
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle enzyme direct biomineralization of ZnS, ZnxCd1−xS and ZnxCd1−xS–ZnS quantum confined nanocrystals†
Abdolhamid Sadeghnejada,
Li Lu b,
Christopher J. Kielyab,
Bryan W. Berger*ac and
Steven McIntosh*a
b,
Christopher J. Kielyab,
Bryan W. Berger*ac and
Steven McIntosh*a
aDepartment of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA 18015, USA. E-mail: mcintosh@lehigh.edu; berger@lehigh.edu
bDepartment of Materials Science and Engineering, Lehigh University, Bethlehem, PA 18015, USA
cProgram in Bioengineering, Lehigh University, Bethlehem, PA 18015, USA
First published on 7th August 2017
Abstract
Biomineralization is an intriguing route towards the low temperature, aqueous phase, green synthesis of inorganic functional nanomaterials. Herein we describe the biomineralization and optical properties of ZnxCd1−xS and ZnxCd1−xS–ZnS quantum confined nanocrystals that have potential application in optoelectronics. The reported biomineralization process is reduced to perhaps its simplest form wherein a single recombinant cystathionine γ-lyase (CSE) enzyme is responsible for catalyzing mineralization within an otherwise inert solution and plays a role in controlling the alloy composition. The biomineralized nanocrystals are sphalerite structured with average diameter below 3 nm. Biomineralization of a passivating ZnS shell on Zn0.73Cd0.27S core nanocrystals is achieved through subsequent addition of Zn precursor. This shell growth increases the photoluminescence quantum yield to 7% and increases the radiative decay time to 97.6 ns compared with 40.9 ns for the core materials.
Introduction
Functional inorganic nanomaterials are utilized in an increasingly diverse range of applications. The synthesis of these materials is typically accomplished in the organic phase at elevated temperature with the addition of a reactive chemical precursor.1 While these conventional approaches enable fine control over nanomaterial composition, crystallinity, size and often shape, and allow us to achieve highly active functional materials, this synthesis methodology inherently leads to significant environmental and thus economic implications for scale-up. In contrast, biological systems mineralize inorganic materials in the aqueous phase under ambient conditions. This has led to a significant interest in developing synthetic biomineralization or bioinspired synthesis routes to generate functional nanomaterials.2,3Nanoparticle biomineralization has been observed to occur within a wide range of bacterial biofilms,4 where the particles are bound within the extracellular matrix of the film and would require substantial purification prior to any technological application. Bacterial ZnS biomineralization has been observed to occur naturally at sites contaminated with high levels of Zn.5–7 For example, Labrenz and Banfield7 reported bacteria-induced ZnS precipitation within anaerobic biofilms formed in mine drainage systems. In addition, Gramp et al. and Bai et al. have both reported on ZnS biomineralization in a laboratory setting utilizing bacteria isolated from natural environments.8,9 In all of these cases, the ZnS is biomineralized under anaerobic conditions and the mineralization process occurs over a period of days or longer. The requirement to (i) purify the nanoparticles from the biofilm bacteria responsible for their synthesis, (ii) use anaerobic processing conditions, and (iii) endure long synthesis times all negatively impact the potential to apply these approaches at an industrial scale.
Perhaps the most widely studied approach to overcoming these barriers, while maintaining aqueous processing conditions, is to utilize a bio-derived or bio-inspired small molecule, typically a peptide, to mediate nanoparticle formation upon addition of a reactive precursor to an aqueous solution. This approach has been successfully applied to a wide range of materials including metals, metal chalcogenides and metal oxides.2
We are pursuing an alternative and more direct approach to biomineralization whereby we isolate, engineer and in a scalable fashion produce a single enzyme that is responsible for the generation of the reactive chemical precursor from an otherwise inert solution and also plays a key role in templating nanomaterial formation.10–14 We have previously demonstrated the application of a putative cystathionine γ-lyase, smCSE, in the direct biomineralization of CdS,10–12 PbS and PbS–CdS core–shell13 quantum confined nanocrystals and an engineered form of silicatein for the biomineralization of CeO2−δ and Ce1−xZrxO2−δ catalytic nanocrystals.14 In this work, we demonstrate the activity role of smCSE towards the biomineralization of ZnS, ZnxCd1−xS and ZnxCd1−xS–ZnS quantum confined nanoparticles.
Experimental
The development, expression and purification of the smCSE enzyme has previously been described in detail elsewhere.10 This enzyme was previously identified as being associated with CdS quantum dots biomineralized utilizing a strain of Stenotrophomonas maltophilia bacteria.12 Briefly, an E. coli codon optimized form of smCSE was expressed within a BL21 strain of E. coli. The E. coli was grown to saturation at 37 °C, prior to dilution to OD600 = 0.8 and induction of expression utilizing 1 mM IPTG followed by incubation for 16 h at 20 °C. Cell lysis was achieved by re-suspending centrifuged cells in lysis buffer and sonicating. Finally, the enzyme was purified utilizing immobilized metal affinity chromatography15 within an imidazole buffer. The purified enzyme was stored at 4 °C in imidazole buffer prior to use.ZnS nanocrystals were grown by incubating a buffered solution (Tris–HCl at pH 9) of zinc acetate (1 mM, Alfa Aesar Puratronic, 99.995% metals basis), L-cysteine (8.25 mM, Spectrum Chemicals, 99.55%), and smCSE enzyme (OD600 = 500) at 37 °C. The UV-vis absorbance spectra of the solutions were collected periodically during this incubation period using a 2600 Shimadzu spectrophotometer with an ISR-2600-Plus integrating sphere. An analogous procedure was utilized for CdS and ZnS–CdS alloy nanocrystal synthesis by varying only the cadmium acetate (1 mM, Alfa Aesar Puratronic, 99.995% metals basis) and zinc acetate molar ratios in solution. The nominal Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cd ratios utilized in the current study were 0:1, 1:1, 2:1, 3:1, 4:1, and 1:0.
:Cd ratios utilized in the current study were 0:1, 1:1, 2:1, 3:1, 4:1, and 1:0.
Following the designated incubation period, the resulting solution was dialyzed (3500 MWCO, Thermo Scientific) against deionized water at 2 °C for one day with the water being refreshed every 6 h. Subsequent ZnS shell biomineralization was achieved using a similar approach. 1 mM zinc acetate, 8.25 mM L-cysteine, and 0.5 OD of smCSE enzyme were added to a dialyzed solution of ZnxCd1−xS core nanocrystals and the solution incubated at 37 °C for 3 h. For comparative purposes, conventional ZnxCd1−xS nanoparticles were chemically precipitated from a 4 mM zinc acetate, 1 mM cadmium acetate and 8.25 mM L-cysteine aqueous solution by addition of 8.25 mM Na2S.
Photoluminescence spectra were collected utilizing a QuantaMaster 400 spectrometer, (Photon Technology International) with excitation wavelength corresponding to the maximum absorbance wavelength. The quantum yields were measured utilizing Coumarin 1 (Sigma-Aldrich) in ethanol as a reference compound following dialysis of the as-synthesized materials.16 Photoluminescence decay times were collected using a Fluorolog Spectrofluorometer equipped with a Deltadiode laser having a peak wavelength of 287 nm.
After synthesis the nanocrystals were sometimes phase transferred into an organic phase of 1/1 v/v oleylamine (98%, Aldrich, primary amine)/1-octadecene (90%, Alfa Aesar) to facilitate sample characterization.11 Phase transfer was accomplished by forcing contact between the aqueous solution and the organic mixture at 70 °C for 3 h by vigorous stirring. The nanoparticles in the organic phase were then precipitated with ethanol, centrifuged at 8500 rpm for 15 min and the resulting pellet was suspended in toluene. Samples for TEM analysis were prepared by drop-casting diluted dialyzed nanocrystal solution onto a holey carbon coated Ni-mesh grid and allowing the solvent to evaporate.
High angle annular dark field (HAADF) imaging and X-ray energy-dispersive spectroscopy (XEDS) were conducted at 200 kV using an aberration corrected JEOL ARM 200CF analytical electron microscope equipped with a JEOL Centurio XEDS system. Spherical equivalent particle size distributions were calculated from HAADF-STEM images using the Gatan Digital Micrograph software. Additional XEDS spectra were collected on centrifuged and dried samples utilizing a Hitachi 4300SE/N FEG-SEM equipped with a light element energy dispersive (EDS) and electron backscatter diffraction (EBSD) camera. Samples were stick to a carbon tape on aluminum holder for SEM analysis at 8 kV.
Results
UV-vis absorption spectra and corresponding photoluminescence spectra obtained from solutions of Zn-acetate, L-cysteine and the smCSE enzyme as a function of incubation time are showed in Fig. 1a. A distinct absorption shoulder at 270 nm was observed after 90 min of incubation. This feature became more distinct and intense with time, yielding a clear absorption peak centered at 280 nm after ∼300 min of incubation. The position of this absorbance peak is consistent with the proposition that ZnS nanocrystals are being biomineralized by the smCSE enzyme in solution.17,18 The reported bulk band gap values of ZnS in the cubic sphalerite form and hexagonal wurtzite form are 3.54 eV (350 nm)19 and 3.94 eV (317 nm),20 respectively. The lower wavelength, higher band gap, absorption maxima for the biomineralized particles is consistent with the formation of quantum confined ZnS nanocrystals. The corresponding emission peak in Fig. 1b shows a large Stoke's shift of ∼115 nm, indicative of photoemission from deep surface traps, and is typical of nanocrystals formed at low temperature in the aqueous phase where surface defects can be significant.21 The fluorescence spectra show decreasing intensity with increasing incubation time, indicative of a gradually decreasing quantum yield (QY). The QY of these as-synthesize pure ZnS nanocrystals was 1.88% for the material formed after 300 min incubation.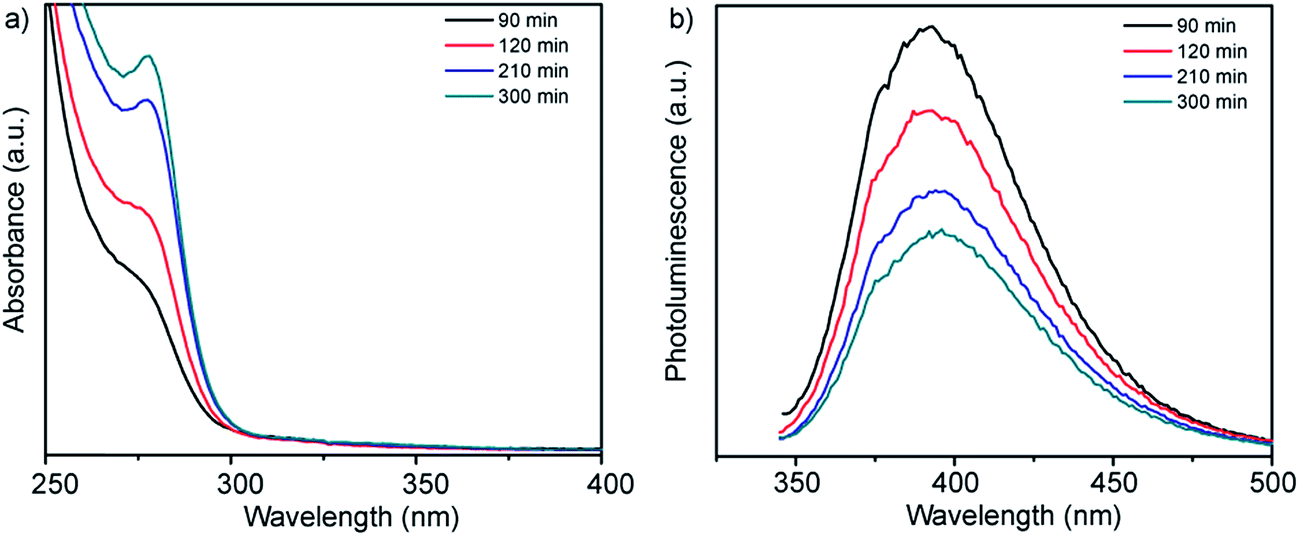 | ||
| Fig. 1 (a) UV-vis absorption and (b) corresponding fluorescence emission spectra of a buffered (pH = 9.0) aqueous solution of Zn-acetate, L-cysteine and smCSE enzyme as a function of incubation time at room temperature. The spectra of solutions incubated in the absence of one or more of these components are provided in Fig. S1† for reference. | ||
These absorbance and fluorescence peaks are not observed for solutions prepared in the absence of any of the primary synthesis ingredients of smCSE, Zn-acetate or L-cysteine, when incubated for 300 min, Fig. S1.† An absorption shoulder at 279 nm and associated photoluminescence peak at 335 nm for solutions containing the enzyme and L-cysteine without any Zn-acetate are in the range expected for tryptophan within the smCSE protein.22 It should be noted that these peaks originating from the enzyme in the reference samples do not change with incubation time.
Direct evidence for the formation of ZnS nanocrystals within the quantum confined size range is provided by HAADF-STEM imaging and XEDS analysis, Fig. 2. The HAADF-STEM images show well defined nanoparticles with clear lattice fringes, Fig. 2a and b. Analysis of the Fast-Fourier Transform (FFT), Fig. 2c, derived from the single nanoparticle shown in Fig. 2b, gives lattice spacings and inter-planar angles that could be consistent with those expected for either the cubic sphalerite ZnS structure viewed along [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ] or the hexagonal wurtzite ZnS structure viewed along [11] or
] or the hexagonal wurtzite ZnS structure viewed along [11] or 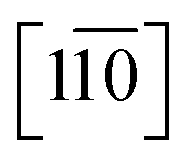 , Fig. S2.†
, Fig. S2.†
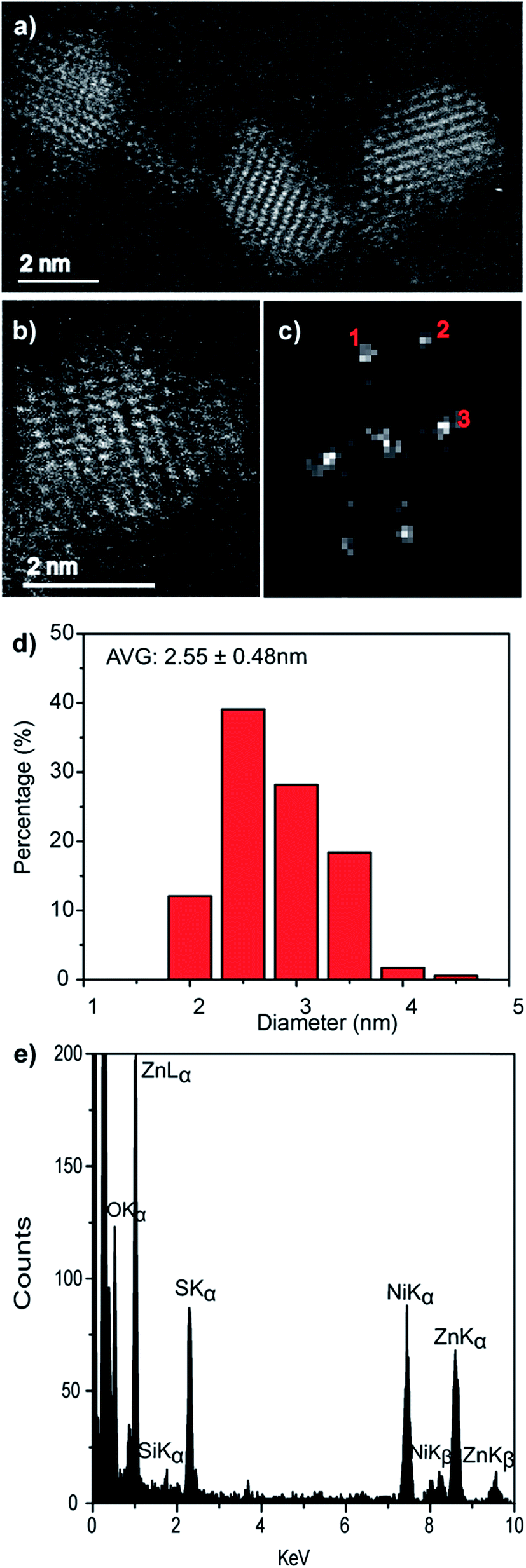 | ||
| Fig. 2 (a and b) Representative HAADF-STEM images, (c) the corresponding FFT pattern from (b), (d) particle size distribution and (e) single particle STEM-XEDS spectrum of biomineralized ZnS nanocrystals. | ||
Particle size analysis of 175 nanoparticles gave a spherical equivalent average diameter of 2.55 ± 0.48 nm, Fig. 2d, indicating that the ZnS nanocrystals diameter is close to the excitation Bohr radius.23 Final confirmation that the nanocrystals are indeed ZnS was provided from STEM-XEDS analysis which showed the presence of both Zn and S in individual particles, Fig. 2e. The Ni and Si peaks in this spectrum are artefacts and originate from the Ni-based TEM grid and the Si-based XEDS detector, respectively.
We have previously reported that smCSE is active towards biomineralization of analogous CdS quantum confined nanocrystals from Cd acetate containing solutions.10 Intriguingly, incubation of smCSE in a mixture of Cd and Zn acetate for 3 h leads to material with absorption and photoluminescence spectra peaks between those expected for the pure CdS and ZnS nanocrystals, Fig. 3. This is consistent with the formation of ZnxCd1−xS alloy nanocrystals where the band gap will lie between that of CdS and ZnS.24 For example, the absorption peak for the ZnxCd1−xS preparation shown in Fig. 3 (with nominal x = 0.8) is at 335 nm, which lies between the 280 nm and 395 nm peaks of the pure ZnS and CdS nanocrystals, respectively. Similarly, the photoluminescence peak from the same sample lies at 450 nm, again lying between the 390 nm and 530 nm peaks of the corresponding ZnS and CdS nanocrystals. SEM-based EDX analysis of the centrifuged and dried agglomerate powder from this sample gave an overall composition of Zn0.73Cd0.27S, Fig. S3,† which is slightly different from the nominal precursor composition of Zn0.80Cd0.20S. A Tauc plot analysis of the optical data gives direct band gap values of 2.8, 3.4, and 4.1 eV for the CdS, Zn0.73Cd0.27S, and ZnS nanoparticles, respectively,25 Fig. S4a.† The quantum yield of the biomineralized Zn0.73Cd0.27S alloy nanoparticles was 5.21%. For comparison, we have previously reported typical quantum yields of 1.8% for the biomineralized CdS nanocrystals.10
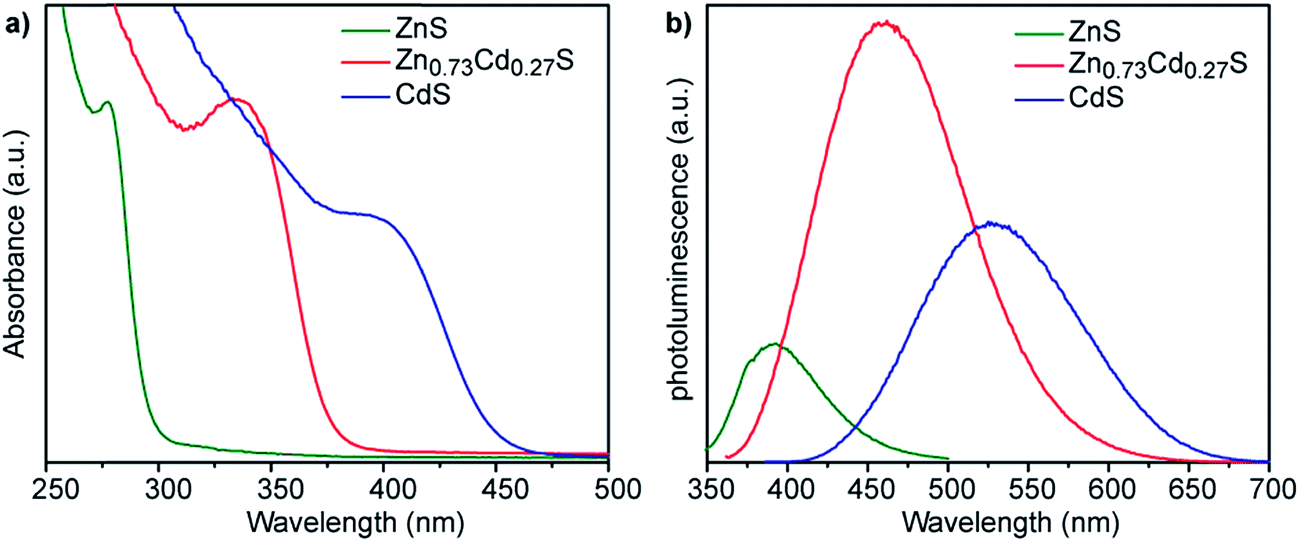 | ||
| Fig. 3 (a) UV-vis absorption and (b) corresponding fluorescence emission spectra of nanocrystals grown with 1 ml mol−1 of either Zn acetate (green lines), Cd acetate (blue lines), or a nominal 4:1 ratio of Zn:Cd acetate (red lines). The alloy composition Zn0.73Cd0.27S was determined from XEDS analysis, Fig. S3.† | ||
As with the pure ZnS nanocrystals, HAADF-STEM imaging demonstrated that the Zn0.73Cd0.27S alloy particles are crystalline, Fig. 4a and b. Analysis of the FFT pattern, Fig. 4c, from a single particle, Fig. 4b, demonstrates a good fit with the sphalerite crystal structure viewed along [001] with d-spacing in good agreement with those calculated using a simple Vegard's law approach for Zn0.79Cd0.21S, Fig. S5.† The size distribution, Fig. 4d, measured from 183 particles indicated an average spherical equivalent diameter of 2.70 ± 0.44 nm. Final confirmation of alloy formation was provided by STEM-XEDS analysis, Fig. 4e, results of an individual particle that demonstrates the co-existence of Cd, Zn and S within a single particle. Quantitative analysis of the XEDS spectrum from this individual particle yielded a composition of Zn0.79Cd0.21S, which is in reasonable agreement with the Zn0.73Cd0.27S average composition obtained from SEM based XEDS analysis of a much larger number of particles.
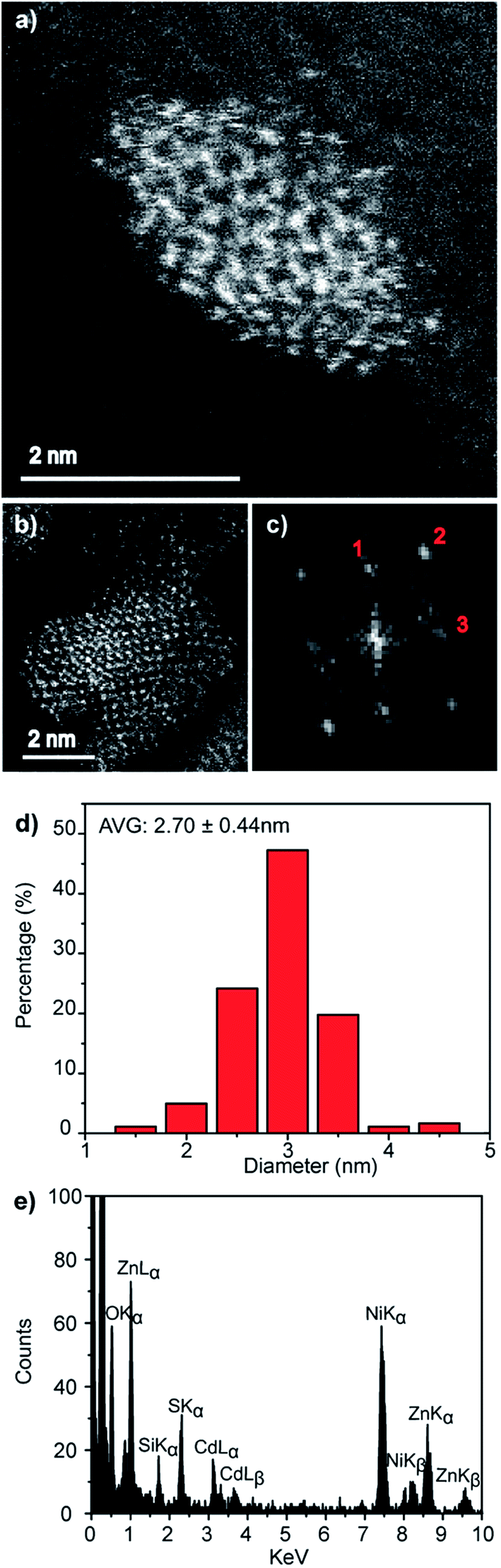 | ||
| Fig. 4 (a and b) Representative HAADF-STEM images, (c) corresponding FFT pattern from (b), (d) particle size distribution and (e) individual particle STEM-XEDS spectrum for Zn0.73Cd0.27S nanocrystals biomineralized from a precursor solution containing a nominal 4:1 Zn:Cd molar ratio. | ||
We also investigated if the alloy composition in the biomineralized ZnxCd1−xS particles can be tuned by varying the Zn:Cd ratio in the precursor solutions. Fig. 5 shows the optical properties of nanocrystals obtained from solutions having nominal 1:1, 2:1, 3:1, 4:1 Zn:Cd ratios following 3 h incubation. All of the materials show clear absorption peaks with peak positions that progressively blue-shift from 385 to 335 nm with increasing Zn content, which is in agreement with the expected trend for ZnxCd1−xS alloy particles that are getting more Zn-rich. The corresponding photoluminescence spectra show a similar trend, with peak position shifting from 528 to 478 nm as the nominal Zn:Cd ratio varies from 1:1 to 4:1. SEM based XEDS analysis of the dried nanocrystal aggregates yielded average compositions of Zn0.38Cd0.62S, Zn0.61Cd0.39S, Zn0.65Cd0.35S, and Zn0.73Cd0.27S, for the nanocrystals biomineralized from precursor solutions with 1:1, 2:1, 3:1, 4:1 Zn:Cd ratios, respectively, Fig. S3.† Comparison of the relative ratios of absorption to photoluminescence intensities of these other compositions to those for Zn0.73Cd0.27S suggests that the quantum yield of these materials will be similar to or lower than the 5.21% determined for Zn0.73Cd0.27S. A Tauc plot analysis, Fig. S4b,† gave direct band gap values of 2.96, 3.02, 3.19, and 3.40 eV, respectively, for this systematic set of samples, with the band gap increasing as the Zn content in the ZnxCd1−xS nanocrystals increases.26,27
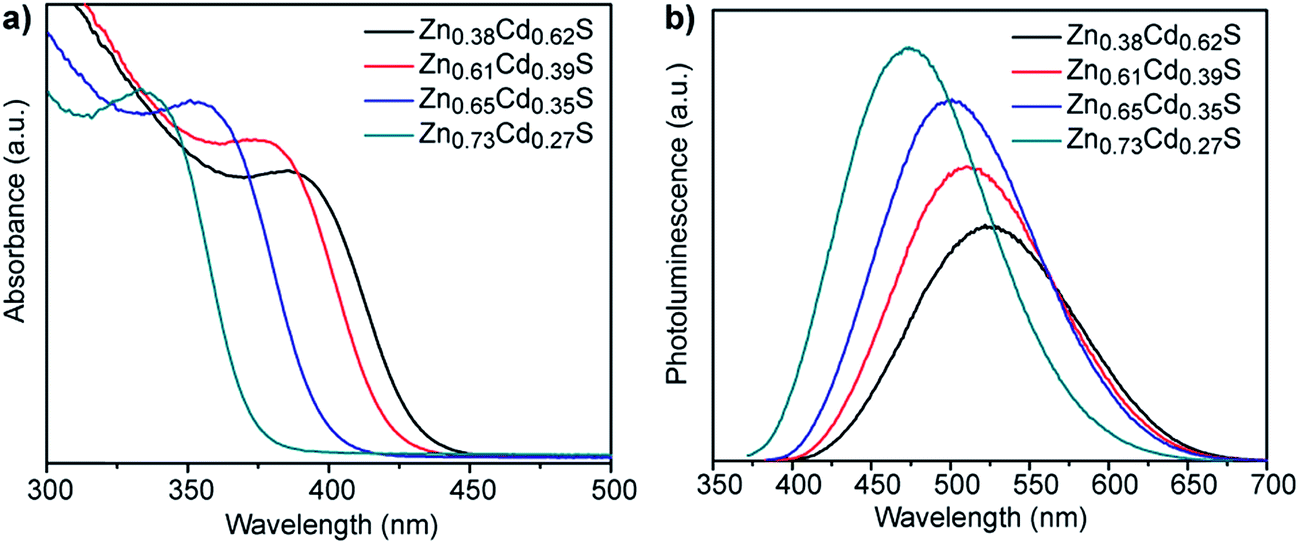 | ||
| Fig. 5 (a) UV-vis absorption and (b) corresponding fluorescence emission spectra of ZnxCd1−xS QDs of various compositions synthesized using different molar ratios of zinc acetate to cadmium acetate. | ||
Intriguingly, while the overall composition trend reflective of the increasing Zn content in solution, the SEM-EDX determined compositions of the biomineralized nanocrystals are not the same as the nominal Zn:Cd precursor ratio in solution. This differs from the route involving direct chemical synthesis from a mixed solution where the measured ZnxCd1−xS alloy composition is a fairly close match to the composition of the precursor solution. For example, direct reaction of Na2S with a 4:1 Zn:Cd molar ratio of the metal acetates in water produces particles with an average composition of Zn0.80Cd0.20S, Fig. S6.† Note that this chemical precipitation is performed in the presence of L-cysteine such that the only difference between the biomineralization and this approach is the presence and use of the enzyme to create the reactive sulfur species in biomineralization. Any influence of L-cysteine-metal complex formation on nanocrystal composition should be the same for both approaches.
ZnS shells are commonly grown onto CdS and ZnxCd1−xS alloy core materials to improve photoluminescent quantum yield.28–30 The higher band gap of ZnS relative to the core aids in confining the exciton in the core while the small lattice mismatch between the materials aids in shell formation.30 Fig. 6a shows the change in absorbance and photoluminescence spectra upon secondary incubation of as-synthesized core shell Zn0.73Cd0.27S particles in a solution of Zn acetate, L-cysteine and smCSE enzyme. The overall decrease is absorption peak intensity of these nanocrystals relative to the pure core material is simply due to dilution upon transfer to the shell growth medium. The measured red-shift in both the absorption and photoluminescence peaks, the 12 nm reduction in Stoke's shift, and the increase in photoluminescence intensity are all consistent with the formation of a ZnS on the surface of the core particles.30 The measured quantum yield of these Zn0.73Cd0.27S–ZnS particles increases to 7.02%, in contrast with the 5.21% determined for the Zn0.73Cd0.27S core particles.
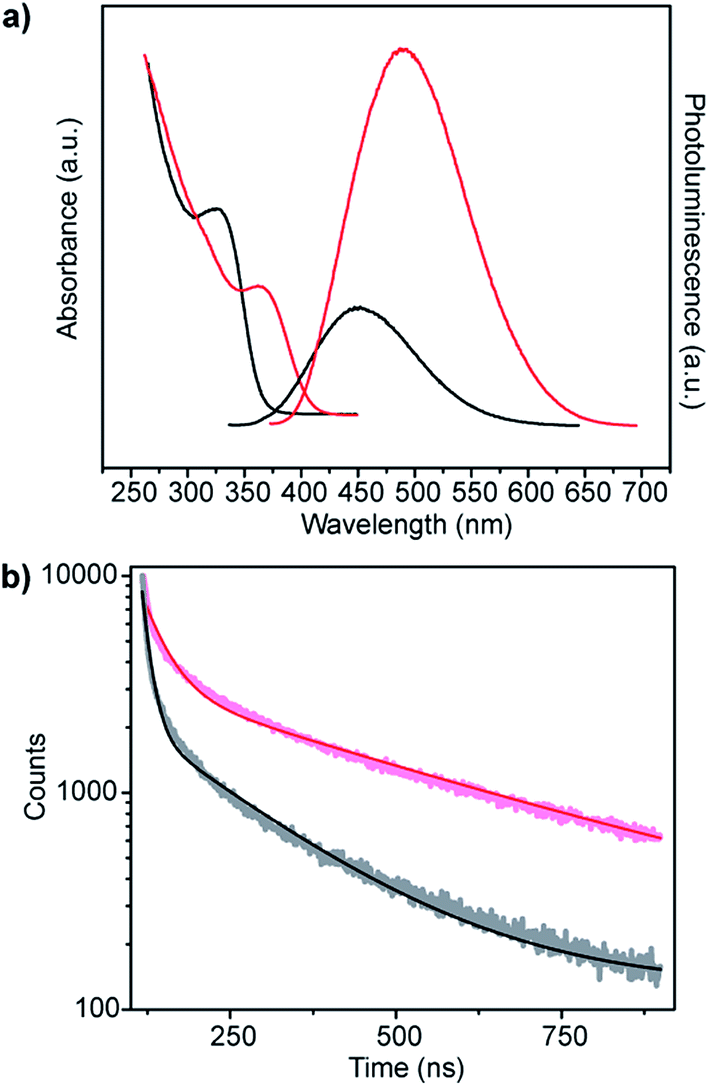 | ||
| Fig. 6 (a) Absorption and corresponding photoluminescence spectra and (b) fluorescence decay-time measurements of Zn0.73Cd0.27S core (black) and Zn0.73Cd0.27S–ZnS (red) biomineralized nanocrystals. | ||
Analysis of the photoluminescence decay profile shown in Fig. 6b provides further evidence for the formation of a passivating ZnS shell on the Zn0.73Cd0.27S core. The double-exponential fitting procedure we and others have employed assigns the faster decay step, τ1, to non-radiative decay and the slower decay step, τ2, to the radiative process.31–33 The radiative recombination rate of the Zn0.73Cd0.27S–ZnS nanocrystals was determined to be 97.6 ns, which is significantly longer than the 40.9 ns measured for the bare Zn0.73Cd0.27S core crystals, Table 1.
![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif)
An electron microscopy analysis of the Zn0.73Cd0.27S–ZnS particles is presented in Fig. 7, with representative HAADF-STEM images shown in Fig. 7a and b. Further analysis of lattice fringes presents in the individual particle shown in Fig. 7b and its corresponding FFT transform, Fig. 7c, gives a reasonable fit to the wurtzite structure viewed along [212] with fringe spacings and interplanar angles in good agreement with those predicted for a Zn0.73Cd0.27S alloy as predicted from a Vegard's law estimate, Fig. S7.† The presence of a ZnS shell is also indicated suggested by a measurable increase in average size to 3.02 nm, Fig. 7d, from the initial 2.70 nm value found for the core-only particles. Taking the ZnS wurtzite c lattice parameter as c 0.626 nm,34 this increase in diameter corresponds to an approximate sub-monolayer ZnS decoration on the core surface. Thus we must be cautious about the use of the word shell to describe what is more accurately described as a surface enrichment of ZnS. STEM-XEDS compositional analysis, Fig. 7e, of an isolated Zn0.73Cd0.27S–ZnS single particle demonstrates the co-existence of Cd, Zn, and S in the nanocrystal. Thus, while a clear shell is not directly discernable in the HAADF-STEM images of the Zn0.73Cd0.27S–ZnS particles due to the small size of the particles and the thin incomplete shell coverage, the increase in quantum yield, average diameter, and photoluminescence time are all consistent with the formation of some sort of passivating surface layer.
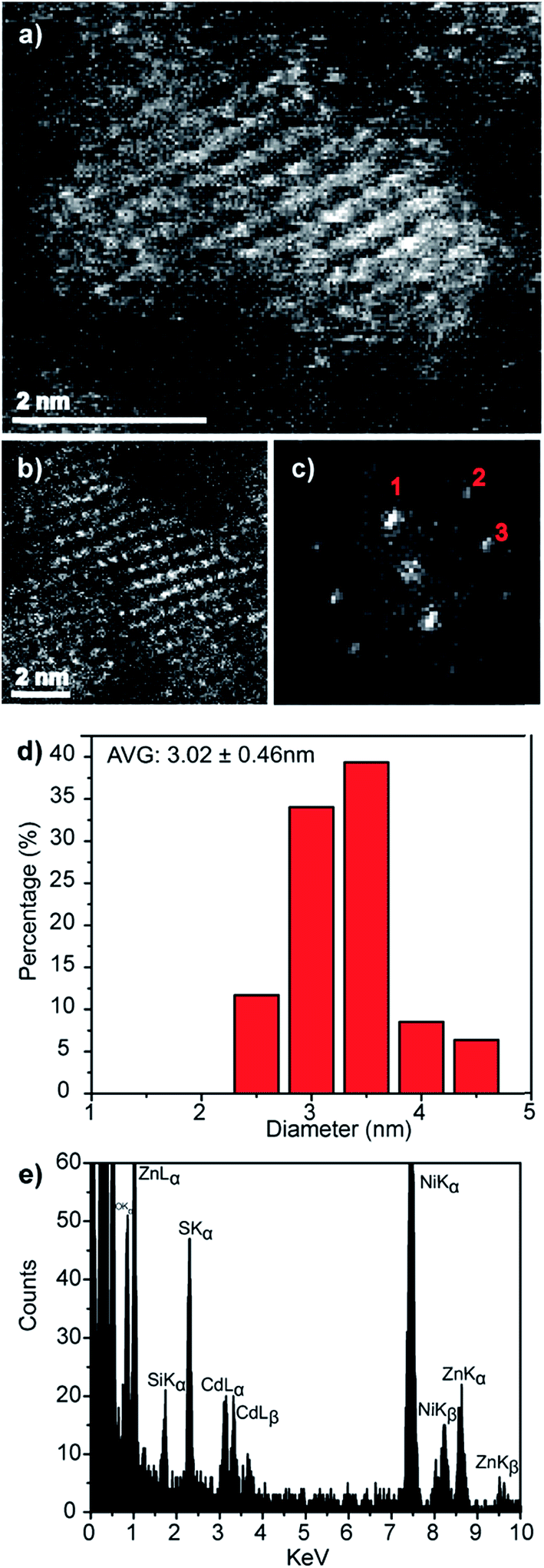 | ||
| Fig. 7 (a and b) Representative HAADF-STEM images of Zn0.73Cd0.21S–ZnS QDs, (c) corresponding FFT from the particle in (b) that can be fitted to the wurtzite structure viewed along the [21] projection, Fig. S7,† (d) corresponding spherical equivalent nanocrystal size distribution from measurements on over 100 particles and (e) XEDS spectrum demonstrating the co-existence of Zn, Cd and S in an individual nanocrystal. | ||
Discussion
The optical properties of the biomineralized ZnS nanocrystals produced in this study are in good agreement with those reported for chemically synthesized ZnS particles of the same size. For example, Ghatak et al. report an absorbance peak at 287 nm for particles with a calculated mean diameter of 2.3 nm, which is within the error limits of our 2.55 ± 0.48 nm ZnS particles which display an absorbance peak at 280 nm.35 Similarly, Li et al. reported an absorption peak at 276 nm for ZnS particles with crystallite size of 2.62 nm, as calculated by a Scherrer analysis of their XRD data.36 The growth in intensity and slight red-shifting of this absorbance peak with increasing incubation time in our biomineralization process is indicative of a gradually increasing average particle size, which is similar to the trend observed in our previous reports for CdS biomineralization.10Manzoor et al. have ascribed the photoluminescence displayed by ZnS nanocrystals to arise primarily from transitions between S vacancies acting as electron traps below the conduction band, and Zn vacancies or surface states.37 The central luminescence peak position of 434 nm reported by Manzoor et al. is higher than the 395 nm peak position noted for our materials. This discrepancy is unlikely to be due to differences in the degree of quantum confinement as the materials from the Manzoor paper had a smaller average crystallite size (2 nm) than those in this work, (2.55 nm), which would be expected to lead to an increase in band gap. Tang et al. synthesized ZnS nanocrystals with a 2.9 nm average crystallite size which displayed dual photoluminescence peaks at 385 and 405 nm due to (i) interaction of the capping agent and surface Zn2+ and (ii) recombination of electrons at surface S vacancy traps with holes at surface Zn vacancies, respectively.38 By way of contrast, Lu et al. ascribed a similar photoluminescence peak at 391 nm, albeit for ZnS crystallites larger than 3.6 nm, to a direct recombination event from the conduction band to Zn vacancies with an additional peak at 422 nm assigned to recombination from a S vacancy to a hole in the valence band.39 The involvement of a surface state in the observed photoluminescence is in good agreement with our data, where the peak position is found to be essentially independent of incubation time. These observations on our biomineralized materials are in good agreement with the work of Tang et al. who also reported a similar lack of shift in photoluminescence peak position with increasing ZnS crystallite size.38
Co-incubation of Zn acetate and Cd acetate causes a decrease in band gap in the resultant ZnxCd1−xS nanoparticles relative to the pure ZnS nanocrystals (Fig. 3), exhibiting a band gap value between that of pure CdS and ZnS, indicative of biomineralization of crystalline alloy nanocrystals.40 This alloy formation is confirmed by the single particle STEM-XEDS data. The composition of these alloy nanocrystals can be controlled to some extent through manipulation of the Zn:Cd precursor ratio in the original reaction solution. Thus, smCSE is in principle capable of catalyzing the formation of ZnxCd1−xS nanocrystals with compositions spanning the entire range from ZnS to CdS. The calculated quantum yield of 5.21% for our biomineralized Zn0.73Cd0.27S material is similar to that reported for aqueous phase chemically synthesized ZnxCd1−xS materials.41
The growth of a wider band gap ZnS shell on the surface of ZnxCd1−xS alloy core nanocrystals leads to surface passivation and an associated increase in both quantum yield and exciton lifetime. Unfortunately, we are unable to form a thick shell on the core materials purely via secondary biomineralization of ZnS. The relatively thin shell layer and the lack of mass-contrast between Cd and Zn prohibits direct imaging of the ZnS outer layer. Indirect evidence for shell growth, rather than Zn substitution to increase the Zn fraction in the ZnxCd1−xS alloy core, comes from the observed increase in quantum yield, average particle diameter, and red-shift in both absorbance and photoluminescence peaks. A blue-shift in the opposite direction would be expected if the Zn content of the core were simply increasing, as is frequently reported42,43 and demonstrated by us in Fig. 5.
The biomineralization mechanism in play here is based on the enzymatic turnover of the sulfur containing amino acid L-cysteine to form H2S, NH3 and pyruvate in solution. smCSE is a member of the cystathionine γ-lyase class of enzymes that are widely reported as active for this reaction.44,45 This H2S then goes on to react with the metal cations in solution to form, in this case, ZnS, CdS or ZnxCd1−xS. The development of this enzymatic biomineralization approach, with reference to CdS biomineralization, is discussed in a previous publication.10 The same mechanism of biomineralization of smCSE is realized in our previous works to react with other metal cations and synthesize nanocrystals such as PbS,13 CulnS2 and (CuInZn)S2.46 However, simply adding H2S, or more easily Na2S, to an aqueous solution of metal cations leads to the formation of bulk material rather than nanocrystals. Nanocrystal formation requires a templating agent to arrest growth. While L-cysteine can play a role as a capping agent, we have previously demonstrated a propensity for the smCSE enzyme itself to also template nanocrystal growth.10
A more active role for the enzyme in the formation of the ZnxCd1−xS nanocrystals reported in this study is supported by the finding that the alloy nanocrystal compositions are not identical to the ratio of precursors in solution. This is in contrast to the result obtained upon direct chemical precipitation via Na2S, even in the presence of L-cysteine, where the ZnxCd1−xS alloy composition is much closer to that of the precursor solution, Fig. S6.† That is, the mechanism of nanocrystal formation is more complex than the indiscriminate precipitation of metal sulfides from solution that typically occurs upon direct precipitation of metal sulfides.47 Developing a deeper understanding of this additional active role of the enzyme in mediating particle synthesis is a focus of ongoing work.
Conclusions
The smCSE enzyme has been demonstrated to be active for the direct co-biomineralization of ZnS and CdS to form quantum confined ZnxCd1−xS nanocrystals in aqueous solution at room temperature under aerobic conditions. The composition of the resulting ZnxCd1−xS alloy nanocrystals can be tuned through adjusting the ratio of cadmium acetate and zinc acetate precursors in solution. However, in contrast to direct chemical precipitation of an alloy material, the ratio of Zn:Cd in the nanocrystals is not the same as the precursor solution indicating an additional role for the enzyme in controlling the particle composition. Sequential biomineralization of ZnS on a ZnxCd1−xS nanocrystal enables the formation of ZnS surface layer on the nanocrystals to improve quantum yield.
Acknowledgements
This material is based upon work supported by the National Science Foundation under the EFRI-PSBR program, Grant No. 1332349.References
- Y. Yin and D. Talapinb, Chem. Soc. Rev., 2013, 42, 2484–2487 RSC.
- C.-Y. Chiu, L. Ruan and Y. Huang, Chem. Soc. Rev., 2013, 42, 2512–2527 RSC.
- M. Sarikaya, C. Tamerler, A. K. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- K. Ikuma, A. W. Decho and B. L. T. Lau, Front. Microbiol., 2015, 6, 1–6 Search PubMed.
- J. W. Moreau, R. I. Webb and J. F. Banfield, Am. Mineral., 2004, 89, 950–960 CrossRef CAS.
- G. Vitor, T. C. Palma, B. Vieira, J. P. Lourenço, R. J. Barros and M. C. Costa, Miner. Eng., 2015, 75, 85–93 CrossRef CAS.
- M. Labrenz and J. F. Banfield, Microb. Ecol., 2004, 47, 205–217 CAS.
- J. P. Gramp, J. M. Bigham, K. Sasaki and O. H. Tuovinen, Geomicrobiol. J., 2007, 24, 609–614 CrossRef CAS.
- H. J. Bai, Z. M. Zhang and J. G. Gong, Biotechnol. Lett., 2006, 28, 1135–1139 CrossRef CAS PubMed.
- R. Dunleavy, L. Lu, C. J. Kiely, S. Mcintosh and B. W. Berger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5275–5280 CrossRef CAS PubMed.
- Z. Yang, L. Lu, C. J. Kiely, B. W. Berger and S. Mcintosh, Ind. Eng. Chem. Res., 2016, 55, 11235–11244 CrossRef CAS.
- Z. Yang, L. Lu, V. F. Berard, Q. He, C. J. Kiely, B. W. Berger and S. McIntosh, Green Chem., 2015, 17, 3775–3782 RSC.
- L. C. Spangler, L. Lu, C. J. Kiely, B. W. Berger and S. Mcintosh, J. Mater. Chem. A, 2016, 4, 6107–6115 CAS.
- C. D. Curran, L. Lu, Y. Jia, C. J. Kiely, B. W. Berger and S. Mcintosh, ACS Nano, 2017, 11, 3337–3346 CrossRef CAS PubMed.
- K. J. Petty, Curr. Protoc. Protein Sci., 2001, 4, 1–16 Search PubMed.
- G. Jones, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294–300 CrossRef CAS.
- C. Unni, D. Philip and K. G. Gopchandran, Opt. Mater., 2009, 32, 169–175 CrossRef CAS.
- N. Kumbhojkar, V. V. Nikesh, A. Kshirsagar and S. Mahamuni, J. Appl. Phys., 2000, 88, 6260–6264 CrossRef CAS.
- B. Bochev and G. Yordanov, Colloids Surf., A, 2014, 441, 84–90 CrossRef CAS.
- T. K. Bergstressr and M. L. Cohen, Phys. Rev., 1967, 164, 1069–1080 CrossRef.
- A. L. Rogach, A. Kornowski, M. Gao and A. Eychmu, J. Phys. Chem. B, 1999, 103, 3065–3069 CrossRef CAS.
- J. T. Vivian and P. R. Callis, Biophys. J., 2001, 80, 2093–2109 CrossRef CAS PubMed.
- J. Trajic, R. Kostic, N. Romcevic, M. Romcevic, M. Mitric, V. Lazovic, P. Balaz and D. Stojanovic, J. Alloys Compd., 2015, 637, 401–406 CrossRef CAS.
- R. Mariappan, M. Ragavendar and V. Ponnuswamy, J. Alloys Compd., 2011, 509, 7337–7343 CrossRef CAS.
- K. Bera, S. Saha, C. Rana and P. c. Jana, Phys. Sci. Int. J., 2016, 10, 1–6 CrossRef.
- N. Safta, A. Sakly, H. Mejri and Y. Bouazra, Eur. Phys. J. B, 2006, 51, 75–78 CrossRef CAS.
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, Prog. Mater. Sci., 2011, 56, 175–287 CrossRef CAS.
- W. K. Bae, M. K. Nam, K. Char and S. Lee, Chem. Mater., 2008, 20, 5307–5313 CrossRef CAS.
- A. Datta, S. K. Panda and S. Chaudhuri, J. Phys. Chem. C, 2007, 111, 17260–17264 CAS.
- J. S. Steckel, J. P. Zimmer, S. Coe-sullivan, N. E. Stott, V. Bulovic and M. G. Bawendi, Angew. Chem., Int. Ed., 2004, 43, 2154–2158 CrossRef CAS PubMed.
- A. C. Berends, F. T. Rabouw, F. C. M. Spoor, E. Bladt, F. C. Grozema, A. J. Houtepen, L. D. A. Siebbeles and C. D. M. Donega, J. Phys. Chem. Lett., 2016, 7, 3503–3509 CrossRef CAS PubMed.
- L. Liu, S. Hu, Y. Pan, J. Zhang, Y. Feng and X. Zhang, Beilstein J. Nanotechnol., 2014, 5, 919–926 CrossRef PubMed.
- S. Cao, J. Zheng, J. Zhao, Z. Yang, M. Shang, C. Li, W. Yang and X. Fang, Adv. Funct. Mater., 2016, 26, 7224–7233 CrossRef CAS.
- X. Liu, Y. Jiang, F. Fu and W. Guo, Mater. Sci. Semicond. Process., 2013, 16, 1723–1729 CrossRef CAS.
- A. Ghatak, G. H. Debnath and P. Mukherjee, RSC Adv., 2015, 5, 32920–32932 RSC.
- Y. Li, Y. Ding, Y. Zhang and Y. Qian, J. Phys. Chem. Solids, 1999, 60, 13–15 CrossRef CAS.
- K. Manzoor, S. R. Vadera, N. Kumar and T. R. N. Kutty, Mater. Chem. Phys., 2003, 82, 718–725 CrossRef CAS.
- H. Tang, G. Xu, L. Weng, L. Pan and L. Wang, Acta Mater., 2004, 52, 1489–1494 CrossRef CAS.
- H. Lu, S. Chu and S. Tan, J. Cryst. Growth, 2004, 269, 385–391 CrossRef CAS.
- J. Ouyang, C. I. Ratcliffe, D. Kingston, B. Wilkinson, J. Kuijper, X. Wu, J. A. Ripmeester and K. Yu, J. Phys. Chem. C, 2008, 112, 4908–4919 CAS.
- C. Xing, Y. Zhang, W. Yan and L. Guo, Int. J. Hydrogen Energy, 2006, 31, 2018–2024 CrossRef CAS.
- M. R. Kim, Y. Kang and D. Jang, J. Phys. Chem. C, 2007, 111, 18507–18511 CAS.
- R. Xie, U. Kolb, J. Li, T. Basche and A. Mews, J. Am. Chem. Soc., 2005, 127, 7480–7488 CrossRef CAS PubMed.
- H. Nakagawa and H. Kimura, Biochem. Biophys. Res. Commun., 1968, 32, 208–214 CrossRef CAS.
- F. C. Brown and P. H. Gordon, Can. J. Biochem., 1971, 49, 484–491 CrossRef CAS PubMed.
- L. C. Spangler, R. Chu, L. Lu, C. J. Kiely, B. W. Berger and S. McIntosh, Nanoscale, 2017, 9, 9340–9351 RSC.
- N. Karar, F. Singh and B. R. Mehta, J. Appl. Phys., 2004, 95, 656–660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05586b |
| This journal is © The Royal Society of Chemistry 2017 |