
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical insights into the reaction mechanism between tetrachloro-o-benzoquinone and N-methyl benzohydroxamic acid†
Weihua Wang*a,
Chao Guoa,
Wenling Fenga,
Qiao Sun b and
Ping Li*a
b and
Ping Li*a
aKey Laboratory of Life-Organic Analysis, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, P. R. China. E-mail: wwh78@163.com; lignip@163.com
bCollaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, School for Radiological and Interdisciplinary Sciences, Soochow University, Suzhou, 215123, P. R. China
First published on 26th June 2017
Abstract
Acquiring the detailed reaction mechanism between halogenated quinones and hydroxamic acids is crucial for better understanding of the potential applications of benzohydroxamic acids in the detoxification of the carcinogenic polyhalogenated quinoid metabolites of pentachlorophenol. In this study, the reaction mechanism between tetrachloro-o-benzoquinone (o-TCBQ) and N-methyl benzohydroxamic acid (N-MeBHA) has been systematically investigated at the B3LYP/6-311++G(d,p) level. It was found that o-TCBQ can react with the anion of N-MeBHA (N-MeBHA−) under mild conditions. As the first step of reaction, a molecular complex is formed between o-TCBQ and N-MeBHA− followed by the nucleophilic attack of the O atom of N-MeBHA− at the C atom attached to the Cl atom of o-TCBQ, resulting in the formation of an unstable intermediate containing an N–O bond. Subsequently, the unstable intermediate decomposes via the homolytic cleavage of the N–O bond to produce N-centered and O-centered radicals. For the O-centered radical, it can isomerize to a C-centered form upon structural relaxation. Finally, these radicals react with each other to form the major C–N bonding products and minor C–O bonding products. In addition, it was found that the reactivity of o-TCBQ with N-MeBHA is higher than that of tetrachloro-p-benzoquinone (p-TCBQ).
1. Introduction
As the major genotoxic and carcinogenic quinoid metabolites of the widely used wood preservative pentachlorophenol (PCP), polyhalogenated quinones can create many hazardous effects in vivo, e.g., acute hepatotoxicity, nephrotoxicity, and carcinogenesis,1,2 where PCP has been classified as a group 2B environmental carcinogen by the International Association for Research on Cancer. Meanwhile, halogenated quinones can also be produced during the oxidation or destruction of polyhalogenated persistent organic pollutants (POPs).3–5 In particular, some halobenzoquinones have been identified as an emerging class of disinfection byproducts in drinking water.6–8 More recently, it was found that the polyhalogenated quinones can react with hydrogen peroxide (H2O2) or organic hydroperoxides to produce hydroxyl and alkoxyl radicals experimentally,9–16 which can partially explain the potential carcinogenicity of polyhalogenated aromatic environmental pollutants.Another species of environmental and biomedical interest are hydroxamic acids, which have been paid extensive attention due to their strong capacity to inhibit transition-metal-mediated oxidative stress and the activity of many enzymes (e.g., metalloproteases and lipoxygenase).17–19 Clinically, the suberoylanilide hydroxamic acid and deferoxamine have been applied for the treatment of cancer and iron-overload diseases. Moreover, hydroxamic acids can effectively detoxify the carcinogenic polyhalogenated quinoid metabolites of pentachlorophenol and other persistent organic pollutants.20–22 For example, benzohydroxamic acid can dramatically accelerate the conversion of the highly toxic tetrachloro-p-benzoquinone (p-TCBQ) to much less toxic chloranilic acid via an unusual double Lossen rearrangement mechanism.20 However, for the N-methyl benzohydroxamic acid, it can react with 2,5-dichloro-1,4-benzoquinone (DCBQ) through a radical mechanism.21,22 Namely, a nucleophilic reaction may take place between DCBQ and N-MeBHA to form a relatively stable initial product followed by its homolytic decomposition to produce N- and O-centered radicals. Especially, for the N-centered radical, it can readily attack the deoxynucleotides, exhibiting its potential biological implications.22 For the O-centered radical, it can isomerize to the C-centered radical, which can be further coupled with the N-centered radical to produce the major C–N bonding product via keto–enol tautomerization. Similarly, the same is also true for the p-TCBQ although no keto–enol tautomerization products have been observed due to the specific geometry of p-TCBQ.22
As the isomer of p-TCBQ and another quinoid metabolites of PCP, o-TCBQ has been involved in the toxicity of the bleached kraft chlorination effluent many years ago.23 However, o-TCBQ has been paid less attention compared with the p-TCBQ.24–30 Can it react with N-MeBHA similar to that of p-TCBQ? If so, what is the detailed reaction mechanism? What are the final reaction products? Obviously, the clarifications of these questions are crucial for the understanding of the reactivity of ortho-halogenated benzoquinones with N-MeBHA as well as the detoxification of the benzohydroxamic acids. Unfortunately, these questions have not been clarified to the best of our knowledge. So, a theoretical investigation on the title reaction appears to be highly desirable in the lack of the relevant experimental studies.
Therefore, to address these questions mentioned above, in this study, the reaction mechanism between o-TCBQ and N-MeBHA has been systematically investigated employing the density functional theory (DFT) method. As a result, the detailed reaction mechanism for the title reaction has been clarified at the electronic level. Besides the major C–N bonding products, the minor C–O bonding products have also been observed. Expectedly, the present results not only can provide scientific proof for the detection and identification of the relevant products of the title reaction, but also can provide useful clues to the design and synthesis of large-size molecules containing C–N bond experimentally.
2. Computational methods
All the species in the whole reaction have been fully optimized at the B3LYP/6-311++G(d,p) level, where the reliability and efficiency of the method in predicting geometries and properties has been verified by a number of systems.31–39 Subsequently, vibrational frequency analysis has been performed at the same level to identify the nature of the optimized structures. Moreover, intrinsic reaction coordinate (IRC)40,41 calculations were performed to further verify that the calculated transition states indeed connected the reactants and products, where the selected results have been given in Fig. S1 of the ESI† for reference.To further testify the reliability of the B3LYP functional, the relevant calculations for the reaction of o-TCBQ with neutral N-MeBHA in the presence of one water molecule and the direct reaction of o-TCBQ with the anion of N-MeBHA have been performed at the M06/6-311++G(d,p) level. As a result, it was found that the calculated results including geometries and energy barriers are consistent with each other at both levels. For the sake of simplicity, the relevant discussions have been given in the ESI† for reference.
To calculate the interaction energy between free radicals, the Boys–Bernardi counterpoise technique has been employed to evaluate the basis set superposition errors (BSSE).42
To investigate the solvent effect on the nucleophilic attack process of N-MeBHA to o-TCBQ, the polarizable continuum model (PCM)43,44 was employed in aqueous solution within the framework of self-consistent reaction filed (SCRF) theory. To explore the possibility of the proceeding of the title reaction in the presence of water molecules, different numbers of explicit water molecules ranging from one to three have been introduced to assist the proton transfer process.
To qualitatively predict which bond to be dissociated in the formed intermediate, ab initio molecular dynamics has been carried out at the B3LYP/6-311G(d,p) level using the atom-centered density matrix propagation (ADMP) molecular dynamics.45–47 The dynamics calculations were performed under condition of constant temperature at 298.15 K. The simulation time is long enough to observe the necessary phenomena with a time step of 0.5 fs.
All the calculations have been carried out using Gaussian 09 program.48
3. Results and discussion
In realistic system, N-MeBHA should exist in its neutral and anionic forms depending on the pH of solution. So the reactions of o-TCBQ with neutral and anionic N-MeBHA have been investigated, respectively.For the sake of simplicity, the symbols MC, IM, TS, and P have been employed to stand for the optimized molecular complexes, intermediates, transition states, and products, respectively. All the Cartesian coordinates of the optimized species have been given in the ESI† for reference.
3.1 The reaction of o-TCBQ with neutral N-MeBHA
Firstly, the reaction of o-TCBQ with neutral N-MeBHA has been investigated. Similar to the reactions of p-TCBQ and o-TCBQ with H2O2,30,49 the nucleophilic attack process of N-MeBHA to o-TCBQ has been mainly explored. As shown in Fig. 1, four reaction modes, namely N1, N2, N3, and N4, have been constructed considering the symmetry of o-TCBQ. A molecular complex MC1 has been located as the first step of the reaction. Moreover, MC1 acts as the common reactant for the four reaction modes, which has been confirmed by the IRC calculations. Subsequently, nucleophilic attack of the hydroxyl O atom of N-MeBHA to the C atom attached to the Cl atom of o-TCBQ occurs, accompanying with the proton transfer from N-MeBHA to the Cl atom attached to the attacked C atom of o-TCBQ. After then, an intermediate containing N–O bond has been produced. As presented in Table 1, the MC1 has been stabilized by about 4.12 kcal mol−1. The energy barriers in the nucleophilic attack processes range from 50.71 to 51.33 kcal mol−1 relative to the separated reactants, implying that it is difficult to occur for the reaction between o-TCBQ and neutral N-MeBHA. Moreover, the calculated Gibbs free energy changes for the formation processes of the intermediates are positive values, suggesting that the whole processes are unfavorable thermodynamically.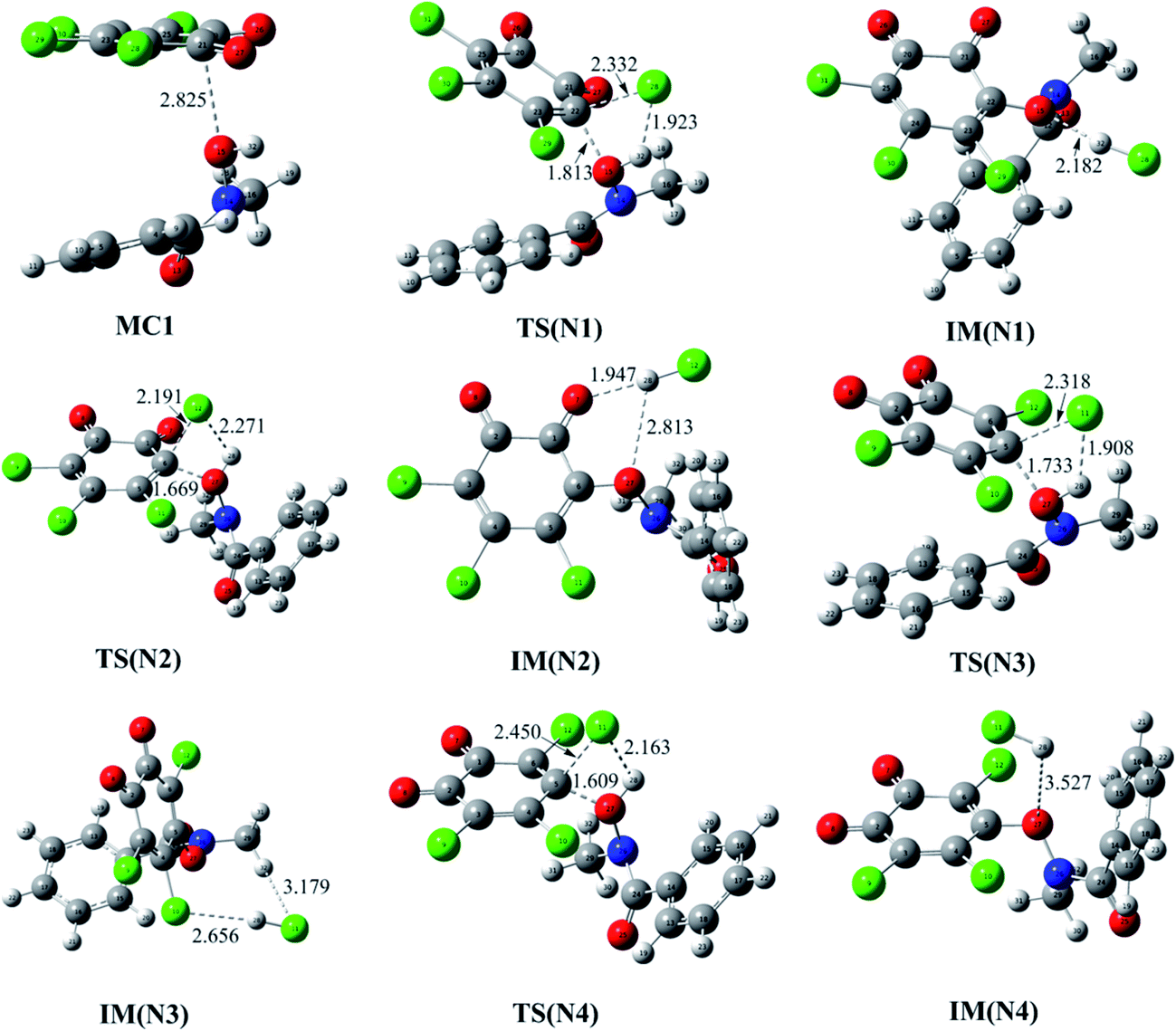 | ||
| Fig. 1 Optimized molecular complexes (MC), transition states (TS), and intermediates (IM) in the different reaction modes of o-TCBQ with isolated neutral N-MeBHA. The selected interatomic distances are given in Å. | ||
| Attack modes | MC | TS | IM |
|---|---|---|---|
| a All the units are in kcal mol−1. The data before and after slash refer to the relative energies, enthalpy changes, and Gibbs free energy changes, respectively. The data in parentheses refer to the results involving one, two, and three water molecules, respectively. | |||
| N1 | −4.12 | 51.33 | −1.15/−0.14/8.72 |
| N2 | −4.12 | 50.78 | −2.10/−1.38/8.30 |
| N3 | −4.12 | 50.71 | −1.29/−0.04/6.23 |
| (−13.68/−22.78/−27.03) | (40.03/27.76/18.21) | (−12.34/−13.08/−23.10) | |
| N4 | −4.12 | 51.00 | −1.55/−0.45/8.34 |
Moreover, given the positive catalytic role of water molecules in the assistance of proton transfer, so the reaction of o-TCBQ with N-MeBHA with the assistance water molecules have been investigated on the basis of the N3 reaction mode. As displayed in Fig. 2, one, two, and three water molecules have been introduced to assist the proton transfer mentioned above. Here, the bridge role of water molecules in the assistance of proton transfer should be highlighted. For example, the introduced single water molecule can accept the hydroxyl proton of N-MeBHA and gives its own proton to the dissociated Cl atom of o-TCBQ simultaneously. Similarly, the same is also true if a second/third water molecule is introduced, where the second/third water molecule accepts the proton of the first/second water molecule and give its proton to the Cl atom of o-TCBQ. As presented in Table 1, compared with the case without involving water molecules, the energy barriers in the nucleophilic attack process have been reduced by 10.68, 22.95, and 32.50 kcal mol−1 to 40.03, 27.76, and 18.21 kcal mol−1 in the assistance of one, two, and three water molecules, respectively. Despite the positive catalytic role of water molecules in reducing the energy barrier, the energy barrier here is still so high that the title reaction is difficult to occur under normal conditions.
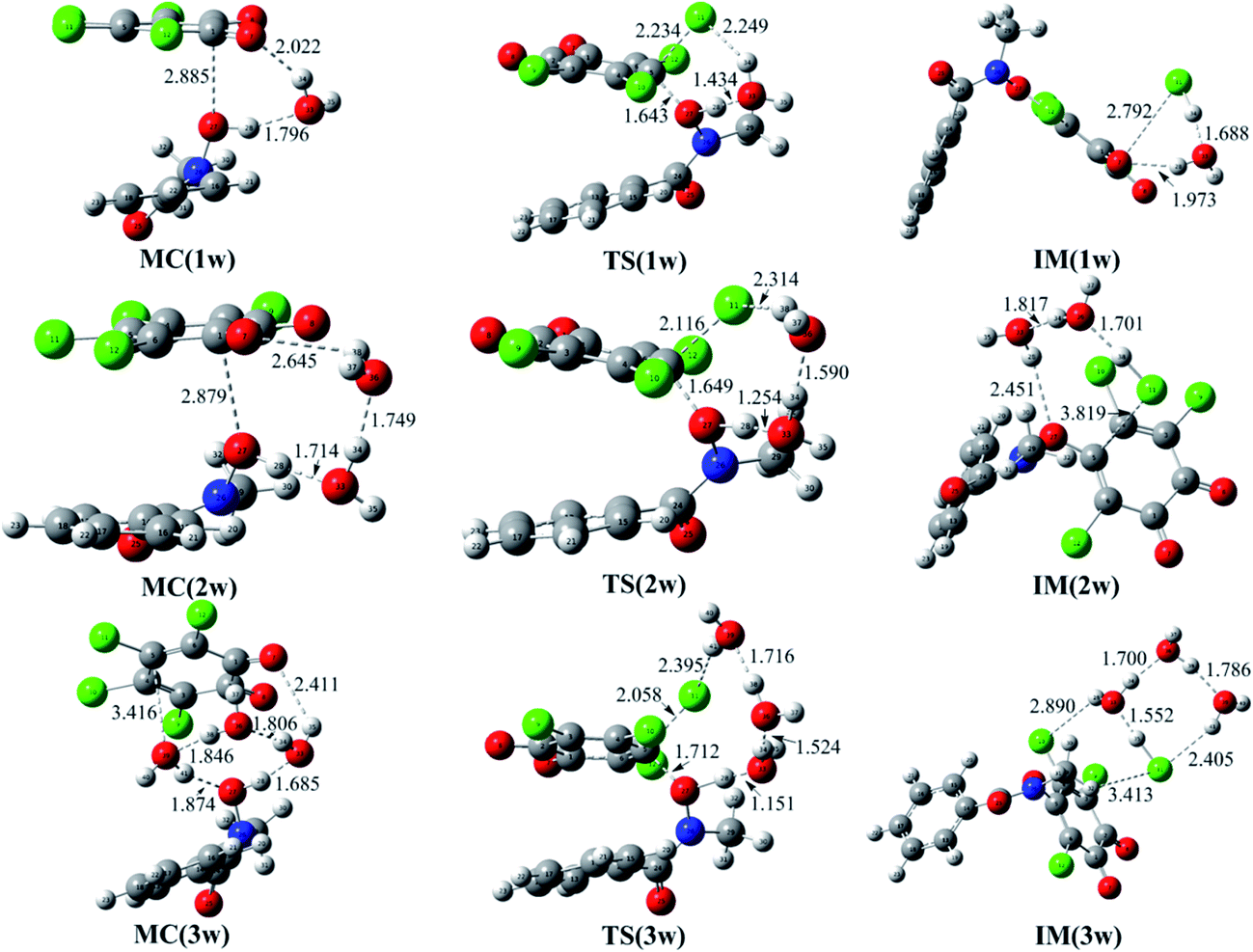 | ||
| Fig. 2 Optimized molecular complexes (MC), transition states (TS), and intermediates (IM) in the reaction of o-TCBQ with neutral N-MeBHA in the assistance of water molecules. The selected interatomic distances are given in Å. | ||
3.2 The reaction of o-TCBQ with N-MeBHA anion
As mentioned above, the reaction of o-TCBQ with neutral N-MeBHA is difficult to take place regardless of the presence of explicit water molecules or not. Given the fact that N-MeBHA− anion can be produced from the acid–base dissociation equilibrium of N-MeBHA and it is a better nucleophile than neutral N-MeBHA, the direct reaction between o-TCBQ and N-MeBHA− has been investigated below to further clarify the reactivity of the title reaction.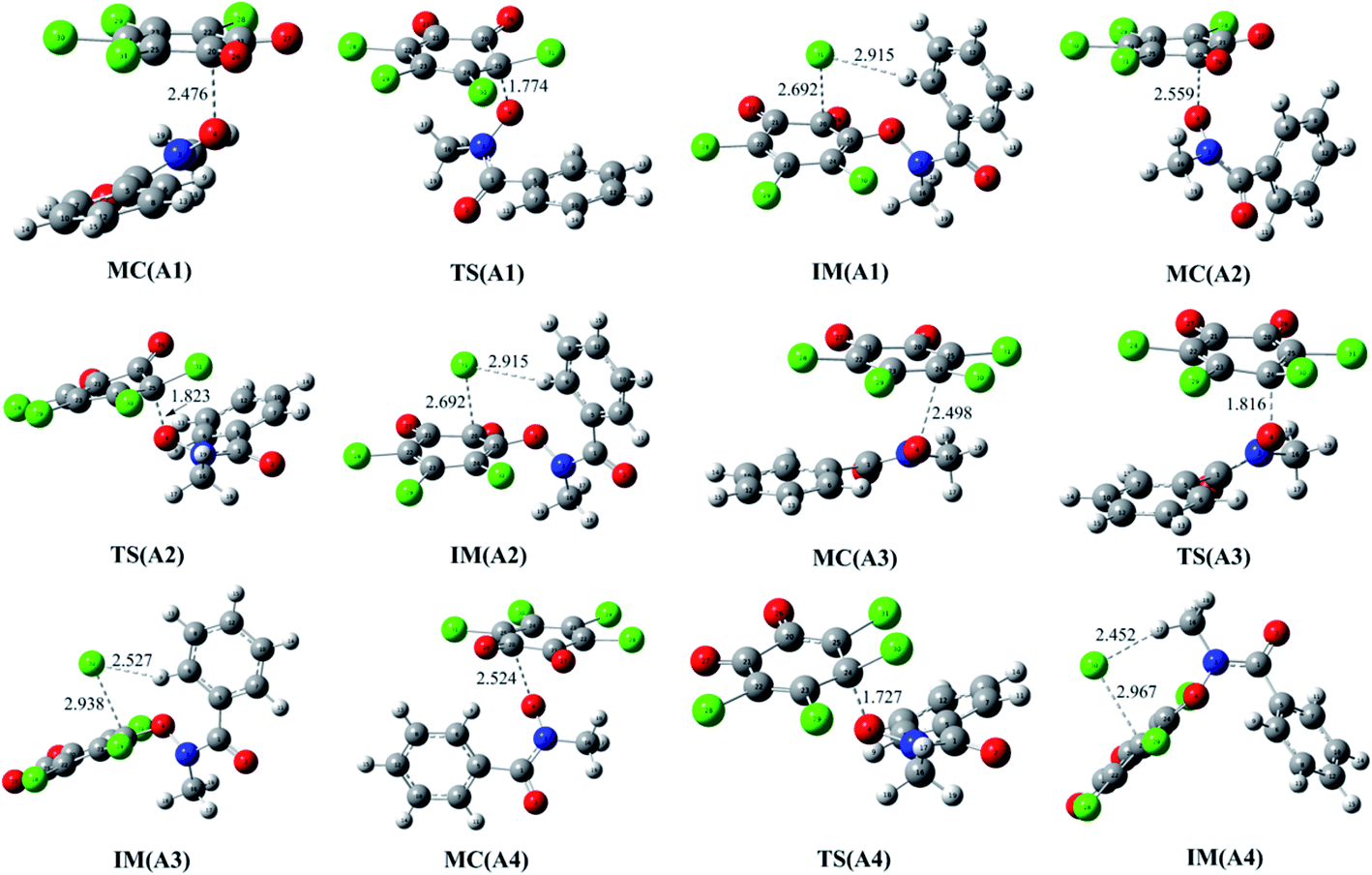 | ||
| Fig. 3 Optimized molecular complexes (MC), transition states (TS), and intermediates (IM) in the different reaction modes of o-TCBQ with N-MeBHA−. The selected interatomic distances are given in Å. | ||
As presented in Table 2, these MCs have been significantly stabilized ranging from 29.44 to 32.24 kcal mol−1 relative to the separated reactants, which are much larger than those of the MCs in the reactions involving neutral N-MeBHA. Moreover, the formation of these MCs is favorable exothermic and spontaneous process thermodynamically from the calculated negative values of the enthalpy and Gibbs free energy changes.
| Attack modes | MC | TS | IM |
|---|---|---|---|
| a All the units are in kcal mol−1. The data before and after slash refer to the relative energies, enthalpy changes, and Gibbs free energy changes, respectively. The data in parentheses refer to the corresponding results in aqueous solution. | |||
| A1 | −32.24/−31.76/−19.99 | −24.30 | −39.33/−38.84/−27.71 |
| A2 | −31.73/−31.18/−20.00 | −22.94 | −39.33/−38.34/−27.72 |
| A3 | −29.44/−29.04/−16.69 | −25.93 | −36.02/−35.52/−23.92 |
| (−9.55) | (−6.51) | (−28.62/−27.92/−19.56) | |
| A4 | −31.73/−31.18/−20.00 | −23.05 | −38.38/−37.80/−26.62 |
| p-A1 | −29.39 | −20.97 | −38.32/−37.83/−26.14 |
| p-A2 | −29.35 | −21.38 | −38.94/−38.48/−26.55 |
| (−9.86) | (−1.51) | (−29.44/−28.75/−21.08) | |
Moreover, given the fact that the reaction takes place in solution generally, so the solvent effect on the above reaction has been included on the basis of the reaction mode A3 possessing the lowest energy barrier. As a result, the original energy barrier has been increased by about 19.42 kcal mol−1 in aqueous solution, which is still lower by 6.51 kcal mol−1 in energy relative to the separated reactants. Actually, the energy barrier is only 3.04 kcal mol−1 relative to the molecular complex, which is comparable to that of the reaction of o-TCBQ with H2O2.30 Therefore, it is easy to occur for the title reaction in solution.
In addition, the reaction of p-TCBQ with N-MeBHA− has also been investigated for comparison. Similar to o-TCBQ, as shown in Fig. 4, the corresponding molecular complexes and transition states have also been located, where two reaction modes (p-A1 and p-A2) have been constructed due to the symmetry of p-TCBQ. As presented in Table 2, the energy barriers for p-A1 and p-A2 are −20.97 and −21.38 kcal mol−1 relative to the separated reactants. Moreover, the energy barrier has been increased to −1.51 kcal mol−1 if solvent effect is considered. Here, the low barrier is consistent with the fact that p-TCBQ can readily react with N-MeBHA under mild conditions.22 For comparison, the reaction profiles for the reactions of N-MeBHA− with o-TCBQ and p-TCBQ have been shown in Fig. 5. Obviously, the lower energy barrier for the reaction of N-MeBHA− with o-TCBQ suggests its higher reactivity than that of p-TCBQ.
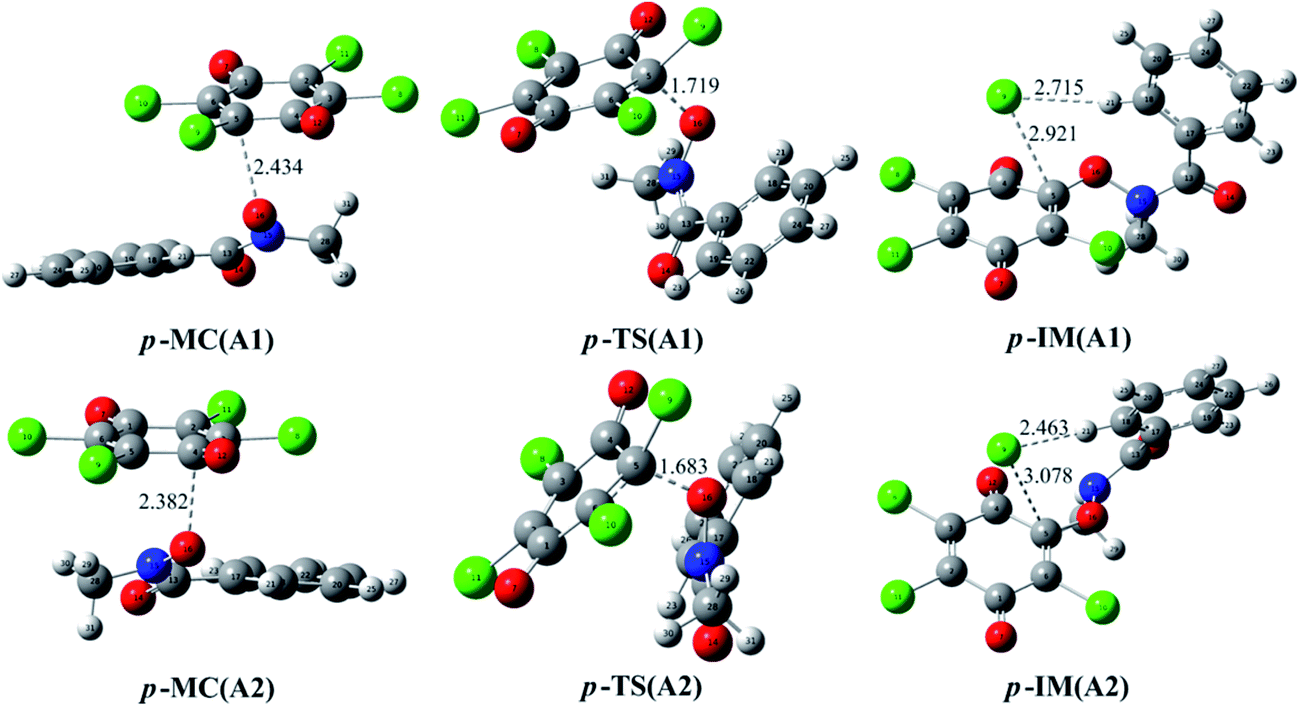 | ||
| Fig. 4 Optimized molecular complexes (MC), transition states (TS), and intermediates (IM) in the different reaction modes of p-TCBQ with N-MeBHA−. The selected interatomic distances are given in Å. | ||
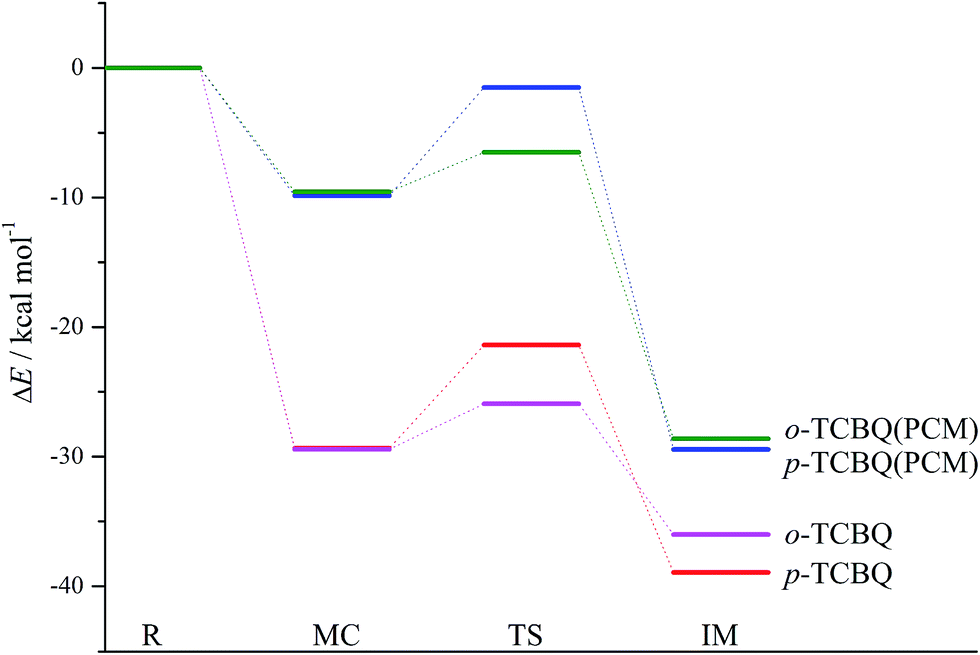 | ||
| Fig. 5 The reaction profiles for the nucleophilic attack processes of N-MeBHA− to o-TCBQ and p-TCBQ in the gas phase and in aqueous solution. | ||
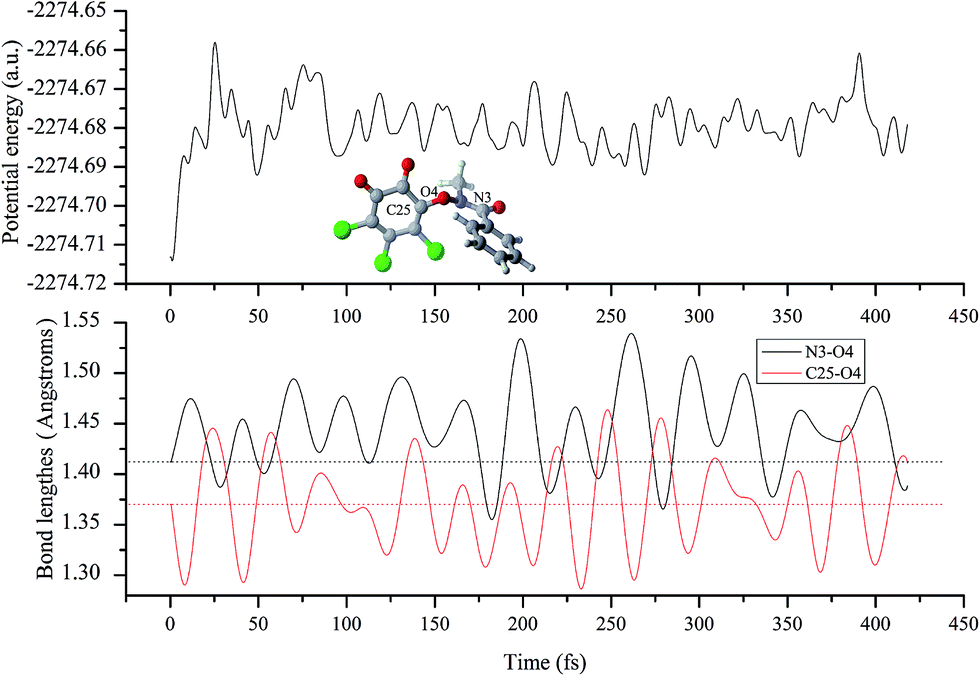 | ||
| Fig. 6 The variations of the potential energy and selected bond lengthes during the molecular dynamics for IM(A2) after the dissociation of Cl−. The dotted lines refer to the equilibrium distances in IM(A2). | ||
Subsequently, to confirm the decomposition mode of N3–O4 bond, its bond decomposition enthalpy (BDE) has been investigated on the basis of the IM(A2) since no transition state has been located for the cleavage of the N3–O4 bond. For the heterolytic cleavage of the N–O bond, the BDE is 140.82 and 165.23 kcal mol−1 for the decomposed species between N+ and O− fragments or the N− and O+ fragments, where the corresponding geometries have been given in Fig. S2 of the ESI† for reference. However, for the homolytic cleavage of the N–O bond, the BDE is only 6.50 kcal mol−1, which is much smaller than that of the heterolytic cleavage. Therefore, the decomposition of IM should occur via the homolytic cleavage of the N–O bond. For the sake of simplicity, the produced N- and O-centered radicals are named as ˙N(CH3)–COAr and o-CBQ˙, respectively.
As displayed in Fig. 7, for the ˙N(CH3)–COAr radical, the unpaired electron is mainly distributed around the N atom before and after structural relaxation. At the same time, small amounts of the unpaired electron have also been observed on the O atom. As for the o-CBQ˙ radical, the spin density redistribution from the O atom involved in the N–O cleavage to its adjacent C* atom attached to the Cl atom occurs upon structural relaxation. Therefore, the N and O atoms in ˙N(CH3)–COAr radical and the C* atom in o-CBQ˙ radical should be the active sites for the subsequent reactions.
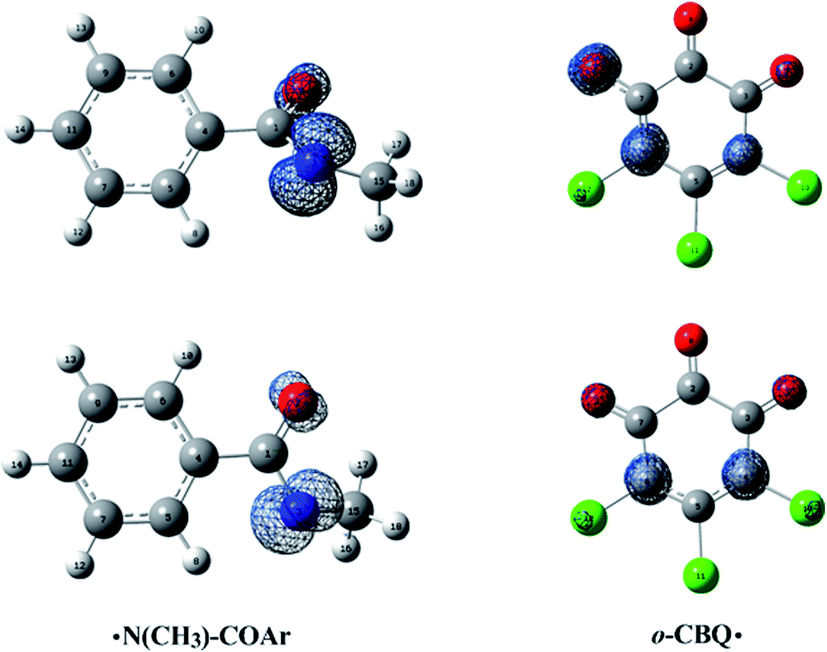 | ||
| Fig. 7 Spin density maps for the free radicals before and after structural relaxation (top and bottom) in the homolytic cleavage of the N–O bond of IM(A2) after the dissociation of Cl−. The isodensity contours are 0.01 electron bohr−3. | ||
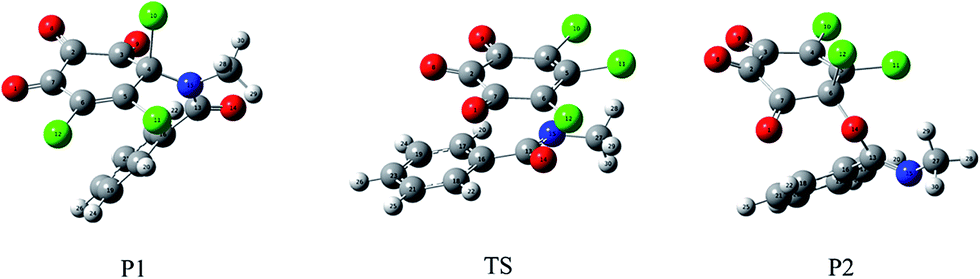 | ||
| Fig. 8 The formed C–N bonding and C–O bonding products for the title reaction and the transition state between them. | ||
| Complexes | ΔEInt | ΔH | ΔG | ΔERel |
|---|---|---|---|---|
| a All the units are in kcal mol−1. | ||||
| P1 | −31.17 | −36.07 | −20.12 | 0.00 |
| P2 | −17.10 | −21.27 | −6.34 | 14.53 |
Moreover, as shown in Fig. 8, a transition state has been located between P1 and P2, which is confirmed by the IRC calculation. The forward and reverse energy barriers are 43.04 and 28.52 kcal mol−1, where the direction from P1 to P2 is defined as the forward reaction. Obviously, it is difficult to interconvert between P1 and P2. Given the fact that no transition state has been observed in the formation processes of P1 and P2, so the major products for the title reaction should be the C–N bonding types as well as the minor C–O bonding types.
On the basis of the above analyses, the reaction mechanism for the title reaction can be summarized as follows. As shown in Fig. 9, o-TCBQ can react with the anionic form of the N-MeBHA. As the first step of the reaction, a molecular complex has been formed followed by the nucleophilic attack of N-MeBHA− to o-TCBQ to form an unstable intermediate containing N–O bond. After that, the unstable intermediate decomposes homolytically via the N–O bond to form the N- and O-centered radicals. For the O-centered radical, it can isomerize to the C-centered radical upon structural relaxation. Finally, these radicals interact with each other to form the C–N bonding products predominately as well as minor C–O bonding products. Certainly, relevant experiments are required to confirm the present results.
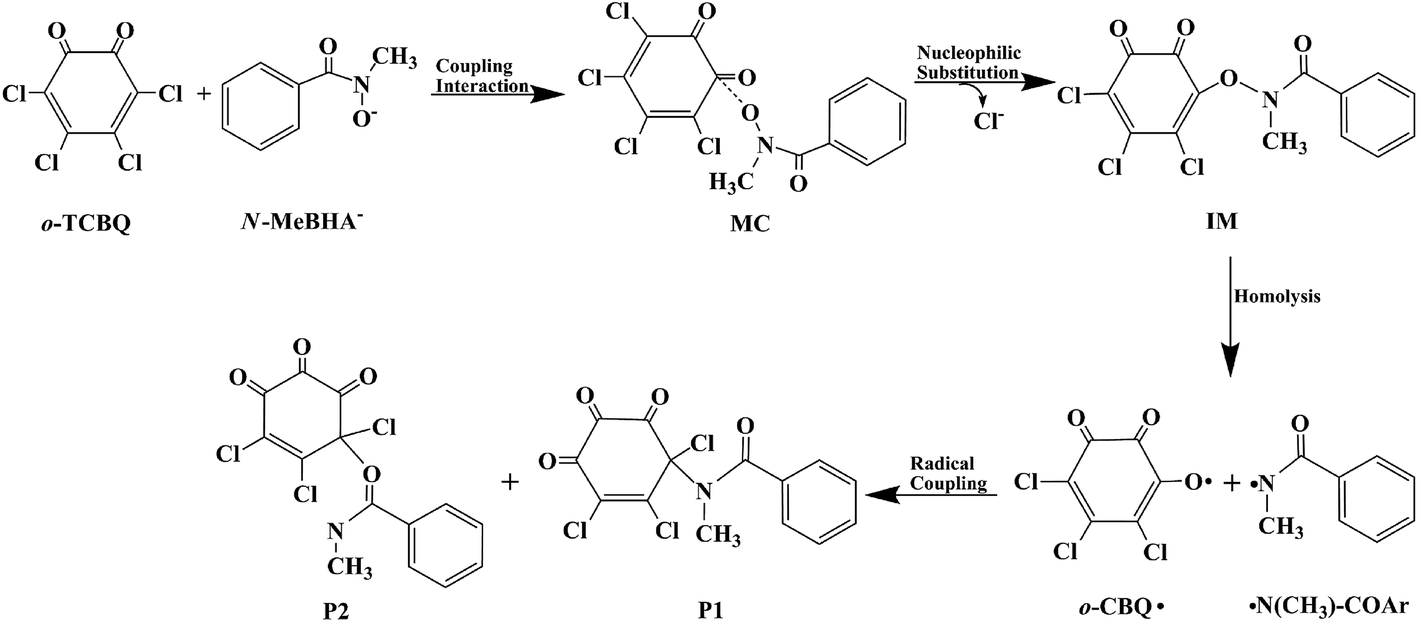 | ||
| Fig. 9 The proposed reaction mechanism for the reaction of o-TCBQ with N-MeBHA−. | ||
4. Conclusions
In this study, the reaction mechanism between o-TCBQ and N-MeBHA has been systematically investigated at the B3LYP/6-311++G(d,p) level. It was found that o-TCBQ can directly react with the anion of N-MeBHA through the following reaction mechanisms. Namely, a molecular complex has been located as the first step of the title reaction followed by the nucleophilic attack process, resulting in the formation of an unstable intermediate containing N–O bond. Subsequently, the unstable intermediate decomposes homolytically via the cleavage of N–O bond to produce the N- and O-centered radicals. For the O-centered radical, it can easily isomerize to the C-centered radical. Finally, the C-centered radical reacts with the N-centered radical to produce the major C–N bonding product, accompanying with the formation of minor C–O bonding product. In addition, it was found that the reactivity of o-TCBQ with N-MeBHA is higher than that of p-TCBQ. Hopefully, the present results can provide helpful information for the detection and identification of the reaction products experimentally. Certainly, relevant experiments are required to further confirm the present results.Acknowledgements
This work is supported by NSFC (21577076, 21303093, and 21003082) and the NSF of Shandong Province (ZR2014BM020).References
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Chem. Res. Toxicol., 2000, 13, 135–160 CrossRef CAS PubMed.
- Y. Song, B. A. Wagner, J. R. Witmer, H. J. Lehmler and G. R. Buettner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9725–9730 CrossRef CAS PubMed.
- B. Meunier, Science, 2002, 296, 270–271 CrossRef CAS PubMed.
- S. S. Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K. Schramm and T. J. Collins, Science, 2002, 296, 326–328 CrossRef PubMed.
- A. Sorokin, J. L. Seris and B. Meunier, Science, 1995, 268, 1163–1166 CAS.
- Y. L. Zhao, F. Qin, J. M. Boyd, J. Anichina and X. F. Li, Anal. Chem., 2010, 82, 4599–4605 CrossRef CAS PubMed.
- F. Qin, Y. Y. Zhao, Y. L. Zhao, J. M. Boyd, W. J. Zhou and X. F. Li, Angew. Chem., Int. Ed., 2010, 49, 790–792 CrossRef CAS PubMed.
- J. H. Li, W. Wang, B. Moe, H. L. Wang and X. F. Li, Chem. Res. Toxicol., 2015, 28, 306–318 CrossRef CAS PubMed.
- B. Z. Zhu, N. Kitrossky and M. Chevion, Biochem. Biophys. Res. Commun., 2000, 270, 942–946 CrossRef CAS PubMed.
- B. Z. Zhu, H. Zhao, B. Kalyanaraman and B. Frei, Free Radical Biol. Med., 2002, 32, 465–473 CrossRef CAS PubMed.
- B. Z. Zhu, B. Kalyanaraman and G. B. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17575–17578 CrossRef CAS PubMed.
- B. Z. Zhu and G. Q. Shan, Chem. Res. Toxicol., 2009, 22, 969–977 CrossRef CAS PubMed.
- B. Z. Zhu, L. Mao, C. H. Huang, H. Qin, R. M. Fan, B. Kalyanaraman and J. G. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16046–16051 CrossRef CAS PubMed.
- B. Z. Zhu, H. T. Zhao, B. Kalyanaraman, J. Liu, G. Q. Shan, Y. G. Du and B. Frei, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3698–3702 CrossRef CAS PubMed.
- B. Z. Zhu, G. Q. Shan, C. H. Huang, B. Kalyanaraman, L. Mao and Y. G. Du, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11466–11471 CrossRef CAS PubMed.
- C. H. Huang, F. R. Ren, G. Q. Shan, H. Qin, L. Mao and B. Z. Zhu, Chem. Res. Toxicol., 2015, 28, 831–837 CrossRef CAS PubMed.
- Y. Yu, J. Wong, D. B. Lovejoy, D. S. Kalinowski and D. R. Richardson, Clin. Cancer Res., 2006, 12, 6876–6883 CrossRef CAS PubMed.
- P. A. Marks and R. Breslow, Nat. Biotechnol., 2007, 25, 84–90 CrossRef CAS PubMed.
- N. N. Li, D. Zhao, M. Kirschbaum, C. Zhang, C. L. Lin, I. Todorov, F. Kandeel, S. Forman and D. F. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4796–4801 CrossRef CAS PubMed.
- B. Z. Zhu, J. G. Zhu, L. Mao, B. Kalyanaraman and G. Q. Shan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20686–20690 CrossRef CAS PubMed.
- B. Z. Zhu and C. H. Huang, Free Radical Biol. Med., 2011, 51, S152 CrossRef.
- C. H. Huang, Molecular mechanisms for novel radical and rearrangement reactions mediated by halogenated quinoid carcinogens, University of Chinese Academy of Sciences, 2013 Search PubMed.
- B. S. Das, S. G. Reid, J. L. Betts and K. Patrick, J. Fish. Res. Board Can., 1969, 26, 3055–3067 CrossRef CAS.
- X. W. Guo and H. Mayr, J. Am. Chem. Soc., 2014, 136, 11499–11512 CrossRef CAS PubMed.
- S. Maddila, V. D. B. C. Dasireddy and S. B. Jonnalagadda, Appl. Catal., B, 2013, 138–139, 149–160 CrossRef CAS.
- M. Freytag, P. G. Jones, R. Schmutzler and M. Yoshifuji, Heteroat. Chem., 2001, 12, 300–308 CrossRef CAS.
- M. Kot and W. Zaborska, J. Enzyme Inhib. Med. Chem., 2006, 21, 537–542 CrossRef CAS PubMed.
- H. R. Zare, M. Eslami, M. Namazian and M. L. Coote, J. Phys. Chem. B, 2009, 113, 8080–8085 CrossRef CAS PubMed.
- C. Guo, W. H. Wang, W. L. Feng and P. Li, RSC Adv., 2017, 7, 12775–12782 RSC.
- P. Li, C. Guo, W. L. Feng, Q. Sun and W. H. Wang, RSC Adv., 2017, 7, 22919–22926 RSC.
- P. Li, Z. Y. Ma, W. H. Wang, Y. Z. Zhai, H. T. Sun, S. W. Bi and Y. X. Bu, Phys. Chem. Chem. Phys., 2011, 13, 941–953 RSC.
- W. L. Feng, C. Ren, W. H. Wang, C. Guo, Q. Sun and P. Li, RSC Adv., 2016, 6, 48099–48108 RSC.
- L. M. Wang and A. L. Tang, Chemosphere, 2011, 82, 782–785 CrossRef CAS PubMed.
- X. B. Wang, Q. Fu and J. L. Yang, J. Phys. Chem. A, 2010, 114, 9083–9089 CrossRef CAS PubMed.
- S. L. Zhuang, H. F. Wang, K. K. Ding, J. Y. Wang, L. M. Pan, Y. L. Lu, Q. J. Liu and C. L. Zhang, Chemosphere, 2016, 144, 1050–1059 CrossRef CAS PubMed.
- S. L. Zhuang, X. Lv, L. M. Pan, L. P. Lu, Z. W. Ge, J. Y. Wang, J. P. Wang, J. S. Liu, W. P. Liu and C. L. Zhang, Environ. Pollut., 2017, 220, 616–624 CrossRef CAS PubMed.
- P. Li, Z. T. Shen, W. H. Wang, Z. Y. Ma, S. W. Bi, H. T. Sun and Y. X. Bu, Phys. Chem. Chem. Phys., 2010, 12, 5256–5267 RSC.
- W. L. Feng, C. Ren, W. H. Wang, C. Guo, Q. Sun and P. Li, Theor. Chem. Acc., 2016, 135, 190 CrossRef.
- W. H. Wang, X. X. Zhang, P. Li, Q. Sun, Z. Li, C. Ren and C. Guo, J. Phys. Chem. A, 2015, 119, 796–805 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 114, 9758–9763 CrossRef CAS.
- S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 115, 10291–10302 CrossRef CAS.
- H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2002, 117, 8694–8704 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
- P. Li, W. H. Wang, Q. Sun, Z. Li, A. J. Du, S. W. Bi and Y. Zhao, ChemPhysChem, 2013, 14, 2737–2743 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05083f |
| This journal is © The Royal Society of Chemistry 2017 |