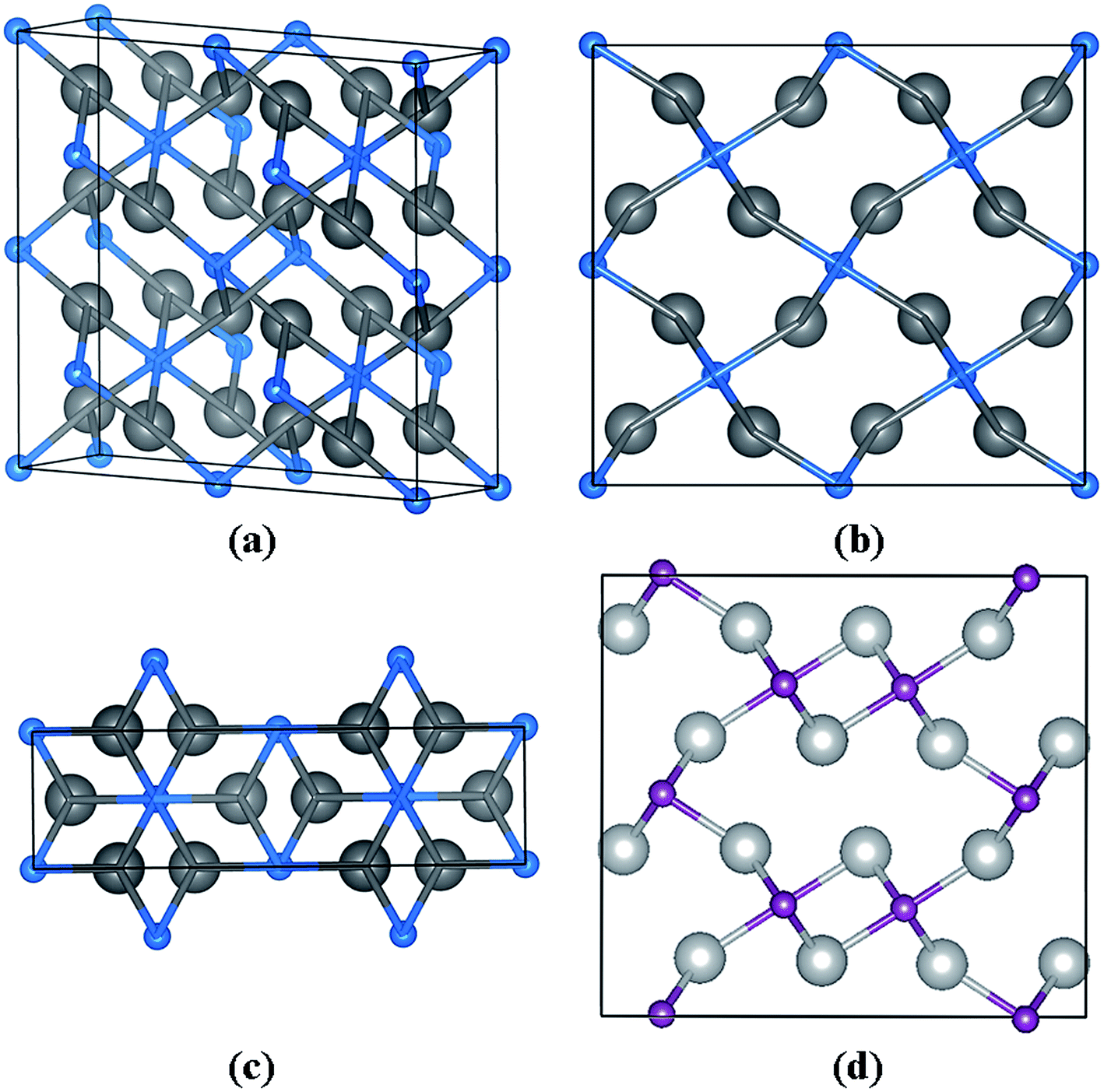
The Pnnm structure, as we predicted, contains two Nb2C in a unit cell at 20 GPa (a = 5.415 Å, b = 4.837 Å, and c = 3.019 Å), in which two inequivalent atoms Nb and C occupy the Wyckoff 8h (0.3462, 0.7632, 0.5000) and 2b (0.5, 0.5, 0.0) sites, as summarized in Table 1 and shown in Fig. 4. Fig. 4(b) along the y-axis and Fig. 4(c) along the z-axis reveal a fundamental structure in the Pnnm phase. Fig. 4(d) shows the structure of Pnma phase along the y-axis. When we compared Fig. 4(b) with (d), it was observed that the carbon atoms in Nb2C-Pnnm could help to achieve better formation of the three-dimensional space grid structure, which avoided the appearance of the interaction between metal atoms.
Table 1 The optimized equilibrium lattice constants a, b, and c (Å), and atomic coordinates for Nb2C-I at 20 GPa
| |
Structure |
Parameters (Å) |
Atom |
x |
y |
z |
| Nb2C |
Pnnm (orthorhombic) |
a = 5.415, b = 4.837, c = 3.019 |
Nb(8h) |
0.3462 |
0.7632 |
0.5000 |
| C(2b) |
0.5000 |
0.5000 |
0.0000 |
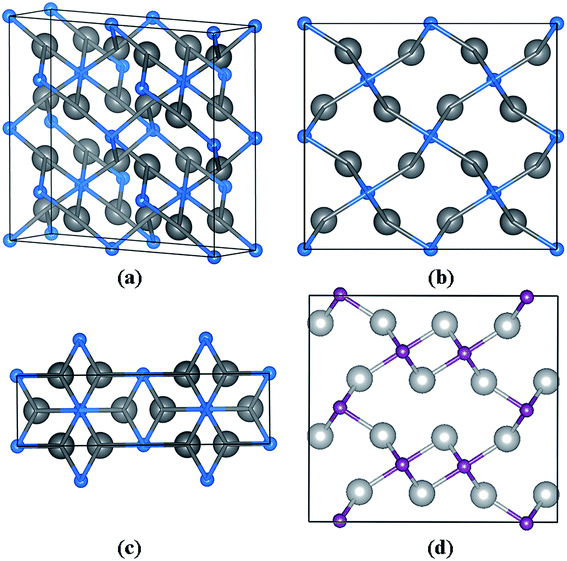 |
| | Fig. 4 Crystal structures of the predicted Nb2C-Pnnm. (a) The structure of Nb2C-Pnnm. (b) The structure along the y-axis. (c) Along the z-axis. The large gray and small blue spheres represent the Nb and C atoms, respectively. (d) The structure of Nb2C-Pnma along the y-axis. | |
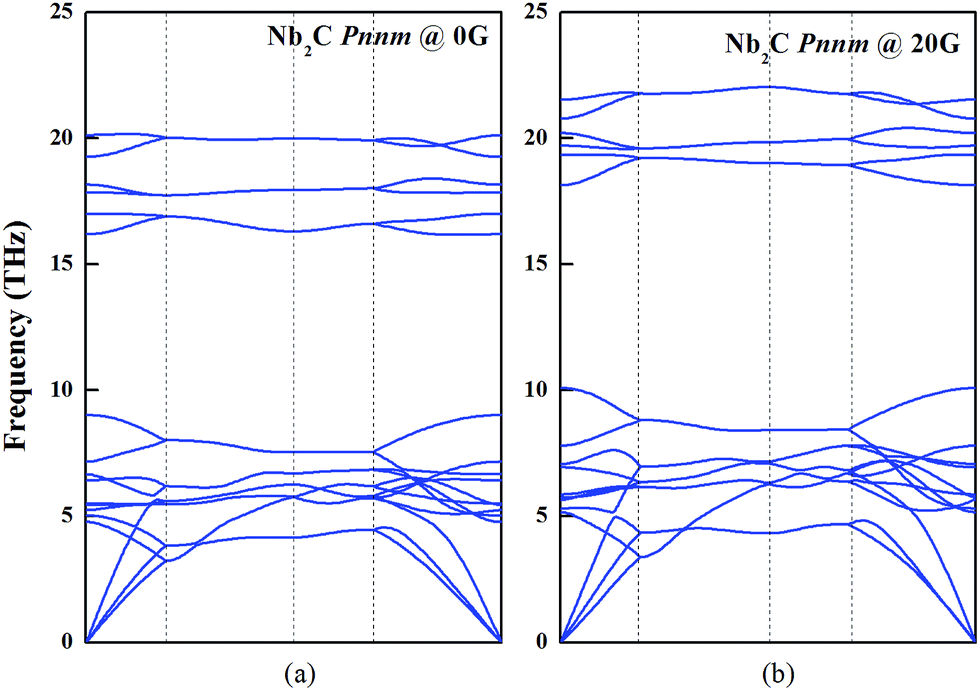
It is well-known that the phonon dispersion curves give information about the global structural stability of the materials. As shown in Fig. 5, we analyzed the phonon dispersion curves to test the lattice dynamical stability of Nb2C-I at 0 and 20 GPa. The calculated phonon curves of Nb2C have no soft mode in the Brillouin zone, indicating that Nb2C is dynamically stable. For the purpose of understanding the mechanical properties, the elastic constants are deemed essential. The elastic constants Cij of the Pnnm phases are listed in Table 2. For the proposed Nb2C-Pnnm, all Cij satisfy Born–Huang criteria,46 which means that the proposed Nb2C-Pnnm is mechanically stable. We determined that the Nb2C-Pnnm could be synthesized under high pressure and preserved under ambient pressure. From Table 2, it can be found that the calculated C33 value is bigger than the values of C11 and C22, indicating that there is relatively high incompressibility along the c-axis. The relative high incompressibility of the proposed Nb2C along the c-axis is perhaps contributed by the existence of four-sided rings, which are continuously along the crystallographic c-axis. In contrast, four-sided rings and eight-sided rings exist alternately along the b-axis or c-axis. Therefore, there is certain disparity between C33 and C11 (or C22).
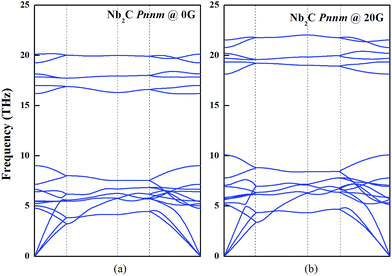 |
| | Fig. 5 Dynamic stability of Nb2C-I. The phonon dispersion curves of Nb2C-I along the high symmetry directions of the Brillouin zone at 0 and 20 GPa. | |
Table 2 Elastic constants Cij, Poisson's ratio ν, bulk modulus B (GPa), shear modulus G (GPa), Pugh's indicator G/B, Young's modulus E (GPa), and Vickers hardness HV of Nb2C-Pnma and Nb2C-Pnnm at 0 GPa
| |
C11 |
C22 |
C33 |
C44 |
C55 |
C66 |
C12 |
C13 |
C23 |
| Pnma |
390 |
405 |
390 |
76 |
119 |
122 |
157 |
129 |
136 |
| Pnnm |
383 |
389 |
415 |
101 |
102 |
137 |
156 |
153 |
127 |
| |
ν |
B |
G |
G/B |
E |
HV |
| Pnma |
0.19 |
236.1 |
178.1 |
0.75 |
426.9 |
26.8 |
| Pnnm |
0.19 |
244.5 |
188.0 |
0.77 |
448.9 |
28.5 |
It is well-known that superhard materials should have high bulk modulus and high shear modulus to resist the volume change and shape change. At 0 and 20 GPa, the bulk modulus of Nb2C-Pnnm is 244 and 310 GPa, respectively, which are larger than the values of Nb2C-Pnma (236 and 304 GPa, respectively). Because the value of the bulk modulus is large, Nb2C can be grouped into incompressible materials. As reported in Table 2, the Poisson's ratio ν of Nb2C-Pnnm is about 0.19. Usually, strong directionality degree of covalent bonding is considered in the materials when the ν value is near 0.2, indicating that the directionality degree of covalent bonding of Nb2C-Pnnm is strong. The Pugh's indicator G/B of Nb2C is 0.77, which is a relatively large value. According to the Pugh's modulus ratio defined by Cheng et al.,47 the Nb2C-I phase is a brittle and hard material with a huge capability to resist elastic plastic deformation. To gain a more comprehensive and profound understanding of the mechanical property, we calculated the Vickers hardness of Nb2C-I. The Vickers hardness HV, estimated by the empirical model, was obtained by the following formula:
| HV = (k2G)0.585 − 3 (k = G/B) |
The calculated hardness of Nb2C-I is 28.5 GPa, which almost matches that of the hard material WC (21.5–33.4 GPa).47,48 Our results suggested that the proposed Nb2C-I could be a potential candidate for ultra-stiff and hard materials.
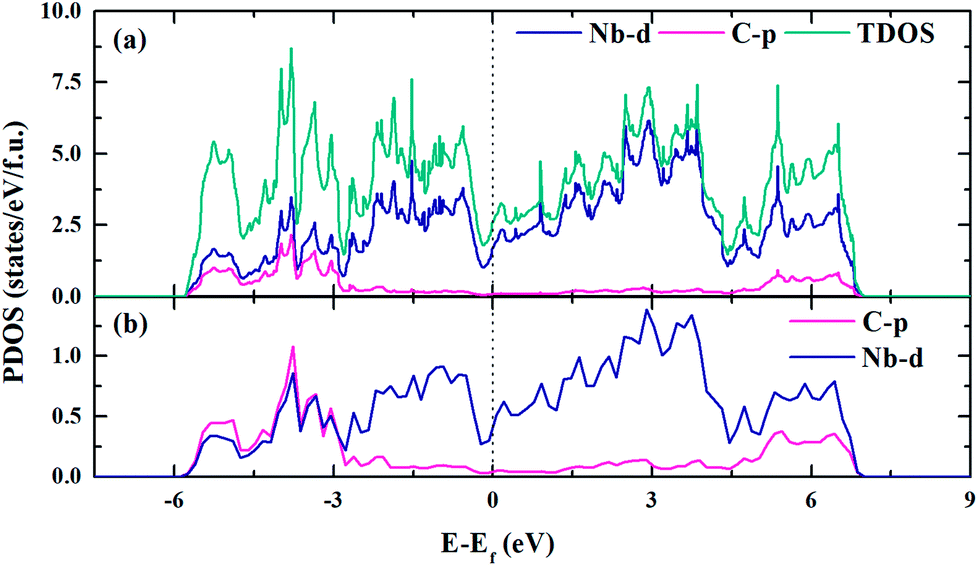
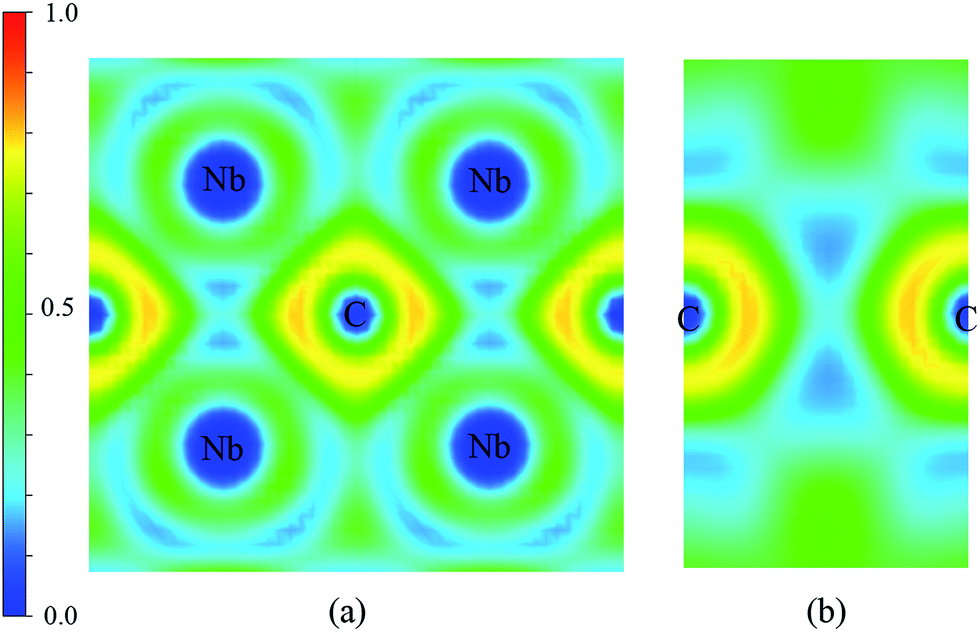
The electronic structure is crucial to understand the origin of physical properties of these carbides. The total and site projected electronic densities of states (PDOS) of Nb2C-I are shown in Fig. 6(a). As the graphic shows, Nb2C is a metal as the d electronics of Nb are mainly attributed to the density near the Fermi energy in our calculation model. To determine the hybridization between C and Nb, projected electronic densities of states of the nearest Nb and C are calculated, as shown in Fig. 6(b). There is an obvious hybridization between C 2p and Nb 4d states, which is a common feature of typical TMC superhard materials. To obtain more information about the bonding character, the electronic localization function (ELF) of Nb2C-I was calculated, as shown in Fig. 7. The electron localization function between Nb atoms and C atoms has a slight increase as compared to that in the background, which indicates that mixed bonds between the two atoms are present in this area, in which the ionic bonds are dominant. As Becke defined,49 the electrons can move freely in the areas of ELF = 0.5. In Fig. 7(a), the green region is almost connected. However, there are disconnected equivalent spheres in Fig. 7(b). In total, the equivalent sphere of ELF = 0.5 is partially connected in the whole crystal, which may enhance the weak metallicity properties.
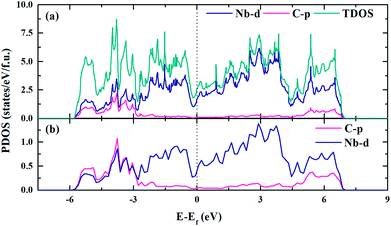 |
| | Fig. 6 Electronic density of states of Nb2C-I. The DOS (a) and PDOS (b) of Nb2C-I. The vertical black dashed line denotes the Fermi level. | |
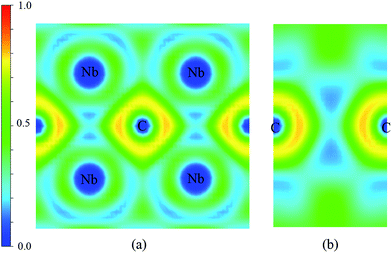 |
| | Fig. 7 Contours of the electronic localization functions (ELF). Electron localization function isosurface maps for (a) Miller indices: (1.67 1 0) and (b) Miller indices: (1 0 0). | |
4 Summary
In summary, we researched the full-scale zero-temperature Nb–C phase diagram using an ab initio evolutionary algorithm implemented in the USPEX code. The P–x phase diagrams of the Nb–C system are constructed. A new structure of Nb2C has been uncovered and denoted as Nb2C-I. The Nb2C-I belongs to the orthorhombic system with the space group Pnnm. Phase stability, mechanical properties, and electronic properties of Nb2C-I were investigated. The Vickers hardness and Young's modulus of Nb2C-I have been calculated to be 28.5 GPa and 448.9 GPa, respectively, which prove that Nb2C-I can be considered as low compressible materials. We considered that the formation of continuous four-sided rings along the c-axis might cause high incompressibility of Nb2C-I in the c-axis direction. A deep analysis of the electronic density of states and chemical bonding indicates that an ionic bond is dominant in the Nb2C crystals.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2016YFB0700505, 2016YFB0701401). Parts of the results described in this paper are obtained on the Era of Computer Network Information Center of Chinese Academy of Sciences.
References
- V. V. Brazhkin, A. G. Lyapin and R. J. Hemley, Philos. Mag. A, 2002, 82, 231–253 CAS.
- N. Mounet and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 205214 CrossRef.
- V. Brazhkin, N. Dubrovinskaia, A. Nicol and N. Novikov, et al., Nat. Mater., 2004, 3, 576–577 CrossRef CAS PubMed.
- J. Qin, N. Nishiyama and H. Ohfuji, et al., Scr. Mater., 2012, 67(3), 257–260 CrossRef CAS.
- J. C. Zheng, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 052105 CrossRef.
- V. L. Solozhenko, D. Andrault and T. G. Fique, et al., Appl. Phys. Lett., 2001, 78(10), 1385–1387 CrossRef CAS.
- Z. Pan, H. Sun and C. Chen, Phys. Rev. Lett., 2007, 98(13), 135505 CrossRef PubMed.
- F. Occelli, D. L. Farber and J. Badro, et al., Phys. Rev. Lett., 2004, 93(9), 095502 CrossRef PubMed.
- W. Weber, Phys. Rev. B: Solid State, 1973, 8, 11 Search PubMed.
- L. E. Toth, Transition Metal Carbides and Nitrides, Academic Press, New York, 1971 Search PubMed.
- S. T. Oyama, Catal. Today, 1992, 15, 179 CrossRef CAS.
- Z. Zhao, K. Bao, F. Tian, D. Duan, B. Liu and T. Cui, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 214104 CrossRef.
- Y. Liang, J. Yang, X. Yuan, W. Qiu, Z. Zhong, J. Yang and W. Zhang, Sci. Rep., 2014, 4, 5063 CrossRef PubMed.
- S. Wang, D. Antonio, X. Yu, J. Zhang, A. L. Cornelius, D. He and Y. Zhao, Sci. Rep., 2015, 5, 13733 CrossRef PubMed.
- H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- E. K. Storms, The Refractory Carbides, Academic Press, New York-London, 1967 Search PubMed.
- E. J. Delgrosso, C. E. Carlson and J. J. Kaminsky, J. Less-Common Met., 1967, 12, 173 CrossRef CAS.
- F. C. Campbell, Elements of Metallurgy and Engineering Alloys, ASM International, Ohio, 2008 Search PubMed.
- V. Javaheri, F. Shahri, M. Mohammadnezhad, M. Tamizifar and M. Naseri, J. Mater. Eng. Perform., 2014, 23, 3558–3566 CrossRef CAS.
- H. W. Hugosson, O. Eriksson, U. Janssion and B. Johansson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 134108 CrossRef.
- H. W. Hugosson, U. Jansson, B. Johansson and O. Eriksson, Chem. Phys. Lett., 2001, 333, 444–450 CrossRef CAS.
- L. Wu, Y. Wang, Z. Yan, J. Zhang, F. Xiao and B. Liao, J. Alloys Compd., 2013, 561, 220–227 CrossRef CAS.
- H. M. Ledbetter, S. Chevacharoenkul and R. F. Davis, J. Appl. Phys., 1986, 60, 1614–1617 CrossRef CAS.
- A. N. Christensen, Acta Chem. Scand., Ser. A, 1985, 39, 803–804 CrossRef.
- K. Yvon and E. Parthé, Acta Crystallogr., Sect. B: Struct. Sci., 1970, 26, 149–153 CrossRef CAS.
- B. Vishwanadh, T. S. R. C. Murthy, A. Arya, R. Tewari and G. K. Dey, J. Alloys Compd., 2016, 671, 424–434 CrossRef CAS.
- J. F. Smith, O. N. Carlson and D. R. R. Avillez, J. Nucl. Mater., 1987, 148, 1–16 CrossRef CAS.
- E. Parthe and K. Yvon, Acta Crystallogr., Sect. B: Struct. Sci., 1970, 26, 153–163 CrossRef CAS.
- E. Rudy and C. E. Brukl, J. Am. Ceram. Soc., 1967, 50, 265–268 CrossRef CAS.
- N. Terao, Jpn. J. Appl. Phys., 1964, 104, 104–111 CrossRef.
- B. Lonnberg and T. Lundstrom, J. Less-Common Met., 1985, 113, 261–268 CrossRef.
- X. Yu, C. R. Weinberger and G. B. Thompson, Comput. Mater. Sci., 2016, 112, 318–326 CrossRef CAS.
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef PubMed.
- A. R. Oganov, C. W. Glass and S. Ono, Earth Planet. Sci. Lett., 2006, 241, 95–103 CrossRef CAS.
- C. W. Glass, A. R. Oganov and N. Hansen, Comput. Phys. Commun., 2006, 175, 713–720 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Bloch, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- A. Togo, Phonopy, https://atztogo.github.io/phonopy/ Search PubMed.
- R. Hill, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349–354 CrossRef.
- Lehrbuch der kristallphysik, ed. W. Voigt, Teubner-Leipzig, New York, Macmillan, 1928, p. 739 Search PubMed.
- A. Reuss, Z. Angew. Math. Phys., 1929, 9, 49–58 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Z. Wu, E. Zhao, H. Xiang, X. Hao, X. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- X. Cheng, H. Niu, D. Li and Y. Li, Intermetallics, 2011, 19, 1275–1281 CrossRef.
- A. Simunek and J. Vackar, Phys. Rev. Lett., 2006, 96, 085501 CrossRef PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05856j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Namin Xiao,
Yongjun Guan and
Xiaosu Yi*
,
Namin Xiao,
Yongjun Guan and
Xiaosu Yi*