
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn alternative, faster and simpler method for the formation of hierarchically porous ZnO particles and their thermoelectric performance†
Raymond V. Rivera Virtudazo a,
Quansheng Guoa,
Rudder Wua,
Toshiaki Takeib and
Takao Mori*a
a,
Quansheng Guoa,
Rudder Wua,
Toshiaki Takeib and
Takao Mori*a
aThermal Energy Materials Group, WPI Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0044, Japan. E-mail: MORI.Takao@nims.go.jp; Fax: +81-29-860-6280; Tel: +81-29-860-4323
bRNFS/Nanofabrication Group, WPI Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0044, Japan
First published on 27th June 2017
Abstract
We devised an alternative, faster and simple method to prepare hierarchically nanoporous ZnO powders (NZnO) via a quick interfacial reaction (double emulsion) from the mixtures of zinc nitrate hexahydrate (solution, Zn(NO3)2·6H2O), n-hexane (C6H14), ammonium bicarbonate (NH4HCO3), Tween 80 (C64H124O26) and Span 80 (C24H44O6), which was calcined at 450 °C. Due to the micro/nanopore structures, the obtained NZnO powders had a larger surface area (∼69.7 m2 g−1) than the commercial ZnO powders. For the thermoelectric evaluation, the textured NZnO pellets (T-NZnOP) were prepared by low pressure spark plasma sintering (SPS) using the newly synthesized NZnO powders (NZnO). In this study, the synthesized NZnO powders that were made into T-NZnOP (sample pellets) showed a significantly lower thermal conductivity with distinctive electrical properties when compared to the bulk commercial ZnO powders. The thermoelectric enhancement can be attributed to the nanopore distribution found in the porous T-NZnOP material which demonstrates the potential usefulness of this method for other porous oxide thermoelectric (TE) materials.
Introduction
Thermoelectric (TE) materials offer a good solution for energy problems by directly converting waste-heat to electric energy and vice versa. In general, the TE performance of the materials are evaluated by the dimensionless figure of merit ZT = σS2T/κ, where S is the Seebeck coefficient (thermopower), σ is the electrical conductivity, T is the absolute temperature and κ = κph + κel is the thermal conductivity (κ) composed of electronic (κel) and phonon contributions (κph).1–5 Some well-known TE materials for refrigeration or power generation are Bi2Te3, PbTe, Si1−xGex (ZT = 1 or better). However, the disadvantages of using these materials are their toxicity (Te based elements), expensive synthesis techniques and poor stability in air or at high temperature.6–8Oxide-TE materials offer a potential solution to the challenges faced by the conventional TE materials. Furthermore, there are several promising high temperature TE applications9 which require high temperature stable materials, such as, borides10 and oxides. Some notable examples of oxide-TE materials are NaxCo2O4 and SrTiO3.5,7,9–11 We believe that these oxide-based materials could provide an inexpensive, environmentally-friendly viable high temperature thermoelectric material. The progress in developing n-type oxide TE materials with comparable ZT is still lacking, which calls for more research efforts in this area. Using ZnO-based thermoelectric materials can be one of the good examples in this strategy.5–7,12–16
ZnO is a promising n-type semiconductor due to its good Seebeck coefficient (∼200 μV K−1) and substantial stability at elevated temperatures (up to 1000 °C).5,7 The Zn–O bond has a covalent character because of the small electronegativity difference which leads to a relatively large carrier mobility.7 However, the simple wurtzite crystal structure and light Zn and O elements result in thermal conductivity as high as ∼100 W m−1 K−1 (ref. 17–19) at RT (for example in ref. 17–19). The reduction of κ becomes a primary (associated with phonon selective scattering4,17,20–22) and essential task towards improving the ZT of ZnO. A low-resistance n-type ZnO sintered body is usually obtained by doping (∼0.5–5 mol%) Al2O3 followed by burning at a high temperature of ∼1673 K (∼1400 °C).7,16
Recently, the porous/textured oxide-TE materials (e.g. nano inclusion in bulk ZnO) were found to reduce κ and play a major role in improving the TE. The reduction of κ was attributed to nanograin, grain boundary, point defects, and porosity (∼90% relative density).2,4,16,23,24 As a consequence, research efforts have mostly been focused on synthesizing/producing hierarchically micro/nanoporous oxide powders (e.g. ZnO) for thermoelectric applications. Therefore, we addressed this issue by developing a simple, faster and eco-friendly method in producing a nanoporous ZnO powders (NZnO). In this study, we used a unique NZnO powder that was synthesized via a simple double emulsion (interfacial reaction) method. Then a low pressured spark plasma sintering (SPS under Ar) was done to establish a nanostructured oxide-based TE material. Low-pressured SPS sintering can fabricate the porous TE sample and at the same time porous nanostructures can generate enhanced TE results.2,4,8
The effect of porosity on TE properties of textured NZnO pellets (T-NZnOP) upon increasing the SPS sintering temperature was investigated. This improvement might be counterbalanced by a decrease in electrical conductivity (σ) which has previously been reported.5 To fully investigate this new approach in producing NZnO powders for TE application, doping was not included in this paper. This investigation is expected to understand and maximize the advantages of existing nanostructure/porous oxide-TE materials in application for TE applications. This also leads to energy saving because the effective nanostructuring of thermoelectric materials is expected to lead to a better efficiency.
Experimental
Chemicals/materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O), ammonium bicarbonate (NH4HCO3, >99%), hexane (C6H14, >99%), Tween 80 (C64H124O26, Mw ∼ 1310), Span 80 (C24H44O6, Mw ∼ 428.62), ZnO commercial powders (ZnO-Com, as reference sample) were obtained from Sigma-Aldrich Company. Ethanol (C2H5OH, >99%) was purchased from Wako Pure Chemical Industries. Ultrapure water was generated by the Milli-Q integral purification system.Synthesis and formation of nanoporous ZnO (NZnO) powders
NZnO was prepared by interfacial reaction at room temperature (RT) similar to the previous report.25,26 NZnO was done as follows: the total volume of the zinc nitrate hexahydrate solution (26.8 g, Zn(NO3)2·6H2O, 90 mmol)‡ was fixed to 36 mL as water phase 1 (WP1). The WP1 was added to the oil phase (OP) consisting of hexane solution (72 mL) with Tween 80 (1.00 g) and Span 80 (0.50 g). The resulting two-phase solution (WP1/OP) was emulsified by a homogenizer for 1 min. The WP1/OP emulsion was immediately poured into the (precipitant) ammonium bicarbonate aqueous solution (2 mol L−1; 250 mL, with stirring) as water phase 2 (WP2). After stirring for 4 h, the WP1/OP/WP2 solution formed a white colloidal suspension. Then the WP1/OP/WP2 colloidal solution was filtered, washed with deionized water/ethanol, vacuum dried at 120 °C for 24 h and then calcined at 450 °C for 4 h aging for the remove the excess surfactants and then formed NZnO powders (see, Fig. S1†). The concentration of the precipitant (WP2) and the ratio concentration of the surfactants (Tween 80 and Span 80) were not changed. The volume ratios of WP1 and WP2 were fixed (WP1/WP2 = 1/7), (see Fig. 1). | ||
| Fig. 1 Schematic illustration for the formation of nanoporous ZnO (NZnO) powders prepared by double emulsion method (interfacial reaction) then low pressured Spark Plasma Sintering (SPS) for thermoelectric (TE) investigation. | ||
Textured NZnO pellet (T-NZnOP) samples sintered using low pressured spark plasma sintering (SPS)
The textured NZnO pellets (T-NZnOP) samples were prepared by sintering using a low pressured Spark Plasma Sintering (SPS) at 550 °C, 750 °C and 950 °C.§ Initially, during low pressured SPS, the pressure was set primarily at 2.8 kN (in RT). Then it was slowly adjusted to 2.2 kN, wherein the heating rate was ∼100 °C min−1 and held for 5 min in an argon atmosphere, followed by cooling at RT.For comparison, the ZnO-Com pellets (T-CZnO-Com) were also prepared by Spark Plasma Sintering (SPS) techniques at 550 °C, 950 °C and 1200 °C as our actual experimental reference datas.¶ During SPS, the pressure was set primarily at ∼2.8 kN (in RT). Then it was slowly adjusted to ∼3.5 kN, wherein the heating rate was ∼100 °C min−1 and held for 5 min in an Ar atmosphere, then cooling at RT.
The sintered sample pellets had a density of ∼91% of the theoretical density after low pressured SPS sintering, especially when sintered at 750 °C and 950 °C. However, a lower density (∼78% of the theoretical density, porous pellets) was made particularly when sintering at the relatively low temperature of 550 °C. Two different pellets from the same sample powders (NZnO) were sintered. After polishing/cleaning all the surfaces of the two different sample pellets, the 1st pellet was used to measure the thermal conductivity (κ) analysis, while the 2nd pellet was cut by a diamond cutter for electrical resistivity (ρ = 1/σ) and Seebeck coefficient (S) measurements simultaneously.
Characterization
Results and discussion
In this research, we adopted the systematic method done by the previous study to maintain the chemical process for the basic formation of hierarchically nanoporous ZnO (NZnO).26,30,31 The zinc nitrate hexahydrate solution, precipitant, and surfactants in WP1, WP2, and OP, respectively, were rapidly mixed, filtered and dried. Then the NZnO powders were formed after annealing at 450 °C. These parameters were maintained to achieve the porous structure orientation and particle size distribution of the NZnO powders (see Fig. S1, S2a.1 and a.2†).We checked the critical weight loss of the as-synthesized (dried) zinc hydroxide carbonate (Zn5(CO3)2(OH)6)31,32 powders by TG/DTA analysis to detect the thermal properties (see Fig. 2a) and the minimum calcination temperature to form ZnO. As shown in Fig. 2a, the prepared Zn5(CO3)2(OH)6 powders decomposed in one relatively sharp step, with peak decomposition temperature at ∼240–250 °C (∼25.2%). Initially, the first percentage weight-loss slightly increased at below 200 °C (∼4%) which was due to the evaporation of physically adsorbed water (H2O) and residual solvent. The second percentage weight-lost became faster, occurred at ∼200–450 °C (∼25.2%). This was due to the decomposition of carbonates groups and the remaining organic substances (surfactant).25,30,31 From these results, the endothermic peak and weight loss was attributed to de-carboxylation and de-hydroxylation.31 For this reason, to satisfy the formation of ZnO structure, calcination was done only at 450 °C (see Fig. S2b†).
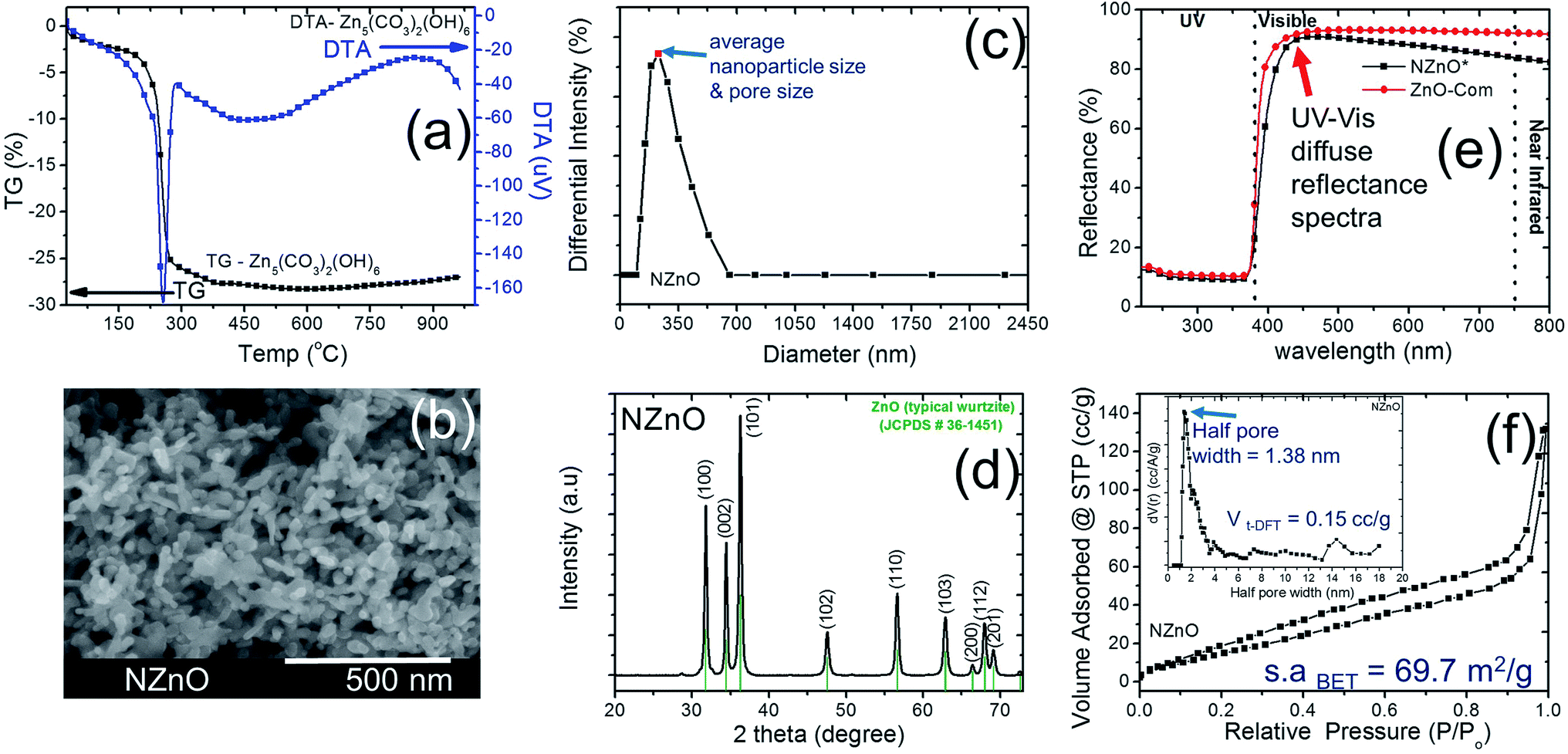 | ||
| Fig. 2 (a) Typical TG/DTA analysis of as-synthesized Zn5(CO3)2(OH)6 powder. (b) SEM image of NZnO particles after calcination at 450 °C. (c) Average particle size distribution of NZnO particles after calcination at 450 °C. (d) Typical X-ray diffraction pattern of NZnO powder (wurtzite structure) after calcination at 450 °C. (e) UV-vis diffuse reflectance spectra of NZnO and ZnO commercial powder (ZnO-Com). (f) N2 adsorption–desorption isotherm for the NZnO particles (inset is the pore size distribution using DFT method). | ||
After calcination, the surface morphology and the stability of the porous structure of NZnO powders were verified using SEM and STEM (Fig. 1 and 2b) micrograph. The images indicate a successful formation of NZnO via a double emulsion and then calcined at 450 °C, (transmission mode (T), Fig. 1) the nano-size particles formed a cluster of nanoporous structured particles (see Fig. 2b and S2d.2–d.4†). The observed surface morphology of the porous NZnO powders matched the results of average PSD. The obtained NZnO powders offered a broad size distribution (combined nanopore and particles size) with a diameter ranging from 200 nm to 700 nm (see, Fig. 2c and S2c†). The main crystal structure of this powders appears to be similar to the wurtzite structure of pure ZnO (JCPDS card no. 36-1451), with lattice constants a = 3.25 Å and c = 5.21 Å (ref. 31–34) (see, Fig. S2a.2†). The diffuse reflectance spectra of the NZnO was validated and compared with commercial ZnO powders using UV-Vis curves as shown in Fig. 2e. The wavelengths at 350 nm and 550 nm in reflection curve demonstrated a protection ability of ultraviolet radiation and a reflection property of visible radiation respectively. The NZnO powders had a good reflectance at visible radiation (about 95%) at 450 nm. Both ZnO powders showed a sudden decrease of reflectance intensity at a wavelength around 445 nm (see Fig. S3c.1 and c.2†). In this case, the observed data is consistent with what has been previously reported for ZnO nano-powders.33,35,36
The aggregation of these nanoparticles leads to the formation of the micro/nanoporous structure of ZnO. In order to confirm the distinct property of the porosity of the NZnO powders, N2 adsorption–desorption isotherms at 77 K using the BET method for surface area (s.aBET) and DFT method for pore (space)-size distribution (inset, PD) were carried out as shown in Fig. 2d.
The characteristic feature of the curves can be identified as the type IV,27 indicating the presence of micro/mesopores of the NZnO powders.27,30 The micro/mesoporosity was further indicated by the pore size distribution curve using DFT method as shown in the inset of Fig. 2d. The analysis shows one peak at ∼1.38 nm and most of the pore sizes are located between 2 and 14 nm, indicating that the material has a narrower pore size distribution with an average pore diameter ∼15 nm. The surface area for the nanoporous NZnO powders is ∼69.7 m2 g−1 based on the BET model and the pore volume was 0.15 cm3 g−1 (DFT method but for BJH method, the pore volume was 0.196 cm3 g−1). The surface area of the NZnO powders is significantly greater than that of commercial ZnO powders (surface area of 10 m2 g−1 to 16 m2 g−1,30 see Fig. S3a.1, a.2 and S3b†). We indicated that the pores are attributed to the interparticle spacing from the self-aggregation of the individual ZnO nanoparticles and the surfactant that helps form micro/mesoporosity.25,26,30,37
In addition, the two surfactants, Tween 80 (C64H124O26) and Span 80 (C24H44O6), are not only aiding the aggregation of nanoparticles into micro/nanoporous but may also act as a capping ligand to limit further growth of ZnO nanoparticles.25,38 As a result, the NZnO powders was formed by the assistance of porous metal–organic frameworks that provide a void foster for the formation of micro/nanoporous structure and the surfactants that aid the self-aggregation of ZnO nanoparticles. However, the specific growth mechanism still need further observation.
The nanostructure of the prepared nanoporous ZnO powder (NZnO) was further characterized by Transmission Electron Microscopy (TEM), High Resolution-Transmission Electron Microscopy (HR-TEM) and Fast Fourier Transformation (FFT) image. Fig. 3 shows the TEM image (a), HR-TEM image (b) and FFT pattern (c) of a single NZnO particle (see, Fig. S6†). The TEM image confirms the nanostructured porous structure of the prepared ZnO powder (see Fig. S6†). The HR-TEM image shows clear and parallel lattice fringes. The lattice spacing is ∼0.261 nm, corresponding to (002) planes of the hexagonal phase of ZnO. The FFT pattern shows that the spots arrange in neat rows, which can be ascribed to wurtzite ZnO and is consistent with the XRD pattern. The HR-TEM and FFT patterns indicate that the prepared porous ZnO particles are single-crystalline, which is similar to the single-crystalline porous ZnO reported in the literature.34,39–41
 | ||
| Fig. 3 (a, b) HR-TEM micrograph of NZnO particles after calcination at 450 °C; (c) enlarged HR-TEM image of selected region marked yellow square from (b) and (d) fast Fourier transformation (FFT) image of the individual NZnO particles. | ||
To investigate the TE performance, the NZnO powders was sintered using low pressured (∼2.2 kN) SPS with different sintering temperatures of 550 °C (T-NZnOP-500C), 750 °C (T-NZnOP-750C) and 950 °C (T-NZnOP-950C) then polished before the TE analysis. Hereafter, the XRD analyses of all the sample textured pellets (T-NZnOP) with increasing sintering temperature (Fig. 4a–c) showed no prominent peaks of impurity other than ZnO structure which confirms that the phase of the material was not affected during low-pressured SPS (see, Fig. S5a and b†). The Bragg reflections for the T-NZnOP belong to the hexagonal (ZnO) wurtzite phase6,7 (JCPDS#36-1451), similar to that of NZnO powders. But the intensity of the (002) reflection is to some extent decreased, especially for the T-NZnOP-950C sample, while for the (100) reflection, the intensity is slightly increased compared to the T-NZnOP-550C (Fig. 4a) and NZnO powders (Fig. 2d and S6†). This indicates a favorable growth along the c-axis of the crystals,42,43 suggesting that the externally applied low-pressure and the pulsed current during sintering can define the texture direction. Hence, we observed this grain texturing in Fig. 4d–i which shows the SEM micrographs of the fractured surface (perpendicular to the pressure direction of the samples) for T-NZnOP-550C (Fig. 4f–i), T-NZnOP-750C (Fig. 4e–h) and T-NZnOP-950C (Fig. 4d–g) using low pressured SPS. The images of T-NZnOP-750C (Fig. 4e) and T-NZnOP-950C (Fig. 4d) showed significant texturing (sintered grains) compared to T-NZnOP-550C (Fig. 4f–i, S5a and S7a,† macropore orientation exist). The bulk relative density of the sintered pellet samples is in the range of ∼78–91%. The density of the specimens is clearly affected by the sintering temperature. But in this case, the NZnO powder had a lower sintering temperature compared to commercial ZnO powders. The grain growth was enhanced even at a low sintering temperature as low as ∼750 °C. This can be attributed to the high surface area of the NZnO powders.44,45 This observation is quite unique compared with the previous findings in the literature, wherein their sintering temperature ranges from 1200 to 1400 °C.4,6,7,46
 | ||
| Fig. 4 (a–c) XRD pattern of T-NZnOP specimens cut perpendicular to the pressure direction with increasing SPS temperature at 823 K (a; 550 °C), 1023 K (b; 750 °C) and 1223 K (c; 950 °C). SEM images of T-NZnOP specimens cut parallel to the pressure direction magnified at 500× (d–f) and 5000× (g–i) with increasing SPS temperature at 823 K (f and i; 550 °C), 1023 K (e and h; 750 °C) and 1223 K (d and g; 950 °C). | ||
The impact of the micro/nanoporous ZnO powders on the TE properties resulted in a distinct enhancement when compared to the previous reported data.2,6,7 The temperature dependence of the electrical resistivity (1/σ, Fig. 5a) and Seebeck coefficient (S, Fig. 5b) for this study's T-NZnOP samples sintered at different temperatures are shown in Fig. 5. As observed in Fig. 5a, the resistivity of all the samples (T-NZnOP) show a decreasing trend with increase of sintering temperature.7,46 This can be explained by the larger grain size (Fig. 4e–h and d–g, higher densities) resulting from increasing the sintering temperature.2,46,47 The high electrical resistance of T-NZnOP-550C* can definitely be attributed to the presence of macropores (Fig. 4f–i), which can strongly contribute to electron scattering. It can be implied that increasing the macro/nanopore structuring and decreasing the sintering temperature can drastically increase the electrical resistivity.46 The temperature dependence of the electrical resistivity of the samples, T-NZnOP-750C and T-NZnOP-950C, with a larger grain growth are similar to that of reported ZnO-based materials with relatively low electrical resistivity, showing small increases as temperature increases (i.e. metallic dependence).12–15
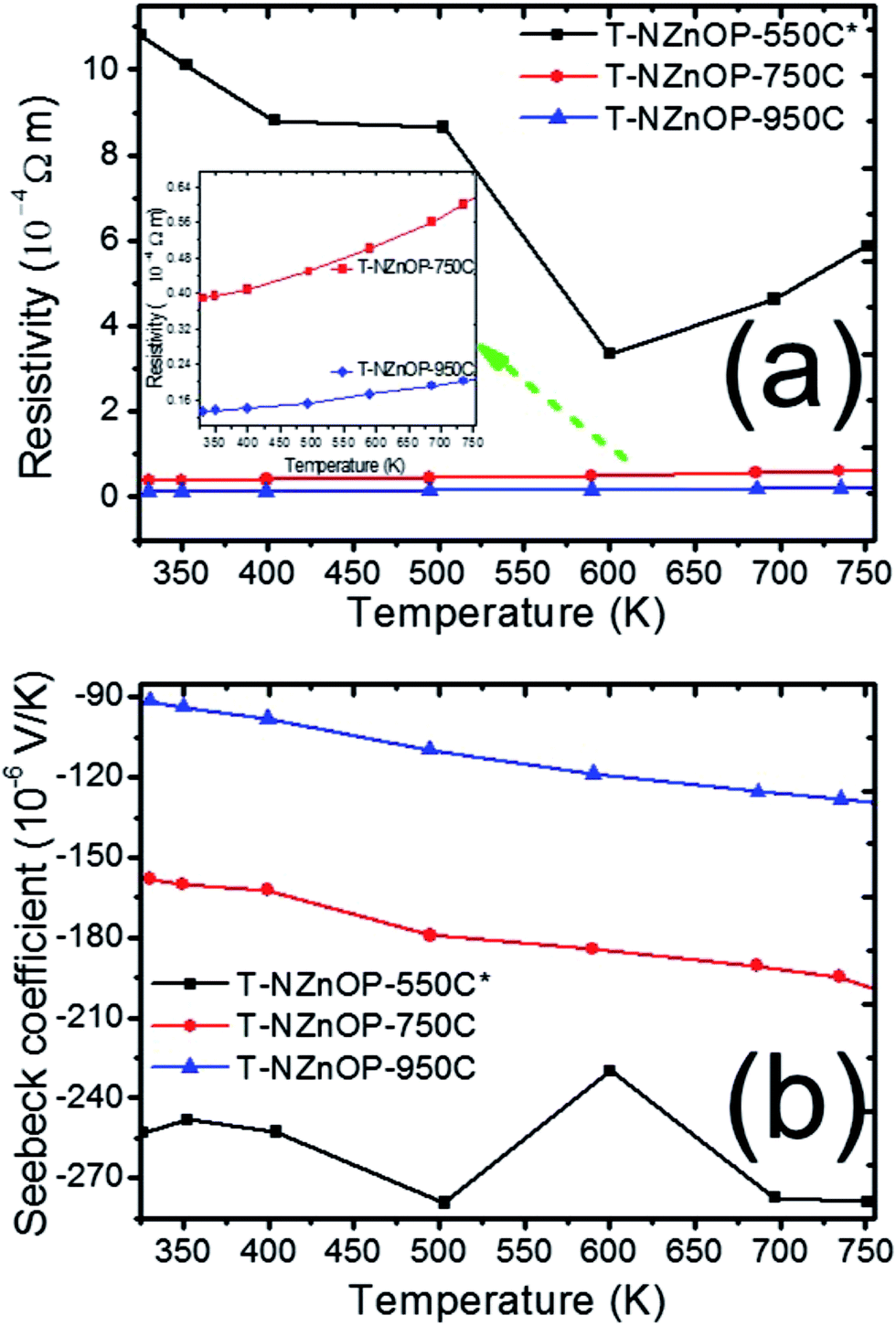 | ||
| Fig. 5 Temperature dependence of (a) resistivity and (b) Seebeck coefficient of T-NZnOP sample sintered at 823 K (550 °C), 1023 K (750 °C) and 1223 K (950 °C). | ||
The sintering dependence observed for the electrical resistivity is also observed for the Seebeck coefficient (S), as shown in Fig. 5b. The negative values indicate that the materials have n-type semiconducting behaviour where the major carriers are electrons.6,7 The absolute value of the Seebeck coefficient (S) tends to decrease with higher sintering temperature of the sample pellets. Furthermore, the magnitude S increases with increasing temperature range of all the samples. The measured absolute value of S for T-NZnOP-550C*, T-NZnOP-750C and T-NZnOP-950C were ∼−253 to −278.9 × 10−6 V K−1, ∼−160 to −204 × 10−6 V K−1 and ∼−91.0 to −135 × 10−6 V K−1 in temperature ranges from 325 to 750 K (Fig. 5b), respectively, which is similar to the reported values.6,7 But the difference is that our samples were sintered at a lower temperature. As expected, we found that the resistivity of T-NZnOP increases (with decreasing sintering temperature) with the increasing absolute value of S.
The effect of the macro/nanopore structuring on the thermal conductivities (κ) of different sintered samples is evident. Fig. 6a illustrates how the RT thermal conductivity (κ) of the samples T-NZnOP-550C*, TNZnOP-750C and T-NZnOP-950C varied upon increasing the sintering temperature. It rapidly increases with the increase in the sintering temperature of the samples. The measured κ of T-NZnOP-550C*, T-NZnOP-750C and T-NZnOP-950C were ∼9.65 to 2.91 W m−1 K−1, ∼46.9 to 8.56 W m−1 K−1 and ∼39.72 to 7.85 W m−1 K−1 in the temperature range of 325 to 750 K, respectively (Fig. 6a).7 This dependence is reasonable because of the grain growth with higher sintering temperatures, wherein the T-NZnOP-750C and T-NZnOP-950C contain grains in the micrometer range (see Fig. 4e–h and d–g), considerably larger than the mean free path of ∼30 nm for phonons in ZnO.2,22,48 The (κ) thermal conductivity value (especially T-NZnOP-750C and T-NZnOP-950C) is considerably lower compared with the previous reported data of normal bulk ZnO-based materials (∼100 W m−1 K−1 at RT).6,7,17–19 We also note that for our present T-ZnOP samples, the sintering temperatures of T-ZnOP were lower compared to typical bulk ZnO-based material wherein their sintering temperature required to reach as high as 1200–1400 °C.6,12–15 Generally, the low (κ) thermal conductivity achieved by the T-ZnOP samples were mainly attributed to the relative suppression of grain growth due to our low-temperature sintering and the formation of macro/nanopore structuring owing to our (synthesized) starting nanoporous ZnO powders. These results clearly validate that the macro/nanopore structuring and interface scattering of the raw powders (ex: NZnO) are very important in reducing the total thermal conductivity in fabricating a TE materials.2,48
 | ||
| Fig. 6 Temperature dependence of (a) thermal conductivity, (b) power factor and (c) dimensionless figure of merit of T-NZnOP specimens sintered at 823 K (550 °C), 1023 K (750 °C) and 1223 K (950 °C). | ||
From the electrical resistivity (ρ = σ−1) and the absolute value of the Seebeck coefficient (S) we can plot the power factor (PF = S2σ),2,5 as displayed in Fig. 6b. With the measurement temperature ranging from 325 to 750 K, the power factor (PF) of T-NZnOP-550C*, T-NZnOP-750C and T-NZnOP-950C were ∼0.59 to 1.32 × 10−4 W m−1 K−2, ∼6.43 to 6.33 × 10−4 W m−1 K−2 and ∼6.34 to 8.17 × 10−4 W m−1 K−2, respectively (Fig. 6b), which is similar to the reported values for undoped bulk ZnO6,7 but done at a lesser pressure with lower sintering temperature by SPS when compared to the referred ZnO datas (ref. 17–19). As observed, with the increase in the sintering temperature of the samples, the PF increases correlates with the decrease in the electrical resistivity.
Compared to the ZnO commercial powder (T-CZnO-Com-1200C, Fig. S8a†) and the previous data on the normal bulk ZnO material (∼100 W m−1 K−1 at RT, for example in ref. 17–19),17–19 our sintered T-NZnOP sample pellets (especially, T-ZnOP-750C, ∼46.9 W m−1 K−1 at RT) showed reduction in thermal conductivity. Understandably, this is due to the defects and grain boundary scattering found in our sample pellets of T-NZnOP that can lower the thermal conductivity6,7,17 as compared to the normal bulk ZnO samples (Fig. S8a†) and referred ZnO datas.17–19 Thus, this leads to ∼15 times (0.07501/0.005; actual porous ZnO/ZnO commercial powder (T-CZnO-Com-950C), Fig. S9c†) or ∼7.4 times (0.07501/0.0102; actual porous ZnO/ZnO referred data, ref. 7, Fig. S10†) improvement of ZT when compared to the actual T-CZnO-Com-1200C, T-ZnO-Com-950C and previous results of pure ZnO results,7 respectively. In addition, the sintered T-NZnOP (especially, T-NZnOP-750C and T-ZnOP-950C) obtained results that exhibited an increase in PF which improved the total ZT values as compared to the previous nanostructured undoped ZnO samples.6,7,49,50
Henceforth, the dimensionless figure of merit (ZT) is as shown in Fig. 6c. The results indicate that the T-NZnOP (especially, T-ZnOP-950C) with micro/nanopore structuring using NZnO powder significantly enhance the TE performance due to increase PF and a reduction in the thermal conductivity if compared with bulk ZnO.17–19 In our results, this corresponds to a maximum ZT value of ∼0.075 for T-NZnOP-950C at ∼750 K. In addition, even for the very low-sintering temperature T-NZnOP-500C* sample with macro/nanopores (Fig. 4f–i) and low power factor, the ZT value is ∼0.03 at ∼750 K, which is still slightly higher compared to actual T-CNZnO-Com-550C (ZT = ∼0.011, Fig. S9c†) and some reported ZT values of based pure ZnO materials.6,7,16
It is evident that the effect of nanoporous ZnO (NZno) powders contributed to the unique micro/nanostructuring of the NZnO pellet (T-NZnOP). A sequence of T-NZnOP micro/nanopore structuring was achieved by increasing the sintering temperature which produces an interesting thermoelectric performance. For instance, the T-NZnOP-550C* samples showed a significantly reduced thermal conductivity with comparative or superior high temperature power factor values as compared to non-nanostructured forms and commercial ZnO powders (Fig. S5, S7–S9†). This can be attributed to the alteration produced by micro/nanopore structuring (with high surface area) of the raw powders (NZnO) and particle–particle contacts. Regarding sintering temperature dependence, the samples sintered at 750 °C and 950 °C exhibited higher thermal conductivity and electrical conductivity compared with the 550 °C sintered sample. This dependence is in accordance with the variation of the microstructural observation.
Conclusions
In summary, our process in producing a nanostructured porous ZnO particles has demonstrated advantage for fabricating a thermoelectric material because of the micro/nanostructured grains which reduce thermal conductivity when compared with the reference ZnO commercial powder. The micro/nanoporous ZnO powders were synthesized via a double emulsion (simple interfacial reaction) and then by a low-pressure SPS for TE evaluation. As a result of the reduced thermal conductivity and a comparable or superior power factor, the textured micro/nanostructured ZnO (T-NZnOP) bulk ceramics showed a good enhancement of ZT up to 750 K, with a maximum ZT = ∼0.075 for T-ZnOP-950C. The ZT value is ∼15 times that of commercial powder sintered under the same conditions. To achieve further higher ZT values, our next plan is to explore doped ZnO samples. Furthermore, this report can open up new ideas for other researchers to improve their TE-oxide materials by utilizing this technique. TE-oxide materials mostly have an advantage because it does not use costly rare earth or any air sensitive elements. This makes the material cost low, makes the processing easier, allows the material to be stored in ambient conditions and still gives a good ZT. We expect that this report will play an important role for improving and developing other porous/nanostructured TE oxide materials and TE devices in the future.Acknowledgements
This research was supported by JST CREST Grant Number JPMJCR15Q6, Japan and JSPS KAKENHI Grant Number JP16H06441. Support from National Institute for Materials Science (NIMS) is also acknowledged. We would like to express thanks to the technical support staff members (Makito Nakatsu and Dr Hidehiko Tanaka) of the International Center for Materials Nanoarchitectonics (MANA) at NIMS for assisting the authors with the use of analytical instruments. We are also thankful to Dr Leila Rubia (De Montfort University, Leicester UK) for her time and professional expertise in proofreading our manuscript.Notes and references
- T. M. Tritt and M. Subramanian, MRS Bull., 2006, 31, 188–198 CrossRef.
- K. Koumoto and T. Mori, Thermoelectric Nanomaterials, Springer, 2013 Search PubMed.
- G. Nolas, J. Sharp and J. Goldsmid, Thermoelectrics: Basic Principles and New Materials Developments, Springer Science & Business Media, 2001 Search PubMed.
- A. U. Khan, K. Kobayashi, D.-M. Tang, Y. Yamauchi, K. Hasegawa, M. Mitome, Y. Xue, B. Jiang, K. Tsuchiya, D. Golberg, Y. Bando and T. Mori, Nano Energy, 2017, 31, 152–159 CrossRef CAS.
- K. Koumoto, R. Funahashi, E. Guilmeau, Y. Miyazaki, A. Weidenkaff, Y. Wang and C. Wan, J. Am. Ceram. Soc., 2013, 96, 1–23 CrossRef CAS.
- J. Hoemke, A. U. Khan, H. Yoshida, E. Tochigi, N. Shibata, Y. Ikuhara and Y. Sakka, J. Ceram. Soc. Jpn., 2016, 124, 515–522 CrossRef CAS.
- E. Guilmeau, P. Díaz-Chao, O. I. Lebedev, A. Rečnik, M. C. Schäfer, F. Delorme, F. Giovannelli, M. Košir and S. Bernik, Inorg. Chem., 2017, 56, 480–487 CrossRef CAS PubMed.
- H. Ning, G. D. Mastrorillo, S. Grasso, B. Du, T. Mori, C. Hu, Y. Xu, K. Simpson, G. Maizza and M. J. Reece, J. Mater. Chem. A, 2015, 3, 17426–17432 CAS.
- T. Mori, JOM, 2016, 68, 2673–2679 CrossRef CAS.
- O. Sologub, L. Salamakha, B. Stöger, Y. Michiue and T. Mori, Acta Mater., 2017, 122, 378–385 CrossRef CAS.
- D. Srivastava, C. Norman, F. Azough, M. C. Schäfer, E. Guilmeau, D. Kepaptsoglou, Q. M. Ramasse, G. Nicotra and R. Freer, Phys. Chem. Chem. Phys., 2016, 18, 26475–26486 RSC.
- I. Terasaki, R. Okazaki and H. Ohta, Scr. Mater., 2016, 111, 23–28 CrossRef CAS.
- Y. Michiue, H. Nishijima, Y. Suzuki and T. Mori, Solid State Sci., 2017, 65, 29–32 CrossRef CAS.
- Y. Michiue, T. Mori, A. Prytuliak, Y. Matsushita, M. Tanaka and N. Kimizuka, RSC Adv., 2011, 1, 1788–1793 RSC.
- M. Ohtaki, T. Tsubota, K. Eguchi and H. Arai, J. Appl. Phys., 1996, 79, 1816–1818 CrossRef CAS.
- K. Koumoto, Y. Wang, R. Zhang, A. Kosuga and R. Funahashi, Annu. Rev. Mater. Res., 2010, 40, 363–394 CrossRef CAS.
- X. Wu, J. Lee, V. Varshney, J. L. Wohlwend, A. K. Roy and T. Luo, Sci. Rep., 2016, 6, 22504 CrossRef CAS PubMed.
- D. I. Florescu, L. Mourokh, F. H. Pollak, D. C. Look, G. Cantwell and X. Li, J. Appl. Phys., 2002, 91, 890–892 CrossRef CAS.
- Ü. Özgür, X. Gu, S. Chevtchenko, J. Spradlin, S.-J. Cho, H. Morkoç, F. H. Pollak, H. O. Everitt, B. Nemeth and J. E. Nause, J. Electron. Mater., 2006, 35, 550–555 CrossRef.
- G. Rogl, A. Grytsiv, P. Rogl, E. Royanian, E. Bauer, J. Horky, D. Setman, E. Schafler and M. Zehetbauer, Acta Mater., 2013, 61, 6778–6789 CrossRef CAS.
- W. Liu, X. Yan, G. Chen and Z. Ren, Nano Energy, 2012, 1, 42–56 CrossRef CAS.
- H. Wu, J. Carrete, Z. Zhang, Y. Qu, X. Shen, Z. Wang, L.-D. Zhao and J. He, NPG Asia Mater., 2014, 6, e108 CrossRef CAS.
- N. Vogel-Schäuble, R. Dujardin, A. Weidenkaff and M. H. Aguirre, J. Electron. Mater., 2011, 41, 1606–1614 CrossRef.
- F. Azough, R. J. Cernik, B. Schaffer, D. Kepaptsoglou, Q. M. Ramasse, M. Bigatti, A. Ali, I. MacLaren, J. Barthel and M. Molinari, Inorg. Chem., 2016, 55, 3338–3350 CrossRef CAS PubMed.
- R. V. R. Virtudazo, M. Fuji and T. Shirai, Mater. Lett., 2011, 65, 3112–3115 CrossRef CAS.
- M. Fujiwara, K. Shiokawa, Y. Tanaka and Y. Nakahara, Chem. Mater., 2004, 16, 5420–5426 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- P. I. Ravikovitch and A. V. Neimark, J. Phys. Chem. B, 2001, 105, 6817–6823 CrossRef CAS.
- A. Kołodziejczak-Radzimska, E. Markiewicz and T. Jesionowski, J. Nanomater., 2012, 2012, 15 Search PubMed.
- M. Bitenc, M. Marinšek and Z. Crnjak Orel, J. Eur. Ceram. Soc., 2008, 28, 2915–2921 CrossRef CAS.
- R. Wahab, S. G. Ansari, Y. S. Kim, M. A. Dar and H.-S. Shin, J. Alloys Compd., 2008, 461, 66–71 CrossRef CAS.
- N. Izu, K. Shimada, T. Akamatsu, T. Itoh, W. Shin, K. Shiraishi and T. Usui, Ceram. Int., 2014, 40, 8775–8781 CrossRef CAS.
- Q. Li, H. Li, R. Wang, G. Li, H. Yang and R. Chen, J. Alloys Compd., 2013, 567, 1–9 CrossRef CAS.
- M. H. Habibi and B. Karimi, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2014, 44, 1291–1298 CrossRef CAS.
- H. Kim, Z. Wang, M. N. Hedhili, N. Wehbe and H. N. Alshareef, Chem. Mater., 2017, 29, 2794–2802 CrossRef CAS.
- Q. Li, E.-t. Liu, Z. Lu, H. Yang and R. Chen, Mater. Lett., 2014, 115–119 CrossRef.
- B. Singh, S. Singh, J. Singh, G. Saini, D. Mehta, G. Singh, S. Tripathi and A. Kaura, Phys. Chem. Chem. Phys., 2015, 17, 30450–30460 RSC.
- A. Barhoum, J. Melcher, G. Van Assche, H. Rahier, M. Bechelany, M. Fleisch and D. Bahnemann, J. Mater. Sci., 2017, 52, 2746–2762 CrossRef CAS.
- H. Li, E.-t. Liu, F. Y. F. Chan, Z. Lu and R. Chen, Mater. Lett., 2011, 65, 3440–3443 CrossRef CAS.
- D.-B. Zhang, H.-Z. Li, B.-P. Zhang, D.-d. Liang and M. Xia, RSC Adv., 2017, 7, 10855–10864 RSC.
- D. Gautam, M. Engenhorst, C. Schilling, G. Schierning, R. Schmechel and M. Winterer, J. Mater. Chem. A, 2015, 3, 189–197 CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- D. S. Sholl, Structural Nanocrystalline Materials. Fundamentals and Applications, ed. C. C. Koch, I. A. Ovid'ko, S. Seal and S. Veprek, Wiley Online Library, 2008 Search PubMed.
- R. K. Sendi and S. Mahmud, Appl. Surf. Sci., 2012, 261, 128–136 CrossRef CAS.
- M. Takata, D. Tsubone and H. Yanagida, J. Am. Ceram. Soc., 1976, 59, 4–8 CrossRef CAS.
- B. Dargatz, J. Gonzalez-Julian, M. Bram, Y. Shinoda, F. Wakai and O. Guillon, J. Eur. Ceram. Soc., 2016, 36, 1221–1232 CrossRef CAS.
- Y. Zhao, B. Chen, A. Miner and S. Priya, RSC Adv., 2014, 4, 18370–18377 RSC.
- Z.-H. Wu, H.-Q. Xie and Y.-B. Zhai, J. Nanosci. Nanotechnol., 2015, 15, 3147–3150 CrossRef CAS PubMed.
- T. Tsubota, M. Ohtaki, K. Eguchi and H. Arai, J. Mater. Chem., 1997, 7, 85–90 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, detailed XRD graphs, EDX, thermogravimetric analysis, SEM, N2 sorption, DFT graphs, BJH graphs and thermal conductivity graphs. See DOI: 10.1039/c7ra05067d |
| ‡ Initially, the molar concentrations were varied (4.5 mol L−1, 2.5 mol L−1 and 1.5 mol L−1). But for simplicity, we focused on 2.5 mol L−1 of Zn(NO3)2·6H2O solution. |
| § After using the low pressured SPS, the sample pellets (T-NZnOP) were labelled according to their different sintering temperatures for 550 °C = T-NZnOP-500C, 750 °C = T-NZnOP-750C and 950 °C = T-NZnOP-950C. Samples labelled T-NZnOP-550C and T-NZnOP-550C* are the same. They are labelled slightly different for the purpose of discussion and to accentuate as the porous pellets. The sintered sample pellets (series of T-NZnOP) had a density of ∼78%, ∼91% and ∼89% of the theoretical density after low pressured SPS sintering especially when sintered at 550 °C, 750 °C and 950 °C, respectively. |
| ¶ All the ZnO commercial powders (ZnO-Com) after SPS, the ZnO-Com sample pellets (T-CZnO-Com) were labelled according to their different sintering temperatures for 550 °C = T-CZnO-Com-500C, 950 °C = T-CZnO-Com-950C and 1200 °C = T-CZnO-Com-1200C, see, Fig. S5 and S7–S9. |
| This journal is © The Royal Society of Chemistry 2017 |