
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic studies en route to the first total synthesis of a naturally occurring quinone from Acorus gramineus, iso-merrilliaquinone, iso-magnoshinin and 2-epi-3,4-dihydro magnoshinin†
Dileep Kumar,
Oindreela Das,
Manvendra Singh Kaurav and
Tabrez Khan *
*
Organic Synthesis Laboratory, School of Basic Sciences, Indian Institute of Technology Bhubaneswar, Argul, Jatni-752050, Odisha, India. E-mail: tabrez@iitbbs.ac.in; Tel: +91 706 4419592
First published on 25th July 2017
Abstract
A unified synthetic strategy based on [4 + 2] cycloaddition of readily accessible coupling fragments was conceptualized for the racemic total synthesis of recently isolated bioactive merrilliaquinone and other closely related quinones such as gramineusquinone B as well as neolignan, magnoshinin. The present article discloses our efforts in this direction which culminated in the total syntheses of gramineusquinone B, iso-merrilliaquinone, iso-magnoshinin and 2-epi-3,4-dihydro magnoshinin.
Introduction
Illicium merrillianum is a small tree or shrub which is endemic to southwestern China and Burma. Besides using the bark, root bark and fruit of this shrub in Traditional Chinese Medicine (TCM) for treating rheumatism,1 it is also found to possess a vast plethora of structurally diverse and unique bioactive natural products.2 Since the onset of the 21st century, Fukuyama and co-workers have been actively engaged in the isolation and bioactivity screening of several anislactone-type sesquiterpenes, allo-cedrane-type sesquiterpenes and seco-prizizaane type sesquiterpenoids from I. merrillianum and other Illicium species.2 Indeed, their pioneering efforts in this arena led to the discovery of potent neurite outgrowth promoters such as merrilactone A,3 11-O-debenzoyl tashironin4 and jiadifenolide5 which garnered significant world-wide attention from the scientific community6–8 with the scope of their application in treatment of neuro-degenerative diseases such as Alzheimer's and Parkinson's.Further, in continuous pursuit for new bioactive compounds as potential leads for drug discovery, very recently Zhang et al. got interested in exploring the leaves and branches of I. merrillianum which ended up with the isolation of some unique abietane diterpenoids9 as well as some bioactive quinoid natural products.10 Perhaps, it was the bioactivity of the (+) antipode of merrilliaquinone (1), Fig. 1, reported by them and accessed via chiral HPLC separation from the racemic natural product that caught our attention. Interestingly, the (+) antipode of merrilliaquinone was found to be several fold more potent and selective towards human hepatoma cells over normal hepatic cells in comparison to the (−) antipode or the racemic form of the natural product.10 Another, interesting fact related to 1 is, it was also reported in the racemic form by Lu et al. as acortatarinowin H from the rhizomes of Acorus tatarinowii Schott just after the report by Zhang et al.11 Further, we were pleasantly surprised to learn that there exist structurally closely related quinones such as 2–4 and neolignans 5, 6 as captured in Fig. 1, all having their origins from herbs used in TCM. For instance, quinones 2 and 3 were of late isolated in optically pure form from the rhizomes of Acorus gramineus, and reported to exhibit decent anti-inflammatory as well as cytotoxicity against certain human cancer cell lines.12 For convenience sake, we have preferred to refer them as gramineusquinone A and B respectively instead of the conventional name assigned by Lee. et al.12 On the other hand, with no bioactivity so far reported gracillisquinone (4) was isolated not long ago from Spallerocarpus gracilli.13 While among the neolignan 5 and 6, magnoshinin with prominent anti-inflammatory activity was reported in 1983 after its isolation from the flower buds of Magnolia salicifolia, more famous as “Shin-i” in TCM.14 Whereas, asarolignan G (6) with anti-neuroinflammatory activity is a very recently surfaced neolignan from the rhizomes of Acorus tatarinowii.15
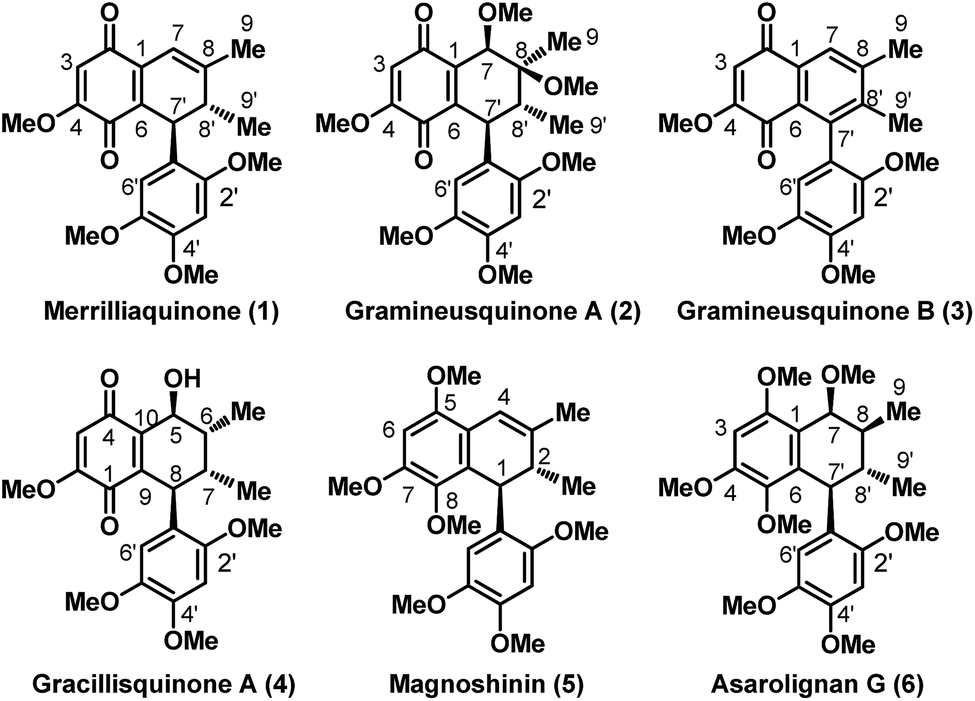 | ||
| Fig. 1 Representative examples of bioactive quinones and neolignans isolated from herbal plants used in Traditional Chinese Medicine (TCM) [indicated numbering as per the isolation paper]. | ||
The impressive bioactivity profile exhibited by 1 though in one of its enantiomeric pure form, as well as close structural resemblance with quinones 2–4, together with a strong prospect to elaborate them to neolignans 5 and 6 engaged our interest towards these natural products. Further, the fact that except for 5 (ref. 16a–c) there exists no synthetic endeavors towards any of these natural products motivated us to embark on a synthetic journey directed towards the racemic total syntheses of these bioactive quinones and neolignans to access them in substantial quantities for thorough clinical assessment. Moreover, initially we aimed to develop a concise unified approach based on [4 + 2] cycloaddition strategy taking inspiration from our earlier success in application of similar approach in context of syntheses of quinoid natural products such as acremine G17 and allomicrophyllone.18
Results and discussion
To test the viability of the approach we had in mind, we initially chose to focus on the development of a racemic synthesis for 1, 3 and 5 before targeting other interested natural product depicted in Fig. 1. In this context, our retrosynthetic plan is as outlined in Scheme 1, which hinges on a Diels–Alder coupling of quinone 9 with either of the arylated diene 10a/b to arrive at the [4 + 2] cycloadduct 11a/b in a highly regioselective fashion. While, the two coupling partner 9 and 10 were envisioned to be accessed through a common and readily available cheap starting material in the form of quinol 7 or its corresponding dimethyl ether 8. A 4–6 step synthetic manipulation on 8 was expected to offer the dienes 10a/b in contrast to a single step access of 9 from 7/8 through known protocol. We anticipated the cycloadduct 11a/b to hold immense potential in context of its elaboration to the targeted natural product. As a simple oxidation of the dihydroquinone in 11b to quinone would end up in the acquisition of 14 isomeric to 1, having C8![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C8′ bond position instead of the C7C8 bond position present in 1, whereas a straightforward oxidative aromatization in the quinone fused cyclohexene ring of 11b would lead to the natural quinone 3. While, reductive methylation and then selective benzylic methylene oxidation at C-7 position of 11a followed by stereoselective installation of the methyl group at C-8′ position was the route we had in mind to arrive at an intermediate like 12 for accessing 5. Similarly, reductive silylation and then selective benzylic methylene oxidation at C-7 position of 11b followed by stereoselective C-8, C-8′ double bond reduction was the route planned to access the functionally well placed intermediate like 13 which could be readily elaborated to either of the natural product 1 or 5 through some straightforward synthetic maneuvers.
C8′ bond position instead of the C7C8 bond position present in 1, whereas a straightforward oxidative aromatization in the quinone fused cyclohexene ring of 11b would lead to the natural quinone 3. While, reductive methylation and then selective benzylic methylene oxidation at C-7 position of 11a followed by stereoselective installation of the methyl group at C-8′ position was the route we had in mind to arrive at an intermediate like 12 for accessing 5. Similarly, reductive silylation and then selective benzylic methylene oxidation at C-7 position of 11b followed by stereoselective C-8, C-8′ double bond reduction was the route planned to access the functionally well placed intermediate like 13 which could be readily elaborated to either of the natural product 1 or 5 through some straightforward synthetic maneuvers.
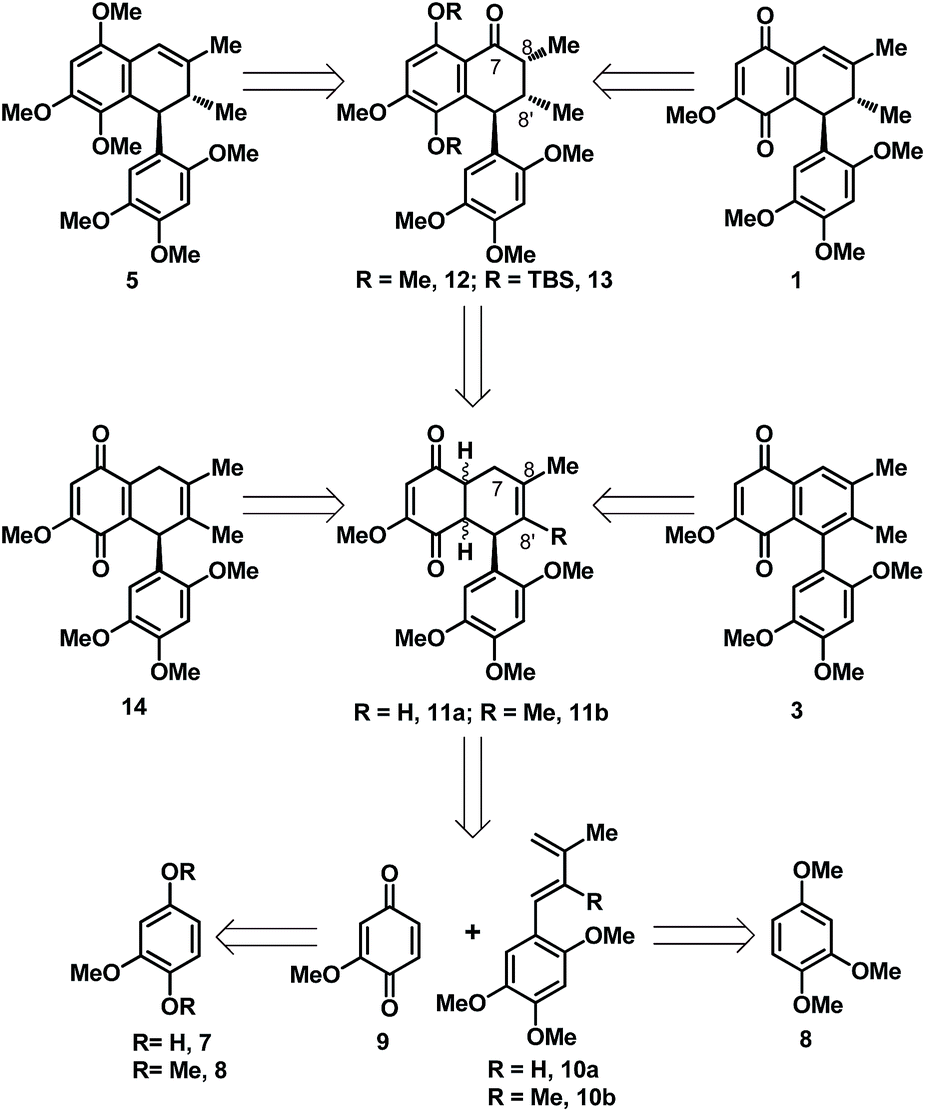 | ||
| Scheme 1 Retrosynthetic plan towards 1, 3 and 5. | ||
Towards the realization of the retrosynthetic plan as outlined in Scheme 1, we commenced our foray with the synthesis of the two coupling partners 9 and 10a. The known quinone 9 (ref. 19) was accessed both via hypervalent iodine mediated oxidation on 8 as well as through NaIO4 mediated oxidation of 7, with comparatively better yields observed in case of latter. While, the synthetic sequence towards the crafting of diene component 10a as depicted in Scheme 2 emanates from the known aldehyde 15 (ref. 20) obtained through a Vilsmeier-Haack formylation on 8. Wittig–Horner olefination of 15 with (carbethoxymethylene)triphenylphosphorane smoothly furnished the trans-cinnamate ester 16 exclusively, which on addition of excess methyllithium resulted in the formation of the cinnamyl alcohol 17. Then, a careful dehydration on 17 in the presence of mesyl chloride and Et3N as the base furnished the desired diene 10a in excellent yield.
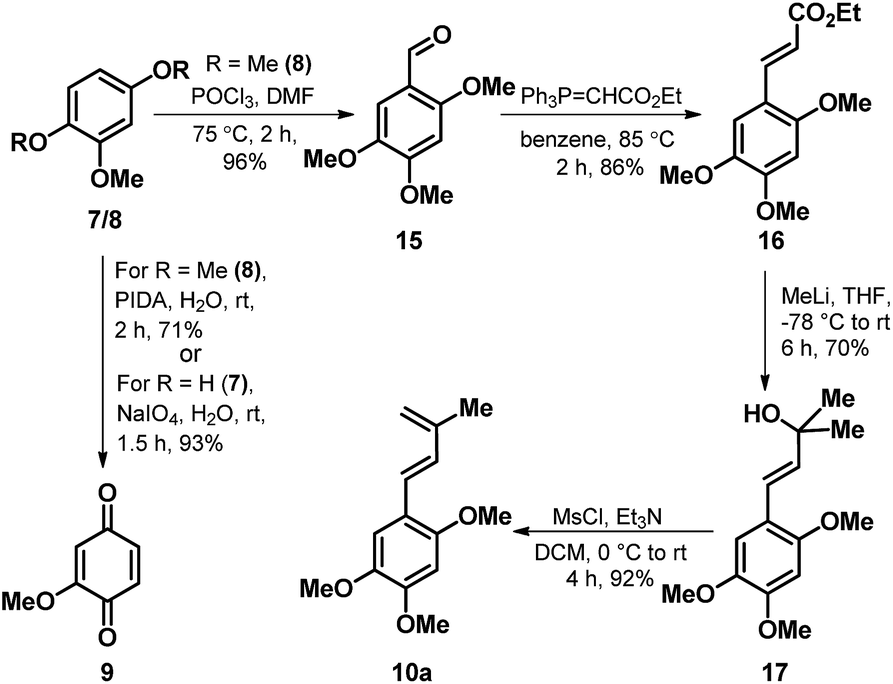 | ||
| Scheme 2 Synthesis of the diene (10a) and dienophile (9). | ||
With both diene and dienophile in hand the next task was to execute the [4 + 2] coupling between the two. Indeed, to our delight as highlighted in Scheme 3, the Diels–Alder coupling between 9 and 10a turned out to be quite eventful as the modest thermal activation of the diene and dienophilic component in dry toluene at 50 °C afforded a readily separable mixture of the bicyclic quinone 18 and naphthoquinone 19 in the ratio of ∼2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and in 59% overall yield. The formation of 18 and 19 is clearly attributable to the spontaneous concomitant partial and complete oxidation in the quinone fused cyclohexene ring of the [4 + 2] cycloadduct 11a respectively. While the structure of 18 was secured through 2D NMR analysis with the key HMBC and nOe correlations as indicated in red and blue arrows respectively (Scheme 3), the close spectral resemblance of naphthoquinone 19 with that of gramineusquinone B (3) hinted it to be the desmethyl derivative of the natural product 3.
:1 and in 59% overall yield. The formation of 18 and 19 is clearly attributable to the spontaneous concomitant partial and complete oxidation in the quinone fused cyclohexene ring of the [4 + 2] cycloadduct 11a respectively. While the structure of 18 was secured through 2D NMR analysis with the key HMBC and nOe correlations as indicated in red and blue arrows respectively (Scheme 3), the close spectral resemblance of naphthoquinone 19 with that of gramineusquinone B (3) hinted it to be the desmethyl derivative of the natural product 3.
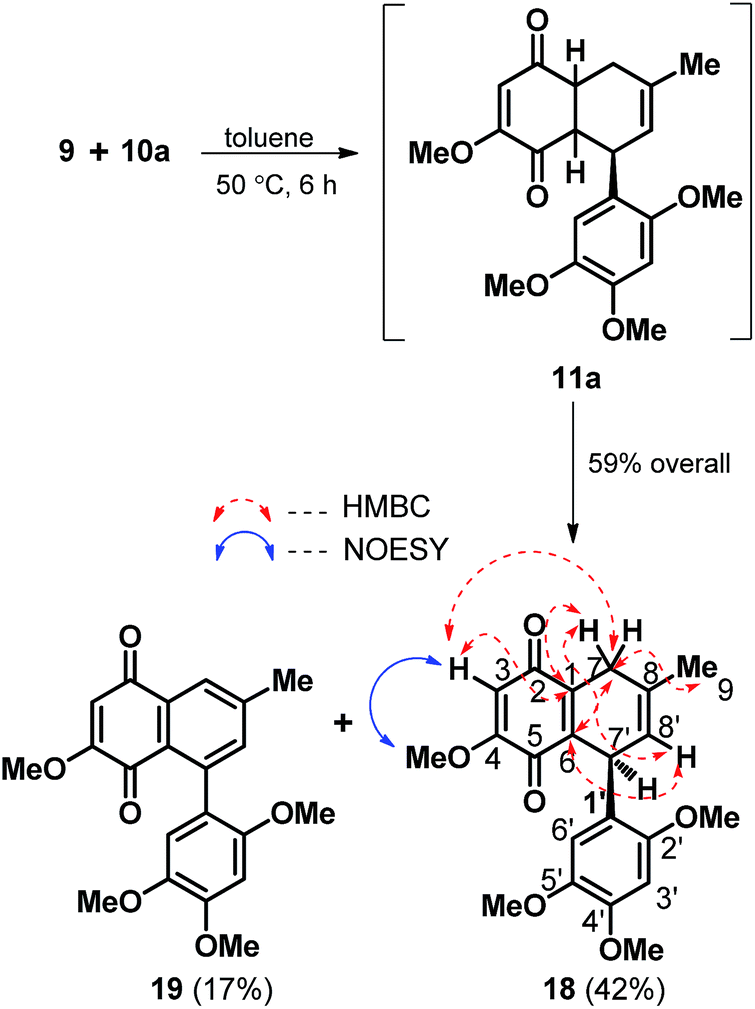 | ||
| Scheme 3 Tandem [4 + 2] cycloaddition and oxidation leading to racemic 18 and desmethyl gramineusquinone B (19). | ||
Encouraged by the outcome of the D–A reaction depicted in Scheme 3, we next turned our attention towards the functionalization of the quinone 18 for its elaboration to the natural products merrilliaquinone (1) and magnoshinin (5). In this regard, we thought of accessing 12/13 by first oxidizing the C-7 methylene position in 18 to arrive at an enone for executing the stereoselective installation of the methyl group at C-8′ position via Gilman-type addition after masking the quinone in the form of methyl/silyl protected quinol. However, failure in our attempts to oxidize the C-7 methylene position both on 18 as well as 18 derived 20, accessed via one-pot reductive methylation of 18 thwarted our plan of synthesizing 12/13. Nevertheless, in our attempts to relocate the double bond from C-8, C-8′ to C-7, C-8 position we explored a modified strategy as depicted in Scheme 4. Indeed, subjection of 18 to the m-CPBA mediated epoxidation offered the epoxy quinone 21 in a diastereoselective manner and in decent yield. But then, the nucleophilic methyl mediated epoxide ring opening on 22 accessed from 21 via reductive methylation as well as via diastereoselective epoxidation of 20 turned out to be a difficult proposition as it failed to offer the desired ring opened product 23 for its further elaboration to 2-epi-magnoshinin (24).
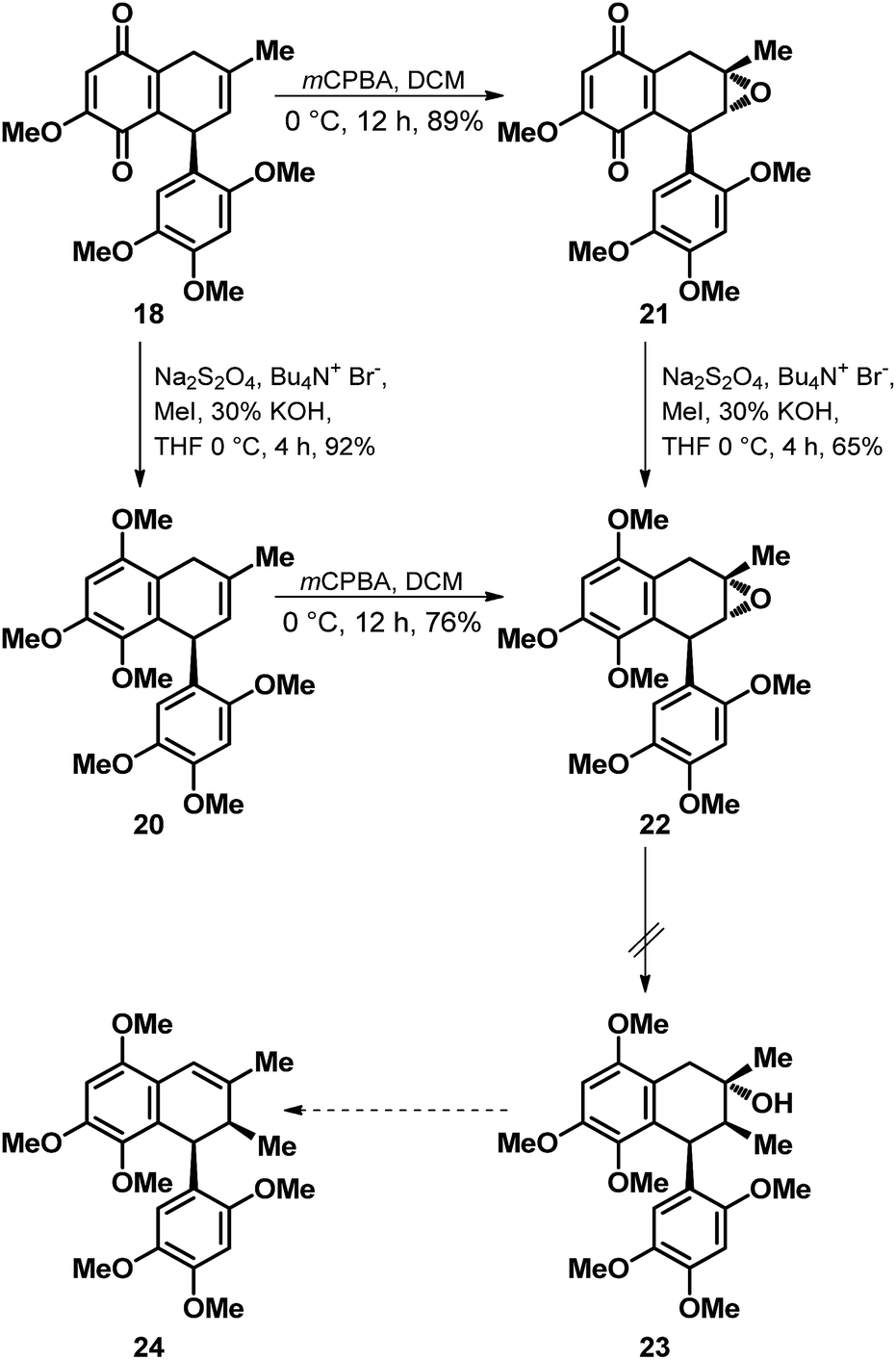 | ||
| Scheme 4 Efforts towards the elaboration of 18 to racemic 2-epi-magnoshinin (24). | ||
Undeterred by our failures in elaborating the cycloadduct 18 to any of the targeted natural product, we next thought of revisiting the strategy in order to access cycloadduct 11b with a pre-installed methyl group at C-8′ position. In this context, through a similar synthetic sequence as earlier demonstrated for preparation of diene 10a (Scheme 2), the diene 10b was synthesized from 15 via the cinnamate ester 25 and tertiary alcohol 26 as depicted in Scheme 5. With access to diene 10b in good yield we readily executed its [4 + 2] coupling with 9 under similar coupling condition as that used for 10a in Scheme 3, but to our surprise with observation of a slight different outcome in terms of product profile. Contrary to the D–A reaction involving 10a, the D–A reaction in case of 10b afforded a mixture of quinone 14 and bicyclic quinol 27 in the ratio of ∼2.6:1 respectively and in an overall yield of 66%. Though 14 could be carefully separated from 27 but the tendency of the quinol 27 to readily undergo spontaneous partial conversion to the quinone form 14 made it difficult to isolate it in the purest form. However, it didn't turn out to be a serious concern as exposure of the mixture of 14 and 27 to NaIO4 afforded a single product 14 which was better serviceable in context of further elaboration to the targeted natural products. Further, the structure of 14 was secured through comparison of its spectral data (1H and 13C NMR) with that of related quinone 18, however for sake of unambiguity we also confirmed the structure through X-ray crystallographic analysis.21 The ORTEP plot of 14 is as highlighted in Scheme 5. In view of the similar chemical constitution of the quinone 14 as that of targeted natural product (1), except for the position of the double bond in the quinone fused cyclohexene ring we presumed appropriate for quinone 14 to be referred as iso-merrilliaquinone.
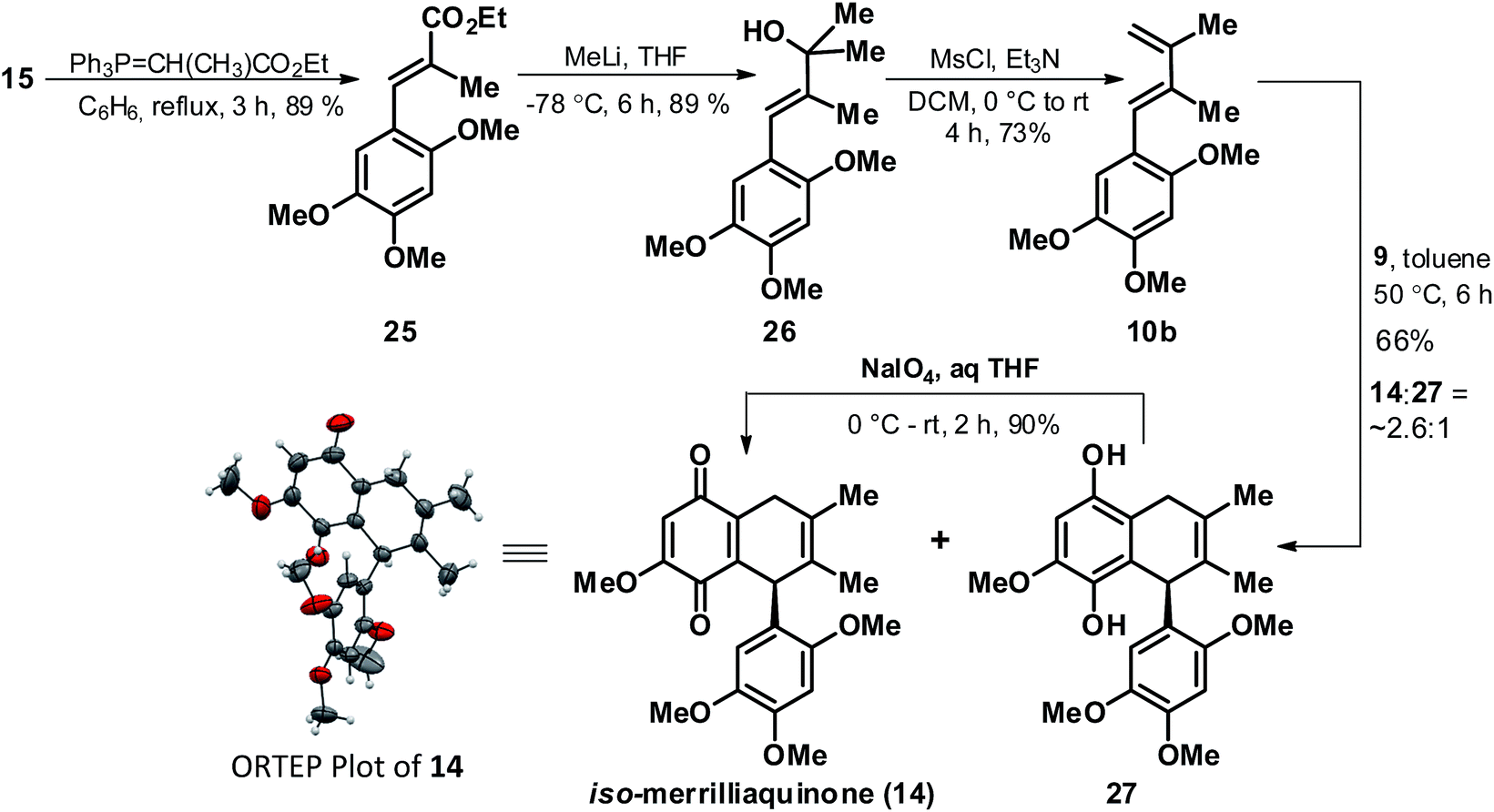 | ||
| Scheme 5 [4 + 2] cycloaddition of 10b and 9 leading to racemic iso-merrilliaquinone. | ||
Towards the end game, foremost keeping in mind the close proximity of 14 to natural product 3, the former was readily subjected to a DDQ mediated aromatization in the quinone fused cyclohexene ring to arrive at gramineusquinone B, 3 in good yields, Scheme 6. The synthetic sample of 3 was found to be spectroscopically (1H and 13C NMR) identical to the reported natural product.12 Then, efforts were channelized towards elaboration of 14 as well as 14 derived 28 (isomeric to magnoshinin, 5) to an intermediate like 12/13 in context of its elaboration to natural product 1 and 5 as per the retrosynthetic plan envisioned in Scheme 1. But this time again, all our investigated conditions proved ineffective in oxidizing the C-7 methylene position in 14 as well as the 14 derived 28. Also, attempts to isomerise the double bond position in 14 as well as 28 proved unfruitful. After repeatedly failing in our attempts to unsymmetrically functionalize the C8C8′ bond both in 14 and 28, we then executed a rather straightforward catalytic hydrogenation on iso-magnoshinin 28 with the hope of accessing the dihydro version of the natural product 5. Indeed the double bond reduction on 28 turned out to be stereoselective offering 29 in decent yields but deviant from the desired stereochemistry at C-8′ position in context of 5. 2D NMR analysis of 28 confirmed the delivery of the two H-atoms on the C8C8′ bond from the side opposite to the phenyl ring at the C-7′ position and hinted 29 to be 8′-epi-7,8-dihydro-magnoshinin or 2-epi-3,4-dihydro-magnoshinin as per the numbering given to the natural product in the isolation paper.14
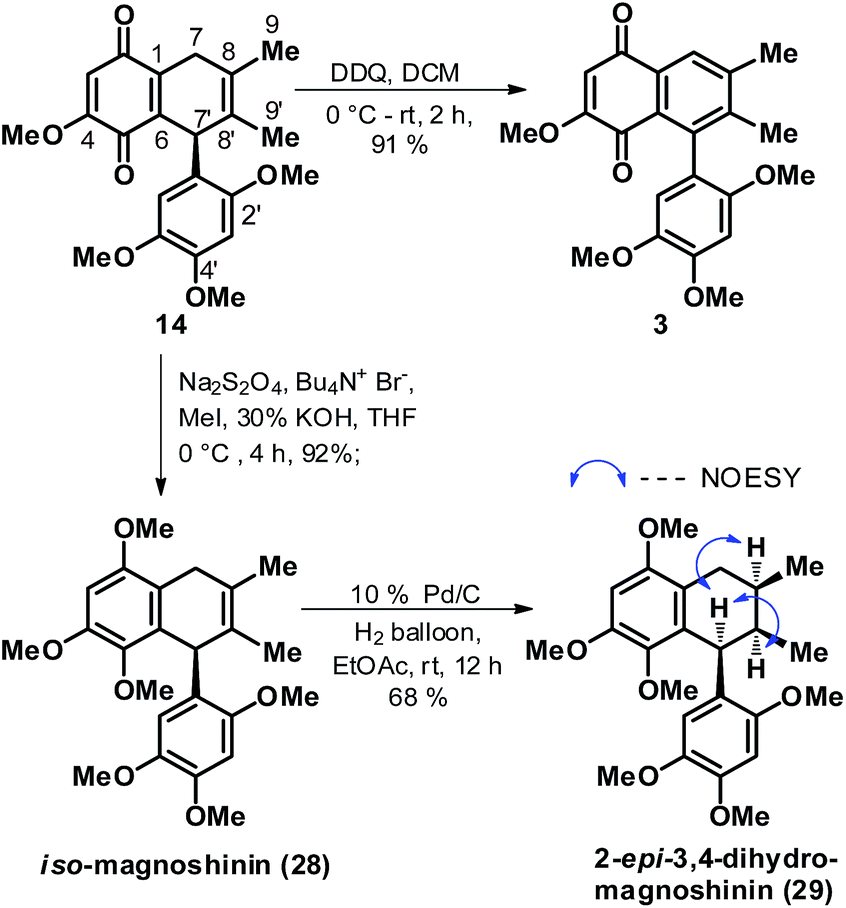 | ||
| Scheme 6 Synthesis of racemic gramineusquinone B (3), iso-magnoshinin (28) and 2-epi-3,4-dihydro-magnoshinin (29). | ||
Conclusions
In conclusion, we have successfully demonstrated the syntheses of gramineusquinone B, iso-merrilliaquinone and iso-magnoshinin as well as 2-epi-3,4-dihydro-magnoshinin from readily accessible fragments through a straightforward Diels–Alder strategy. Efforts are currently underway to overcome the difficulties encountered in the present route for the total syntheses of merrilliaquinone and magnoshinin which may eventually pave the way for other interested bioactive quinones and neolignans.Experimental
All the reagents were purchased from Sigma-Aldrich and other commercial suppliers and used without further purification. While most of the desired solvents supplied by commercial suppliers were dried using the standard drying procedures.22 All nonaqueous reactions were executed under nitrogen atmosphere. Melting points were determined on a Buchi M-560 apparatus and are uncorrected. The 1H and 13C NMR spectra were recorded on 400 and 100 MHz Bruker spectrometer respectively using TMS as an internal standard. The following abbreviations are used for spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, p = quintet/pentet, dd = doublet of doublet, ddd = doublet of doublet of doublet and m = multiplet. The chemical shifts are reported as δ values (ppm) and the coupling constants (J) values are reported in Hz. High Resolution Mass Spectra (HRMS) were obtained using electron spray ionization (ESI) technique and as TOF mass analyzer. IR spectra were recorded on a Bruker FT/IR-460 Plus spectrometer. Reactions monitoring were done using precoated SiO2-gel GF254 glass TLC plates while spot visualizations were done under UV light and spot developing stains like p-anisaldehyde or KMnO4. Purifications were done using column chromatography with 100–200 mesh size SiO2-gel as the stationary phase. Compound 9 (ref. 19) and 15 (ref. 20) were prepared according to the earlier reported procedures.(E)-1,2,4-Trimethoxy-5-(3-methylbuta-1,3-dien-1-yl)benzene (10a)
To a solution of 17 (1 g, 3.968 mmol) in dry DCM (10 mL), 10 mL of triethylamine was added. The reaction mixture was then cooled to −5 °C before adding mesyl chloride (0.46 mL, 5.945 mmol) followed by DMAP (48.31 mg, 0.396 mmol). The reaction mixture was then allowed to warm to rt and stirred for another 1.5 h until complete consumption of starting material and formation of a new spot was indicated by the TLC analysis. The reaction was worked up by diluting it with water followed by extraction in DCM (4 × 40 mL). The organic layer after drying over Na2SO4 and then removal of the solvent under reduced pressure offered a crude oily residue which was purified by stirring the crude residue with 25–30% DCM for 30 min. followed by decantation of the solvent and evaporation under reduced pressure to get the diene 10a (850 mg, 3.628 mmol, 91.5% yield) as an oil. IR (neat): νmax 2931, 1512, 1466, 1398, 1276, 1258, 1207, 1039, 757, 735 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.04 (s, 1H), 6.84 (d, J = 16.4 Hz, 1H), 6.75 (d, J = 16.4 Hz, 1H) 6.50 (s, 1H), 5.07 (d, J = 1.2 Hz, 1H), 5.01 (br s, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.84 (s, 3H), 1.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 151.61, 149.48, 143.47, 142.76, 130.02, 122.87, 118.35, 115.96, 109.41, 97.82, 56.71, 56.52, 56.09, 18.73.Procedure for the Diels–Alder reaction of 9 and 10a
To a solution of dienophile 9 (294 mg, 2.130 mmol) in toluene (5 mL), diene 10a (797 mg, 3.408 mmol) was added and allowed to stir at 50 °C for 6 h. On complete consumption of starting material by TLC analysis the reaction was worked up by evaporating out the entire toluene under reduced pressure and subjecting the resultant crude residue to SiO2 gel column chromatographic purification using petroleum ether/ethyl acetate (7:3) as the eluent to first isolate the partially oxidised DA adduct 18 (332 mg, 0.896 mmol, 42% yield) followed by the aromatized naphthoquinone derivative 19 (135 mg, 0.366 mmol, 17%) with an overall yield of 59% for the D–A coupling.
2-Methoxy-6-methyl-8-(2,4,5-trimethoxyphenyl)naphthalene-1,4-(5H, 8H)-dione (18)
Brown-red solid, mp: 138.6–139.4 °C. IR (neat): νmax 2936, 1673, 1649, 1608, 1510, 1440, 1314, 1221, 1206, 1032 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.50 (s, 1H), 6.45 (s, 1H), 5.88 (s, 1H), 5.48 (d, J = 2.0 Hz, 1H), 4.89 (dd, J1 = 5.6 Hz, J2 = 10.4 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.75 (s, 3H), 3.73 (s, 3H), 3.08 (dd appearing as t, J = 7.0 Hz, 2H), 1.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.49, 180.85, 158.53, 151.31, 148.67, 143.39, 140.57, 139.91, 128.10, 123.18, 122.06, 113.25, 106.82, 98.81, 57.25, 56.92, 56.10 (2C), 35.25, 29.13, 22.70. HRMS (ESI) m/z calcd for C21H23O6 (M + H)+: 371.1489; found: 371.1487.2-Methoxy-6-methyl-8-(2,4,5-trimethoxyphenyl)naphthalene-1,4-dione (19)
Brown-red solid, mp: 167.3–168.4 °C. IR (neat): νmax 2937, 1683, 1649, 1615, 1595, 1516, 1465, 1215, 1033, 852 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 1.6 Hz, 1H), 7.31 (d, J = 1.5 Hz, 1H), 6.68 (s, 1H), 6.57 (s, 1H), 6.10 (s, 1H), 3.94 (s, 3H), 3.84 (s, 6H), 3.65 (s, 3H), 2.49 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 185.44, 179.56, 161.10, 150.64, 149.23, 144.42, 143.05, 139.78, 137.89, 133.07, 127.02, 126.47, 121.37, 113.07, 108.27, 97.37, 56.61, 56.36, 56.32, 56.20, 21.77. HRMS (ESI) m/z calcd for C21H21O6 (M + H)+: 369.1333; found: 369.1366.5,7,8-Trimethoxy-3-methyl-1-(2,4,5-trimethoxyphenyl)-1,4-dihydronaphthalene (20)
To a stirred solution of 18 (20 mg, 0.054 mmol) in THF (2 mL) containing n-Bu4NBr (43.51 mg, 0.135 mmol) at 0 °C were added 30% aq. Na2S2O4 (2.5 mL, 4.310 mmol), MeI (0.42 mL, 6.756 mmol) and aqueous KOH (1 mL) sequentially. After 5 min, the mixture was allowed to warm to rt, and stirring was continued for 1.5 h at the same temperature when complete consumption of starting material along with formation of a new spot was indicated by TLC analysis. The reaction was worked up by extracting it in ethyl acetate followed by drying of the separated organic phase over Na2SO4. Removal of the solvent under reduced pressure afforded a residue which was purified by SiO2 gel column chromatography using hexane/ethyl acetate (7:3) as the eluent to get 20 (20 mg, 0.050 mmol, 92% yield) as an oil. IR (neat): νmax 2934, 2836, 1601, 1507, 1463, 1337, 1235, 1204, 1039, 829 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.54 (s, 1H), 6.44 (s, 1H), 6.28 (s, 1H), 5.61–5.59 (m, 1H), 5.17 (d, J = 4.0 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.62 (s, 3H), 3.29 (s, 3H), 3.22–3.11 (m, 2H), 1.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.46, 150.92, 150.73, 147.71, 143.31, 140.45, 132.70, 129.34, 127.54, 122.90, 117.05, 113.30, 98.46, 95.56, 59.82, 57.21, 56.78, 56.28, 56.10, 55.67, 35.12, 28.89, 23.25. HRMS (ESI) m/z calcd for C23H29O6 (M + H)+: 401.1959; found: 401.1951.
5-Methoxy-1a-methyl-7-(2,4,5-trimethoxyphenyl)-1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene-3,6-dione (21)
To a cooled solution of 18 (15 mg, 0.040 mmol) in DCM (2 mL), m-CPBA (9.3 mg, 0.053 mmol) was added portion wise and allowed to stir for 12 h at 0 °C until complete consumption of starting material along with formation of a new spot was indicated by TLC analysis. The reaction was worked up by evaporating DCM and the resultant crude residue was purified by SiO2 gel column chromatography using hexane/ethyl acetate (3:2) as the eluent to arrive at the epoxide 21 (14 mg, 0.036 mmol, 89% yield) as a pale yellow solid, mp: 139.7–140.8 °C. IR (neat): νmax 2936, 1674, 1655, 1611, 1511, 1461, 1314, 1221, 1032, 806 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.76 (s, 1H), 6.48 (s, 1H), 5.85 (s, 1H), 4.66 (d, J = 1.6 Hz, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H), 3.22 (ddd, J1 = 1.2 Hz, J2 = 2.4 Hz, J3 = 20.0 Hz, 1H), 3.16 (s, 1H), 2.68 (dd, J1 = 2.4 Hz, J2 = 20.0 Hz, 1H), 1.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.20, 181.05, 158.52, 152.13, 149.39, 143.12, 138.53, 137.20, 116.60, 115.04, 106.96, 97.67, 61.44, 57.40, 56.79, 56.38, 56.18, 56.13, 37.58, 29.08, 22.57. HRMS (ESI) m/z calcd for C21H23O7 (M + H)+: 387.1438; found: 387.1423.
3,5,6-Trimethoxy-1a-methyl-7-(2,4,5-trimethoxyphenyl)-1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene (22)
The compound 22 was obtained in 76% yield (11.9 mg, 0.028 mmol) via epoxidation of 20 (15 mg, 0.038 mmol) using m-CPBA (8.0 mg, 0.048 mmol) and employing similar epoxidation procedure as that described for 21. Alternatively it was also obtained in 65% yield (7.0 mg, 0.017 mmol) by reductive methylation of 21 (10 mg, 0.026 mmol) using 30% aq. Na2S2O4 (1.26 mL, 2.172 mmol), MeI (0.2 mL, 3.238 mmol) and aqueous KOH (1 mL) employing similar procedure as that described for 20. IR (neat): νmax 2924, 2851, 1680, 1650, 1612, 1511, 1462, 1214, 1034, 850 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.56 (s, 1H), 6.42 (s, 1H), 6.31 (s, 1H), 5.19 (d, J = 2.0 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 3.64 (s, 3H), 3.42 (d, J = 19.0 Hz, 1H), 3.41 (s, 3H), 3.25 (dd, J = 2.0, 1.0 Hz, 1H), 2.78 (d, J = 18.0 Hz, 1H), 1.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.07, 151.67, 151.32, 148.63, 143.13, 141.06, 130.12, 121.48, 114.06, 113.78, 98.10, 96.06, 62.08, 60.26, 57.02, 56.89, 56.76, 56.16, 56.09, 55.74, 35.55, 28.51, 22.93. HRMS (ESI) m/z calcd for C23H29O7 (M + H)+: 417.1908; found: 417.1941.(E)-1-(2,3-Dimethylbuta-1,3-dien-1-yl)-2,4,5-trimethoxy-benzene (10b)
To a solution of 26 (598 mg, 2.248 mmol) in dry DCM (10 mL), 10 mL of triethylamine was added. The reaction mixture was then cooled to −5 °C before adding mesyl chloride (0.26 mL, 3.372 mmol) followed by DMAP (27.4 mg, 0.224 mmol). The reaction mixture was then allowed to warm to rt and stirred for another 2 h until complete consumption of starting material and formation of a new spot was indicated by the TLC analysis. The reaction was worked up by diluting it with water and extracting in DCM (4 × 40 mL) followed by drying of the separated organic phase over Na2SO4. Removal of the solvent under reduced pressure afforded a residue which was purified by column chromatography using hexane/ethyl acetate (8:2) as the eluent to get 10b (408 mg, 1.643 mmol, 73% yield) as an oil. IR (neat): νmax 2947, 2833, 1605, 1511, 1464, 1316, 1302, 1206, 1177, 1036, 875 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.79 (s, 1H), 6.66 (s, 1H), 6.53 (s, 1H), 5.15 (s, 1H), 5.03 (s, 1H), 3.91 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 2.06 (d, J = 0.4 Hz, 3H) 1.99 (d, J = 0.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 151.90, 148.68, 144.97, 142.47, 136.24, 122.65, 119.02, 114.72, 112.55, 97.50, 56.66, 56.53, 56.12, 21.08, 15.54. HRMS (ESI) m/z calcd for C15H21O3 (M + H)+: 249.1485; found: 249.1460.
2-Methoxy-6,7-dimethyl-8-(2,4,5-trimethoxyphenyl)naphthalene-1,4-(5H,8H)-dione (iso-merrilliaquinone, 14)
To a solution of dienophile 9 (20 mg, 0.145 mmol) in toluene, diene 10b (58 mg, 0.232 mmol) was added and allowed to stir at 50 °C for 6 h. On complete consumption of the dienophile and with formation of two new spots of 14 and 26 by TLC analysis, the reaction was worked up by evaporating out the entire toluene under reduced pressure. To the resultant crude residue was then added NaIO4 (19.9 mg, 0.093 mmol) in aq. THF (1.5 mL) at 0 °C. The reaction was allowed to warm to rt over 2 h until the complete conversion of mixture of 14 and 26 to a single spot of 14 was indicated by TLC analysis. The reaction was worked by extracting it in EtOAc (4 × 15 mL), washing the organic phase with brine and drying over Na2SO4. Removal of the solvent under reduced pressure followed by purification of the crude residue by SiO2-gel column chromatography using petroleum ether/ethyl acetate (7:3) as the eluent afforded 14 (32.8 mg, 0.085 mmol, 59% overall yield) as an orange solid, mp: 197.8–198.3 °C. IR (neat): νmax 2935, 2851, 1674, 1652, 1636, 1609, 1512, 1351, 1227, 1033 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.49 (s, 1H), 6.47 (s, 1H), 5.84 (s, 1H), 4.76 (t, J = 5.6 Hz, 1H), 3.88 (s, 3H), 3.83 (s, 3H), 3.73 (s, 6H), 3.22–3.08 (m, 2H), 1.76 (s, 3H), 1.56 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.64, 180.95, 158.42, 151.95, 148.62, 143.30, 140.50, 139.96, 126.81, 122.85, 121.59, 113.36, 106.71, 98.65, 57.46, 56.73, 56.08, 56.01, 39.47, 31.07, 18.51, 16.20. HRMS (ESI) m/z calcd for C22H25O6 (M + H)+: 385.1646; found: 385.1675.
Gramineusquinone B (3)
To stirred solution of 14 (10 mg, 0.026 mmol) in DCM (1 mL) at 0 °C, DDQ (5.9 mg, 0.026 mmol) was added. The reaction mixture was then allowed to warm to rt and stirred for another 5 h until complete consumption of starting material and formation of a new spot was indicated by the TLC analysis. The reaction was worked up by removing the solvent under reduced pressure and then subjecting the resultant residue to SiO2 gel column chromatographic purification using hexane/ethyl acetate (7:3) as the eluent to arrive at 3 (9 mg, 0.023 mmol, 91% yield) as a red solid, mp: 172.6–173.3 °C. IR (neat): νmax 2937, 2842, 1685, 1648, 1617, 1517, 1213, 1085, 1031, 856, cm−1; 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 6.63 (s, 1H), 6.40 (s, 1H), 6.06 (s, 1H), 3.95 (s, 3H), 3.80 (s, 3H), 3.76 (s, 3H), 3.65 (s, 3H), 2.45 (s, 3H), 2.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 185.52, 179.83, 160.77, 150.39, 148.94, 143.80, 143.76, 143.63, 139.41, 130.74, 127.21, 127.04, 120.48, 112.72, 108.03, 97.91, 56.57, 56.43, 56.23, 56.16, 21.47, 16.92. HRMS (ESI) m/z calcd for C22H23O6 (M + H)+: 383.1489; found: 383.1481.
5,7,8-Trimethoxy-2,3-dimethyl-1-(2,4,5-trimethoxyphenyl)-1,4-dihydronaphthalene (iso-magnoshinin, 28)
Reductive methylation on 14 (20 mg, 0.052 mmol) using 30% aq. Na2S2O4 (2.5 mL, 4.310 mmol), MeI (0.42 mL, 6.756 mmol) and aqueous KOH (1 mL) through similar procedure as described for preparation of 20, afforded compound 28 (20 mg, 0.048 mmol, 92% yield) as pale yellow solid. IR (neat): νmax 2932, 1602, 1509, 1462, 1439, 1336, 1316, 1238, 1203, 1033 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.51 (s, 1H), 6.41 (s, 1H), 6.40 (s, 1H), 5.04 (br s, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 3.64 (s, 3H), 3.32 (s, 3H), 3.30 (br s, 2H), 1.78 (s, 3H), 1.64 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.27, 151.23, 150.78, 147.61, 143.20, 140.03, 134.46, 127.73, 127.11, 122.74, 116.70, 113.73, 97.90, 95.21, 59.81, 56.96, 56.55, 56.22, 56.04, 55.68, 39.47, 30.98, 19.03, 16.66. HRMS (ESI) m/z calcd for C24H34NO6 (M + NH4)+: 432.2381; found: 432.2353.5,7,8-Trimethoxy-2,3-dimethyl-1-(2,4,5-trimethoxyphenyl)-1,2,3,4-tetrahydronaphthalene (2-epi-3,4-dihydro-magnoshinin, 29)
To stirred solution of 28 (16 mg, 0.0386 mmol) in MeOH (1 mL) was added 10% Pd/C (1.6 mg). The mixture was then allowed to stir at room temperature under hydrogen balloon pressure for 6 h until complete consumption of starting material was indicated by TLC analysis. The reaction mixture was then filtered through celite, washed with EtOAc (4 × 5 mL) and the filtrate was concentrated under reduced pressure. The resultant crude residue was purified by flash column chromatography (petroleum ether/EtOAc = 8:2) on silica gel to afford the desired 2-epi-3,4-dihydro-magnoshinin, 29 (11 mg, 0.026 mmol, 68% yield). IR (neat): νmax 2932, 2834, 1596, 1509, 1462, 1395, 1333, 1233, 1204, 1039 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.56 (s, 1H), 6.42 (s, 1H), 6.23 (s, 1H), 4.80 (d, J = 6.0 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 3.79 (s, 3H), 3.60 (s, 3H), 3.20 (s, 3H), 2.62 (dd, J1 = 4.5 Hz, J2 = 17.0 Hz, 1H), 2.29 (ddd, J1 = 1.2 Hz, J2 = 12.0 Hz, J3 = 17.0, Hz, 1H), 2.14–2.08 (m, 1H), 1.97–1.92 (m, 1H), 1.06 (d, J = 7.0 Hz, 3H), 0.52 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 152.56, 151.04, 150.84, 146.97, 142.35, 142.16, 133.84, 126.26, 120.45, 113.80, 98.11, 95.64, 59.59, 57.28, 56.57 (2C), 56.11, 55.64, 40.31, 35.91, 32.80, 26.60, 19.61, 10.00. HRMS (ESI) m/z calcd for C24H36NO6 (M + NH4)+: 434.2537; found: 434.2571.
Acknowledgements
D. K. is grateful to UGC, India for the Junior Research Fellowship (JRF). T. K. thank IIT Bhubaneswar for the financial and infrastructural support. DST, SERB (EMR/2014/000826) is also acknowledged for providing funding to the lab.Notes and references
- Y. H. Liu, Flora Republicae Popularis Sinicae, Science Press, Beijing, 1996, vol. 30, p. 227 Search PubMed.
- For a review please refer: Y. Fukuyama and J.-M. Huang, Stud. Nat. Prod. Chem., 2005, 32, 395–427 CrossRef CAS and ref. cited therein.
- J. M. Huang, Y. Fukuyama, C. S. Yang, H. Minami and M. Tanaka, Chem. Pharm. Bull., 2000, 48, 657–659 CrossRef CAS PubMed.
- M. Kubo, C. Okada, J.-M. Huang, K. Harada, H. Hioki and Y. Fukuyama, Org. Lett., 2009, 11, 5190–5193 CrossRef CAS PubMed.
- J.-M. Huang, R. Yokoyama, C.-S. Yang and Y. Fukuyama, J. Nat. Prod., 2001, 64, 428–431 CrossRef CAS.
- For few selected synthesis of merrilactone A please refer: (a) V. B. Birman and S. J. Danishefsky, J. Am. Chem. Soc., 2002, 124, 2080–2081 CrossRef CAS PubMed; (b) M. Inoue, T. Sato and M. Hirama, J. Am. Chem. Soc., 2003, 125, 10772–10773 CrossRef CAS PubMed; (c) G. Mehta and S. R. Singh, Angew. Chem., Int. Ed., 2006, 45, 953–955 CrossRef CAS PubMed; (d) W. He, J. Huang, X. F. Sun and A. J. Frontier, J. Am. Chem. Soc., 2007, 129, 498–499 CrossRef CAS PubMed; (e) W. He, J. Huang, X. F. Sun and A. J. Frontier, J. Am. Chem. Soc., 2008, 130, 300–308 CrossRef CAS PubMed.
- For few selected approaches towards 11-0-debenzoyl tashironin please refer: (a) A. Polara, S. P. Cook and S. J. Danishefsky, Tetrahedron Lett., 2008, 49, 5906–5908 CrossRef CAS PubMed; (b) G. Mehta and P. Maity, Tetrahedron Lett., 2011, 52, 1753–1756 CrossRef CAS; (c) G. Mehta and P. Maity, Tetrahedron Lett., 2011, 52, 1749–1752 CrossRef CAS.
- For few selected approaches towards jiadifenolide please see (a) Y. Shen, L. Li, Z. Pan, Y. Wang, J. Li, K. Wang, X. Wang, Y. Zhang, T. Hu and Y. Zhang, Org. Lett., 2015, 17, 5480–5483 CrossRef CAS PubMed; (b) H.-H. Lu, M. D. Martinez and R. A. Shenvi, Nat. Chem., 2015, 7, 604–607 CrossRef CAS PubMed; (c) J. Xu, L. Trzoss, W. K. Chang and E. A. Theodorakis, Angew. Chem., Int. Ed., 2011, 50, 3672–3676 CrossRef CAS PubMed; (d) G. Mehta, H. M. Shinde and R. Senthil Kumaran, Tetrahedron Lett., 2012, 53, 4320–4323 CrossRef CAS.
- X. Tian, R. Yue, S. Zhang, Y. Shen, J. Ye, L. Shan, H. Li, B. Wen, X. Xu and W. Zhang, Eur. J. Org. Chem., 2014, 4753–4758 CrossRef CAS.
- X. Tian, L. Li, J. Pei, R. Yue, X. Fang, J. Zhang, W. He, L. Shan, Y. Shen and W. Zhang, RSC Adv., 2015, 5, 75857–75862 RSC.
- Y. Lu, Y. Xue, S. Chen, H. Zhu, J. Zhang, X.-N. Li, J. Wang, J. Liu, C. Qi, G. Du and Y. Zhang, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- S. Y. Lee, E. Moon, S. Y. Kim, S. U. Choi and K. R. Lee, Biosci., Biotechnol., Biochem., 2013, 77, 276–280 CrossRef CAS PubMed.
- N.-Y. Chen, J. Shi and T. Chen, Planta Med., 2000, 66, 187–188 CrossRef CAS PubMed.
- T. Kikuchi, S. Kadota, K. Yanada, K. Tanaka, K. Watanabe, M. Yoshizaki, T. Yokoi and T. Shingu, Chem. Pharm. Bull., 1983, 31, 1112–1114 CrossRef CAS.
- D.-P. Qin, X.-L. Feng, W.-Y. Zhang, H. Gao, X.-R. Cheng, W.-X. Zhou, Y. Yu and X.-S. Yao, RSC Adv., 2017, 7, 8512–8520 RSC.
- For the synthesis of magnoshinin and other selected lignan synthesis please refer: (a) S. Kadota, K. Tsubono, K. Makino, M. Takeshita and T. Kikuchi, Tetrahedron Lett., 1987, 28, 2857–2860 CrossRef CAS; (b) S.-I. Yoshida, T. Ogiku, H. Ohmizu and T. Iwasaki, Synlett, 1994, 11, 895–898 CrossRef; (c) B. L. Yvon, P. K. Datta, T. N. Le and J. L. Charlton, Synthesis, 2001, 10, 1556–1560 Search PubMed; (d) Q. Xiao, J. J. Jackson, A. Basak, J. M. Bowler, B. G. Miller and A. Zakarian, Nat. Chem., 2013, 5, 410–416 CrossRef CAS PubMed.
- G. Mehta, T. B. Khan and S. Y. C. Kumar, Tetrahedron Lett., 2010, 51, 5116–5119 CrossRef CAS.
- G. Mehta and T. B. Khan, Tetrahedron Lett., 2010, 51, 6590–6593 CrossRef CAS.
- (a) K. A. Parker, D. M. Spero and K. A. Koziski, J. Org. Chem., 1987, 52, 183–188 CrossRef CAS; (b) H. Tohma, H. Morioka, Y. Harayama, M. Hashizume and Y. Kita, Tetrahedron Lett., 2001, 42, 6899–6902 CrossRef CAS.
- Y.-L. Lin, C.-S. Wu, S.-W. Lin, J.-L. Huang, Y.-S. Sun and D.-Y. Yang, Bioorg. Med. Chem., 2002, 10, 685–690 CrossRef CAS PubMed.
- CCDC 1548143 contain the crystallographic information for 14.†.
- D. L. F. Armarego, D. D. Perrin, Purification of Laboratory Chemicals, IV edn, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1548143. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05148d |
| This journal is © The Royal Society of Chemistry 2017 |