
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oriented perylene incorporated optically anisotropic 2D silica films†
Deepa Sriramulua,
Shuvan Prashant Turagab,
Andrew Anthony Bettiolb and
Suresh Valiyaveettil *a
*a
aMaterials Research Laboratory, Department of Chemistry, National University of Singapore, 3 Science Dive 3, Singapore 117543. E-mail: chmsv@nus.edu.sg
bDepartment of Physics, National University of Singapore, 2 Science Drive 3, Singapore 117542
First published on 27th June 2017
Abstract
Controlling the assembly of molecules in thin films is essential for developing functional materials for sensitive and smart coatings. Highly oriented heterogeneous and optically anisotropic 2D silica films were prepared at the liquid–liquid interface using octadecylsilane and perylenesilane as starting materials. The film formation involved the organization of molecules assisted by van der Waals interactions and network formation from silica polymerization. All films were fully characterized using a wide range of instruments. Wetting behavior of the thin film was established using contact angle measurement. The octadecylsilica (O-Si) film showed a water contact angle of ∼107° on the hydrophobic side and ∼70° on the hydrophilic side. The films prepared from perylenesilane (P-Si) and mixtures of perylenesilane and octadecylsilane (POx-Si) through hydrolysis, were fully characterized and showed higher contact angle than the O-Si films. As expected, an increase in the concentration of octadecylsilane in POx-Si film led to the disruption of the π–π stacking of perylene groups, followed by changes in optical properties of the film, which were established using spectroscopic techniques. Such bifunctional anisotropic films can be used for creating interesting functional coatings on different substrates.
1 Introduction
Interfacial polymerization or polycondensation has gained popularity over bulk polymerization in recent years owing to its wide range of applications in membrane technology1,2 and catalytic applications.3 Polyamides4 and polyureas5 are widely used for preparing thin or unimolecular films using interfacial polymerization due to their high flexibility and thermal stability.6,7 Interfacial polymerization is a process involving polycondensation of reactants at the interface of two immiscible phases (i.e. air/water or liquid–liquid) under normal conditions of temperature and pressure to yield a thin dense polymer film.8–10 Such controlled polymerizations are widely used to prepare 2D organic polymers but only a few studies have been carried out to form structurally organized films from organic/inorganic molecules as building blocks.11,12Porous silica films are used for antireflective coatings13 and as membranes for molecular separations.14,15 General methods to prepare silicate films include the chemical vapor deposition method (CVD)16–18 or the sol–gel method followed by dip-coating,19,20 spin coating21,22 and spray coating.23 A simple and cost effective methodology is needed to prepare homogeneous silica film for practical applications. Liquid–liquid interface conditions have been exploited for the synthesis of silica films from methyltrimethoxysilane24 and mesoporous silica films were prepared for various applications.25
In this study, structurally oriented, optically anisotropic silica films were prepared at the liquid–liquid interface polymerization and orientation dependent optical and wetting properties were measured (Scheme 1). A unique advantage of this method is the ability to incorporate desired molecules at different concentrations without changing the direction of molecular orientations (i.e. ⊥ to the plane of the film) during the synthesis of silica film at the liquid–liquid interface. The factors such as concentration, molecular structure, intermolecular interaction and the aggregation properties are used to control the organization and distribution of individual molecules inside the film. Also, we investigate the structure–property relationship of silica films prepared from octadecyl silane and perylene silane molecules. Octadecylsilica films (O-Si) showed hydrophobic properties on one side and hydrophilic properties on the other side. Molecular orientation dependant optical properties of perylene silica film (P-Si) and the hybrid octadecyl–perylenesilica films (POx-Si) were investigated in detail.
 | ||
| Scheme 1 Molecular structure and schematic illustration of the general structure and formation of (a) O-Si and (b) P-Si and (c) POx-Si at the interface of two immiscible liquids. | ||
2 Results and discussion
2.1. Mechanism of silica film formation at the liquid–liquid interface
Silica film formation at the chloroform–water interface was achieved through condensation of silane precursors (Fig. S1†) in the presence of a catalyst. The liquid–liquid interface offers added advantages of forming a heterogeneous well-oriented silica film using different monomers. Octadecyltricholorsilane was dissolved in chloroform and PEI or ammonia was dissolved in water. In the presence of the base catalysts, the chlorosilane or alkoxysilane groups were hydrolyzed to silanols (–Si–OH) at the interface between organic and aqueous phase, followed by the condensation of Si–OH groups to form two dimensional Si–O–Si network structures. Such interface reaction leads to well aligned silica films with hydrophobic organic groups are placed in an oriented manner in the CHCl3 layer.Octadecylsilica (O-Si) films were compared using different concentrations of octadecyltrichlorosilane and 2 wt% PEI as a catalyst for a fixed time of 20 min. PEI was used as a preferred catalyst over aqueous ammonia, due to the rapid polymerization of octadecyltrichlorosilane at the chloroform–water interface in presence of ammonia, preventing stable film formation at the interface. Silica film formation was observed within 20 min upon the addition of octadecyltrichlorosilane in chloroform layer and PEI catalyst in the aqueous layer. The film formed was used for further characterizations. The observed high rate of formation of the film could be due to high reactivity of trichlorosilane which readily reacts with water to form Si–OH group and undergo condensation stabilized by polyethylenimine catalyst.26
FESEM images of the film at the chloroform side (Fig. 1a and S2a†) and aqueous side (Fig. 1b) generated at the interface using octadecylsilane were recorded. Minimum differences in morphology of the film surface facing chloroform side (hydrophobic) and water side (hydrophilic) were observed. Microstructure analysis of hydrophilic side of the film was found to be smoother with RMS roughness of 64.4, as compared to hydrophobic side which had a RMS roughness of 84.4. Hydrolysis and condensation of silane precursors are much faster in the presence of water and silica film was formed at the interface of water and chloroform. Owing to the presence of abundant water molecules for the hydrolysis of octadecyltrichlorosilane, there is little time for the individual clusters to grow before they crosslink with each other leading to a smooth surface (Fig. 1b). As the film thickness increases, the network hinders supply of water molecules to the rest of the unhydrolyzed silane precursors in the chloroform layer. The individual clusters grew in size before crosslinking with each other leading to a formation of rough surface of the film (Fig. 1a).
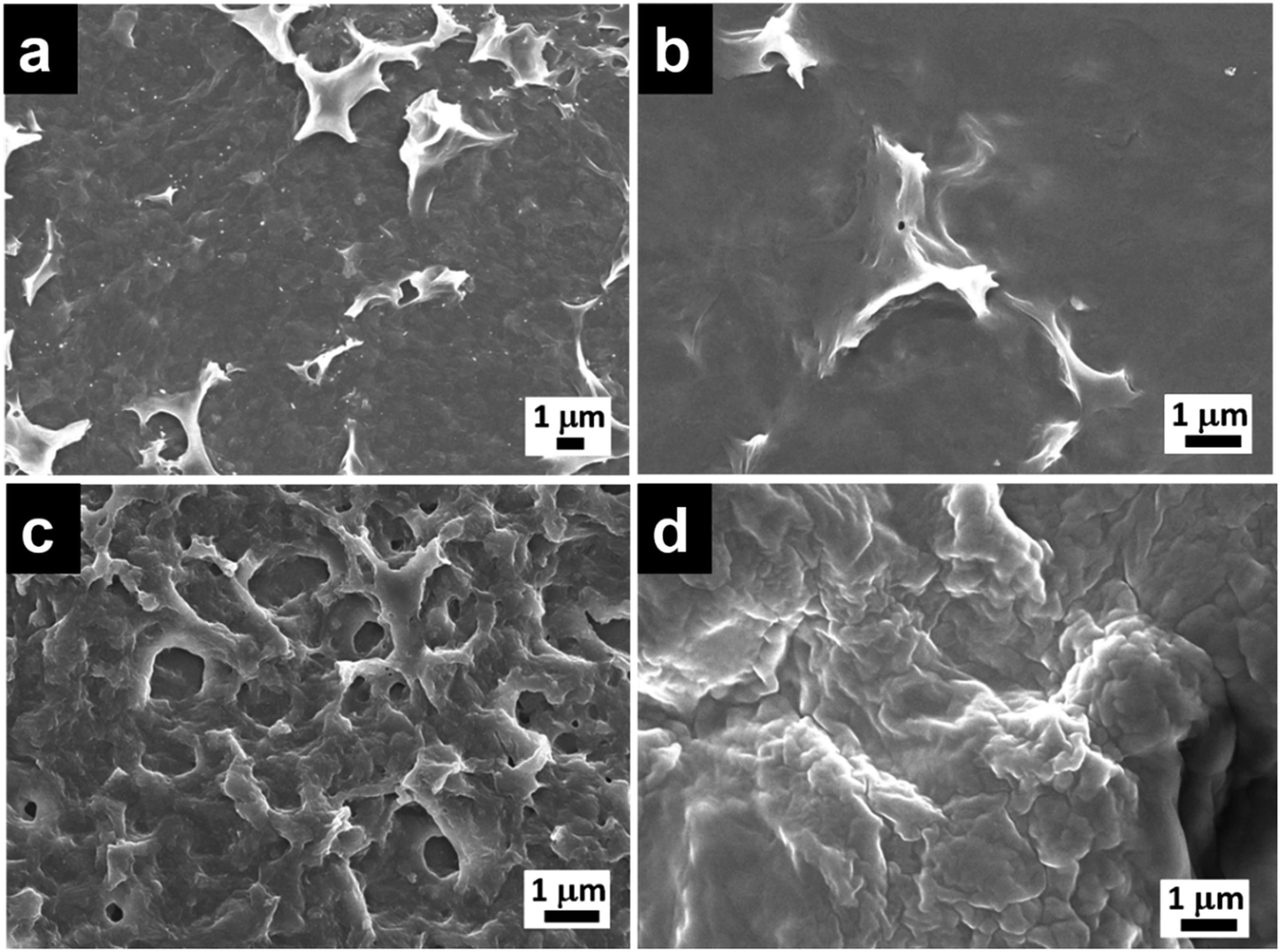 | ||
| Fig. 1 FESEM micrographs of O-Si film (a) organic side and (b) aqueous side, morphologies on the organic side of (c) P-Si and (d) PO100-Si film. | ||
Similarly, perylenesilane with triethoxy group formed a film at the interface after 1 day using ammonia as the catalyst. FESEM image of P-Si film shows a rough surface which is different from the surface morphology of O-Si film (Fig. 1c and S2b†). P-Si film showed a surface roughness of ∼115 on both sides of the silica films. This is due to packing and aggregation of rigid planar perylene molecules leading to highly disordered films as compared to the film formed from flexible and linear octadecyltrichlorosilane.
Silica films obtained by mixing N-(2-ethylhexyl)-N′-(3-(triethoxysilyl)propyl)perylene-3,4,9,10-tetracarboxylic acid diimide and octadecyltrimethoxysilane in molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:10, 1:100 and 1:1000 showed changes in morphology upon increasing the concentration of octadecyltrimethoxysilane (Fig. S2c–f†) when compared to silica film formed from pure octadecylsilane (Fig. 1a) or perylenesilane (Fig. 1c) precursors. At higher concentration of octadecylsilane such as PO100-Si (1:100) and PO1000-Si (1:1000), silica films formed were continuous and showed a smooth morphology as compared to silica film containing lower concentrations of octadecylsilane, PO1-Si and PO10-Si films. At higher concentration of octadecyltrimethoxysilane, the flexible octadecyl group tend to fill in the gaps formed via the aggregation of the perylene units and form a smooth film surface (Fig. 1d).
:1, 1:10, 1:100 and 1:1000 showed changes in morphology upon increasing the concentration of octadecyltrimethoxysilane (Fig. S2c–f†) when compared to silica film formed from pure octadecylsilane (Fig. 1a) or perylenesilane (Fig. 1c) precursors. At higher concentration of octadecylsilane such as PO100-Si (1:100) and PO1000-Si (1:1000), silica films formed were continuous and showed a smooth morphology as compared to silica film containing lower concentrations of octadecylsilane, PO1-Si and PO10-Si films. At higher concentration of octadecyltrimethoxysilane, the flexible octadecyl group tend to fill in the gaps formed via the aggregation of the perylene units and form a smooth film surface (Fig. 1d).
2.2. Fourier transform infrared spectroscopy (FT-IR) analysis
O-Si and P-Si films formed were further analyzed by Fourier transform infrared spectroscopy (FT-IR) (Fig. 2). A few peaks corresponding to Si–O bonds are observed in the low frequency region, 700–1400 cm−1. Asymmetric stretching vibration of Si–O–Si can be observed at 1025 cm−1 for both O-Si and P-Si film, which confirm silica network formation (Fig. 2a). A broad peak at 3400 cm−1 and at 1600 cm−1 corresponds to –OH group from silica or adsorbed water. Symmetric and asymmetric –CH2 stretching of O-Si film are observed at ∼2851 cm−1 and 2919 cm−1. The position of the –CH2 asymmetric and symmetric stretching peak maxima provides qualitative information about the conformational order of the –CH2– groups on the alkyl chains. For disordered random structures, the frequency of the –CH2– asymmetric stretching band is close to that of a liquid alkane (2924 cm−1) and for well packed alkyl chains, the frequencies are shifted to lower wavenumbers (2915–2918 cm−1).27 The observed peak at 2919 cm−1 indicates high populations of octadecyl chains are packed in a well ordered trans-conformation. | ||
| Fig. 2 FT-IR spectra of O-Si and P-Si films (a) and POx-Si films of (i) PO1-Si, (ii) PO10-Si, (iii) PO100-Si and (iv) PO1000-Si formed using a perylenesilane:octadecylsilane molar ratio of 1:1, 1:10, 1:100 and 1:1000 (b) at CHCl3/water interface. | ||
In addition to the peaks corresponding to silica network, P-Si film showed the presence of carbonyl stretching vibrations of imide peaks around 1700 and 1600 cm−1 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic stretching vibrations around ∼1590 cm−1, as previously reported for other perylenediimide derivatives.28 Interestingly, IR spectra of POx-Si (using different molar ratio of perylene and octadecylsilane) film, showed increase in the intensity of –CH2 alkyl stretching peaks with increase in concentration of octadecylsilane from 1:1 (PO1-Si) to 1:1000 (PO1000-Si) and decrease in diimide peak corresponding to perylene molecules (Fig. 2b).
C aromatic stretching vibrations around ∼1590 cm−1, as previously reported for other perylenediimide derivatives.28 Interestingly, IR spectra of POx-Si (using different molar ratio of perylene and octadecylsilane) film, showed increase in the intensity of –CH2 alkyl stretching peaks with increase in concentration of octadecylsilane from 1:1 (PO1-Si) to 1:1000 (PO1000-Si) and decrease in diimide peak corresponding to perylene molecules (Fig. 2b).
2.3. Thermogravimetric analysis (TGA)
Thermogravimetric analyses of silica films were performed using a heating rate of 10 °C min−1 in air (Fig. 3, DTA of all silica films are given in Fig. S3†). Three weight loss regions were observed for O-Si film. The first weight loss observed below 150 °C is due to loss of physically adsorbed water.29 Second weight loss (∼37.9%) at 200–400 °C region is due to the loss of alkyl chain. The third loss of 43% at 400–800 °C region is due to the degradation of all organic contents and complete dehydroxylation of Si–OH group to form Si–O–Si network. The 13% residual weight is attributed to the SiOx. | ||
Fig. 3 Thermogravimetric analysis of ( ) O-Si, ( ) O-Si, ( ) P-Si film and ( ) P-Si film and ( ) PO100-Si film using a heating rate of 10 °C min−1 in air. ) PO100-Si film using a heating rate of 10 °C min−1 in air. | ||
The P-Si film shows a slight decrease in weight loss (5%) in the region of 100–200 °C which could be due to loss of physisorbed water from the film. The second degradation in the region of 300–500 °C with 53.2% weight loss is assigned to the degradation of alkyl groups, partial degradation of perylene diimide and unreacted ethoxy groups on silane functional group. The third weight loss of 26.2% at 500–800 °C region corresponds to degradation of all carbon contents, Si–C, and C–N groups and the remaining 15% residual weight is attributed to the SiOx.30,31
PO100-Si film showed a TGA trace almost similar to O-Si film, with 40.3% weight loss around 150–400 °C region is due to the loss of alkyl chain moieties from the material and unreacted ethoxy groups on silane functional group and about 30.6% weight loss in the region 400–550 °C due to the loss of perylene groups. The observed weight loss (9.1%) at 550–800 °C accounts for degradation of remaining carbon content present in the film. The 17% residual weight is attributed to silica from the film.
2.4. X-ray diffraction studies
Small angle X-ray diffraction (SAXD) and wide angle X-ray diffraction curve (WAXD) of O-Si film are given in Fig. 4a, which show diffraction peaks at 1.72°, 5.11° (SAXD) and single strong peak around 21.53° (WAXD). Applying Braggs law, 2dsinθ = λ, with λ = 1.5406 Å gave corresponding d-values of 51.3 Å, 17.28 Å and 4.12 Å, respectively. The d-spacing at 4.12 Å, is attributed to intermolecular interactions and van der Waals's forces observed in densely packed alkyl chains.32 Peak observed at 51.3 Å is closer to the reported value of d = 52.4 Å for a bilayer structure of octadecylsilyl groups.29
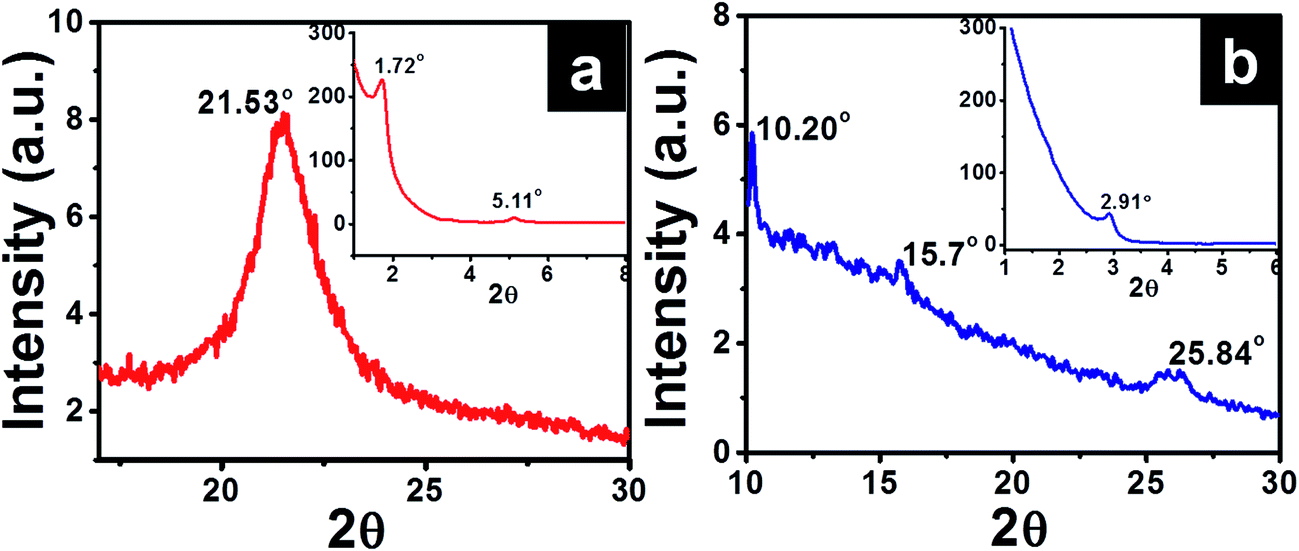 | ||
| Fig. 4 XRD spectra of (a) O-Si and (b) P-Si film showing a wide angle X-ray diffraction (inset – small angle X-ray diffraction pattern). | ||
XRD profiles of P-Si film (Fig. 4b) showed diffraction peaks at 2θ = 2.91°, 10.20°, 15.7° and 25.84° (corresponding d-spacing of 30.33 Å, 8.67 Å, 5.64 Å and 3.44 Å, respectively). The observed peak with d = 8.67 Å is similar to the width of perylene core (calculated value = 9.2 Å).33,34 Similar ordering was also observed for columnar packing of alkyl substituted perylene diimide liquid crystals.33,34 Diffractions corresponding to 5.64 Å and 3.44 Å are attributed to π–π stacking of cofacially stacked perylene cores.35 Similarly high resolution TEM images and selected area electron diffraction pattern (inset in Fig. 5) observed for P-Si film (Fig. 5a) and PO100-Si (Fig. 5b) indicate a close packed perylene crystalline lattice in the film.
 | ||
| Fig. 5 TEM images of P-Si film (a) and PO100-Si film (b), (inset: selected area electron diffraction pattern of the corresponding film). | ||
2.5. Contact angle measurement
Hydrophobic and hydrophilic properties of the surface of the silica films collected separately on a coverslip are measured (Fig. S4†) using contact angle setup. Morphology of water drop was retained on the surface with alkyl chains (chloroform side) and spreading of water was observed on the silica side (facing the water side) of the film (Fig. 6, inset). The observed differences can be attributed to the distribution of long alkyl groups on the organic side and the presence of PEI and –Si–OH moieties on the water side of the film.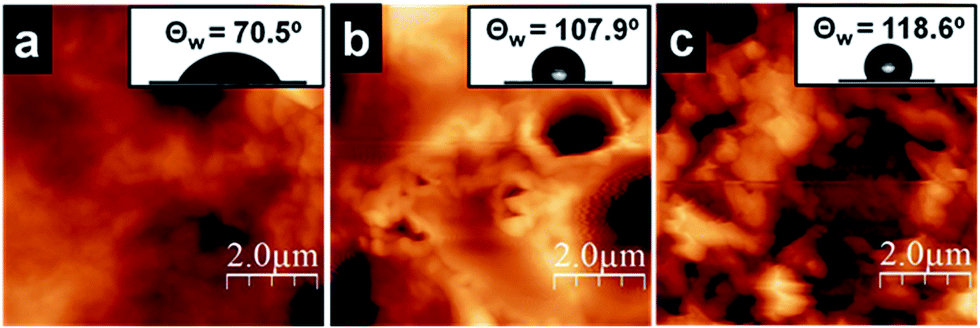 | ||
| Fig. 6 AFM micrographs of the (a) aqueous side and (b) organic side of O-Si film, (c) aqueous of the P-Si film. Inset: corresponding contact angle of water on the surface of the films. | ||
Static contact angle measurements on silica films were performed using a goniometer with 1 μL water droplets. The wetting properties of silica film facing water side and chloroform side are shown in Fig. 6. One side of the O-Si film is hydrophilic (θ ∼ 70.5°, Fig. 6a, inset) and the other side of the film is hydrophobic (θ ∼ 107.9°, Fig. 6b, inset). It is expected to see such variations owing to the differences in functional groups present on both sides of the film. However, P-Si film surface showed a water contact angle θ of ∼118° (Fig. 6c, inset), which is greater than O-Si film. This can be attributed to the close packing of perylene molecules on the silica film surface. But, hydrophilic and hydrophobic side of the P-Si film did not show significant differences in contact angle, which needs to be further explored to understand the distribution of organic moieties before and after transferring the film on the cover slip. It is hypothesized that the perylene being a planar, rigid aggregating molecule, the large aggregate formation leads to defects on the surface, which then minimizes the difference in functionality on both side of the film.
Silica films formed by using perylenesilane and octadecylsilane precursors at different molar ratios, showed an increase in contact angle with an increase in the concentration of octadecyl chains incorporated in the film (Table 1). The increase in hydrophobicity observed can be attributed to the introduction of C18 alkyl groups, which could fill the defects created by the aggregation and packing of perylene molecules on the surface. But contact angle for PO100-Si (∼116°) is lower than that of PO1-Si and PO10-Si. With increase in concentration of octadecylsilane, the packing of octadecyl groups tend to be more dominant and compact, which then reduces the availability of alkyl chains needed to fill the defect caused by the perylene aggregates.
| Silane filmsa | Static contact angle (θ) | |
|---|---|---|
| Chloroform side/deg | Water side/deg | |
| a Where, O-Si and P-Si refers to film formed from octadecasilane and perylenesilane alone. PO1-Si, PO10-Si and PO100-Si film formed by mixing perylenesilane and octadecasilane in certain molar ratios such as 1:1, 1:10 and 1:100, respectively. |
||
| O-Si | 107.9 | 70.5 |
| P-Si | 118.0 | 118.6 |
| PO1-Si | 125.8 | 125 |
| PO10-Si | 129.4 | 128 |
| PO100-Si | 116.4 | 105.7 |
2.6. Photophysical studies using silica films
Photophysical studies of P-Si film obtained from interfacial polymerization and drop casting of perylenesilane on quartz plate were done using spectroscopic techniques.Optical images of dropcasted films from unreacted perylene silane precursor using FITC filter and P-Si films are given in Fig. S5.† P-Si film showed a well-ordered homogeneous film morphology. Interestingly, optical micrographs of PO1-Si, PO10-Si, PO100-Si and PO1000-Si in bright field and under FITC filter showed significant changes from red to light green color with increasing the concentration of octadecyl groups in the film (Fig. S6†). This is further supported by UV-Vis absorption and emission spectra of the films (Fig. 7). The absorption spectrum of perylenesilane in chloroform solution showed corresponding monomeric peaks at 526 nm, 489 nm and 458 nm, due to π–π* transition of the perylene rings, designated as 0–0, 0–1 and 0–2 transition (Fig. 7a).36 Compared to solution state absorption spectra, the spectra of the films are broad and less structured. Inverse increase in intensity of A0–1 absorption peaks are observed in all silica films compared to perylenesilane in solution with respect to A0–0. P-Si film showed merging of peaks at 489 nm and 458 nm and a broad featureless absorption spectra indicating face to face stacked H-aggregates of perylene molecules in the film. Similar peak pattern was also observed for PO10-Si film, which indicates perylene molecules inside the silica films are aggregated via strong intermolecular π–π interaction. Upon increasing the concentration of octadecylsilane in PO100-Si and PO1000-Si, (0–1), vibronic band at (458 nm) showed a relative increase in intensity as compared to (0–0) transition at 520 nm and also vibronic peaks are clear and less broad in appearance. Such relative increase in intensity of (0–1) vibrionic transition is typically observed for perylene molecules stacked cofacially forming H-aggregates.37 Thus, ratio of absorption intensity for A0–0/A0–1 increased from 0.78 for P-Si to 0.93 for PO1000-Si film indicating that extent of aggregation decreases with increase in concentration of octadecylsilane in the hybrid silica film.38
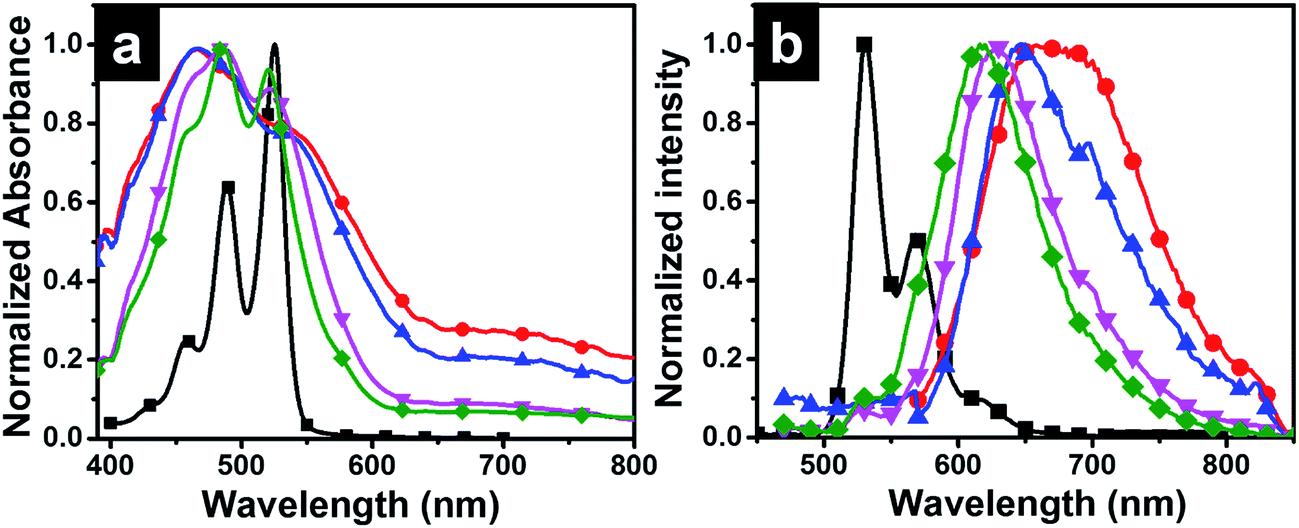 | ||
Fig. 7 Normalized absorption (a) and emission (b) spectra of ( ) perylenesilane solution in THF (0.1 mM), and solid state spectra of ( ) perylenesilane solution in THF (0.1 mM), and solid state spectra of ( ) P-Si, and mixed perylenesilane–octadecylsilane films ( ) P-Si, and mixed perylenesilane–octadecylsilane films ( ) 1:10 (PO10-Si), ( ) 1:10 (PO10-Si), ( ) 1:100 (PO100-Si), ( ) 1:100 (PO100-Si), ( ) 1:1000 (PO1000-Si) formed at the interface. Excitation wavelength was at 350 nm. ) 1:1000 (PO1000-Si) formed at the interface. Excitation wavelength was at 350 nm. | ||
The emission spectrum of perylenesilane in chloroform solution (Fig. 7b) showed characteristic non-aggregated peaks at 532, 567 and 617 nm.36 Significant changes in fluorescence properties were observed in silica films synthesized at the interface of two liquids. P-Si film showed a broad emission spectrum with a red shift in the maximum at 670 nm (Fig. 7b). The longer wavelength band confirmed the presence of cofacial π–π interaction between perylene groups in the solid state which is in agreement with the data obtained from powder X-ray diffraction studies (Fig. 4b). Intense excimeric peaks were observed for PO1-Si at 650 nm and 698 nm. Also, with increasing the concentration of octadecyl group with respect to perylenesilane, a blue shift in emission maximum was observed from 650 nm to 617 nm (PO1000-Si), which clearly indicates the reduction in π–π interaction of perylene groups in presence of long octadecyl groups during film formation. PO1000-Si film showed a major excimeric emission at 618 nm, whereas film formed using 1:1 mixture of the two silane precursors (PO1-Si) showed emission at 698 nm. However, the excimer emission of PO1-Si film showed a bathochromic shift when compared to PO1000-Si film, which implies that strong π–π interactions and well organized molecular aggregates in PO1-S facilitate better exciton migrations in PO1-Si film.39
In order to further understand the arrangement of perylene molecules inside the film formed at the interface of two liquids, optical anisotropy of the films was measured using polarized UV-Vis absorption (Fig. 8) and polarized angle dependent emission spectra (Fig. 9).
 | ||
Fig. 8 Polarized UV-Vis absorbance spectra of drop casted film of perylenesilane monomer (A), interfacial silica films of P-Si (B) and PO100-Si (C) on quartz plate and the plot of peak absorbance vs. polarization angle (D) of drop casted film of perylenesilane ( ), interfacial film of P-Si ( ), interfacial film of P-Si ( ) and PO100-Si ( ) and PO100-Si ( ) as a function of polarization angle of the incident light. ) as a function of polarization angle of the incident light. | ||
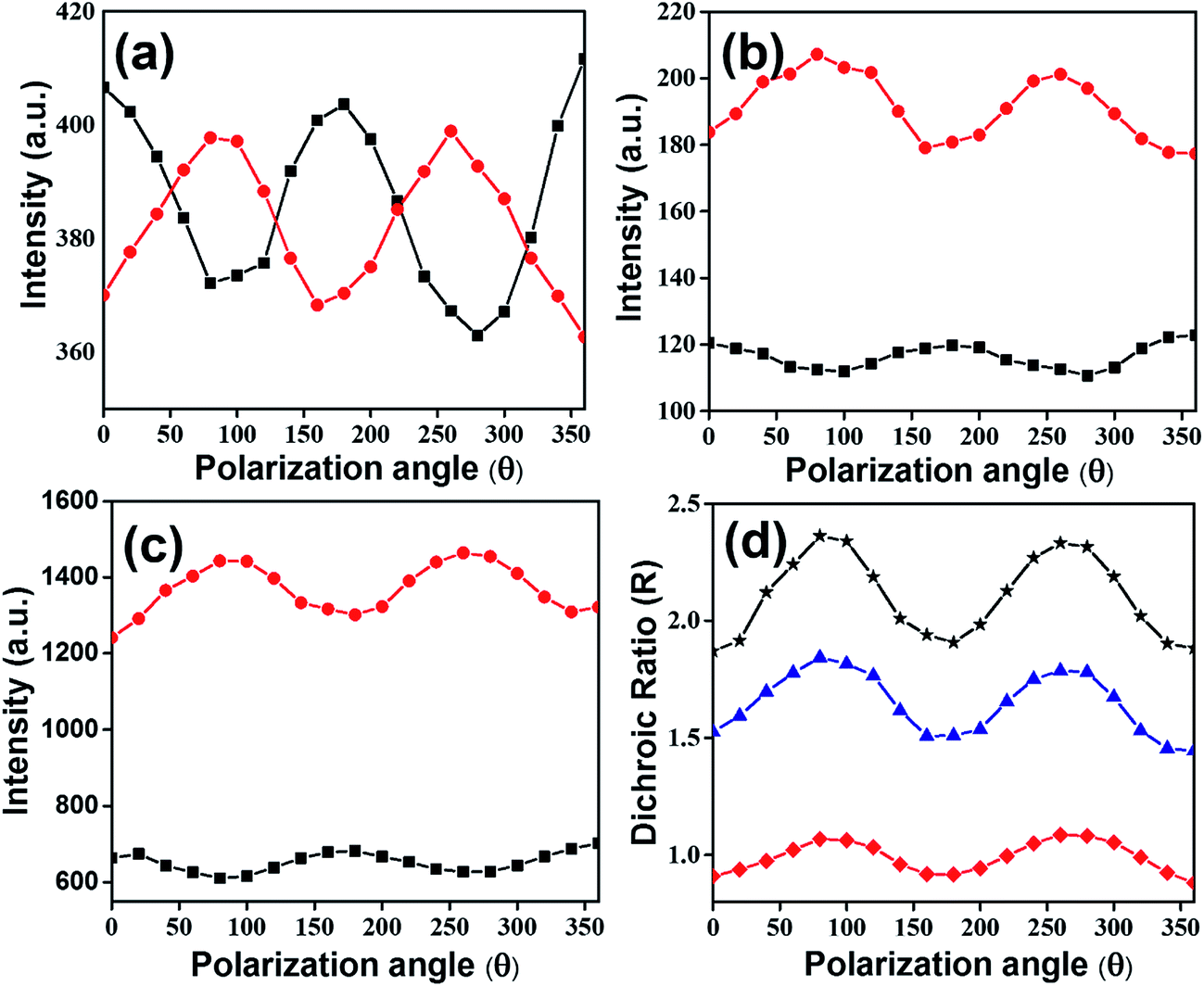 | ||
Fig. 9 Polarization angle dependence emission spectra of drop casted film of perylenesilane monomer (a), interfacial films of P-Si (b) and PO100-Si (c), where incident light is parallel (I∥,  ) and perpendicular (I⊥, ) and perpendicular (I⊥,  ) to plane polarized light. Plot of dichroic ratio with polarization angle (d) of drop casted films of perylenesilane ( ) to plane polarized light. Plot of dichroic ratio with polarization angle (d) of drop casted films of perylenesilane ( ), interfacial film of P-Si ( ), interfacial film of P-Si ( ) and PO100-Si ( ) and PO100-Si ( ). Excitation wavelength used was 403 nm. ). Excitation wavelength used was 403 nm. | ||
The measurements of absorbance anisotropy of the films were performed by changing the position of the polarizer of the incident light every 10° within the range of 0° to 90°. As expected low degree of packing was observed in the dropcasted film of perylenesilane (Fig. 8A) as compared to P-Si (Fig. 8B) and PO100-Si films (Fig. 8C). The degree of packing was measured by calculating absorbance ratio A = A0–0/A0–1. The absorbance ratios for the drop casted film from perylenesilane, P-Si and PO100-Si were 0.6, 0.8 and 0.9, respectively. However, slight variation in the absorbance ratio was also observed with the changes in polarization angle from 0° to 90°, indicating polarization dependent absorbance properties of the film. When the sample was held normal to incoming polarized light, polarization dependence was maximum for PO100-Si film and least polarization dependence was observed for drop casted film of perylenesilane and interfacial film of P-Si (Fig. 8D).
The lack of polarization dependence for the dropcasted film indicates that precursor perylenesilane molecules are randomly oriented on the glass slide.40 Further, in order to study the alignment of dye molecules in P-Si, and PO100-Si films, polarized emission spectra were recorded by rotating the incident polarizer (from 0° to 360°) at every 20° (Fig. 9). Significant differences in emission intensities of the polarized light parallel and perpendicular to the plane of polarization were observed for PO100-Si film (Fig. 9c) when compared to P-Si film (Fig. 9b) and drop casted film of perylenesilane precursor (Fig. 9a). Among the three films investigated, the least differences in parallel and perpendicular emission intensities were observed for drop casted films of perylenesilane (Fig. 9a) owing to low molecular order inside the film.
Polarization ratio was calculated using a formula (eqn (1)).41
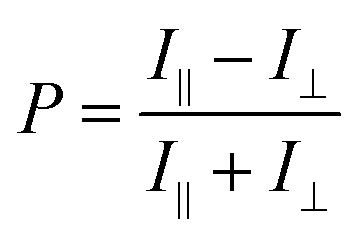 | (1) |
The alignment of perylene molecules can be quantitatively measured using the dichroic ratio (D) which is defined as the ratio of intensities of the dye molecule parallel (I∥) and perpendicular (I⊥) to the incident polarized light, as shown in eqn (2).42 D value is used to calculate the order parameter (S), as shown in eqn (3).43 Dye molecules are said to be fully aligned if the calculated order parameter, S = 1 and completely random alignment when S = 0.44
 | (2) |
 | (3) |
Dichroic ratio of two silica films and the drop casted film of perylenesilane are depicted graphically with respect to polarization angle in Fig. 9d. The estimated values of D and S are summarized in Table 2.
| Sample | Absorbance ratio, A0–0/A0–1 | λemi (nm) | Polarization degree (P) | Dichroic ratio (D) | Order parameter (S) |
|---|---|---|---|---|---|
| Perylenesilane dropcast film | 0.6 | 654 | 0.02 | 0.91–1.06 | −0.03–0.02 |
| P-Si film | 0.8 | 660 | 0.05 | 1.5–1.84 | 0.14–0.22 |
| PO100-Si film | 0.9 | 650 | 0.2 | 1.9–2.3 | 0.23–0.30 |
Drop casted film of perylenesilane showed the least order parameter (S ∼ 0.02), indicating a fully random arrangement of perylene molecules. This is expected due to the random organization of perylene molecules in the drop casted film. On the other hand, introduction of long octadecyl groups in excess with respect to perylene molecules (PO100-Si) provides an induced higher degree of order in the film, formation and organization of large aggregates of perylene is retarded, which then lead to alignment of molecules inside the silica film.
2.7. POx-Si film as fluorescent sensor for sensing aniline vapors
Perylene tetracarboxylic diimide is widely used in optical sensing due to its high photostability, and good electron deficient properties.45 Recently, perylene incorporated nanofibers were used as a fluorescent sensor to sense amine vapors. As a proof of concept, silica films prepared in this study using different combination of perylenesilane and octadecylsilane (PO1-Si, PO10-Si, PO100-Si) were exposed to aniline vapors (30 min and 24 h). Efficient fluorescence quenching was observed from all three silica films. Percentage quenching efficiency was calculated from the fluorescence data using the following eqn (4).
 | (4) |
Fluorescence quenching can be attributed to photoinduced electron transfer from HOMO of electron rich aniline to HOMO of electron poor perylene molecules. However, due to changes in aggregation of perylene, the vapor sensing for aniline is slightly different for different silica films (Fig. 10). Silica film formed using a combination of 1:1 mixture (PO1-Si) showed higher quenching efficiency of ∼81% than PO100-Si (69%). Decrease in concentration of perylene in the film led to changes in structure, solubility of amines in the film and interaction of amines with perylene molecules inside PO100-Si films. The observed fluorescence quenching can be attributed to strong interaction between electron deficient perylene molecules with electron rich aniline molecules.46,47
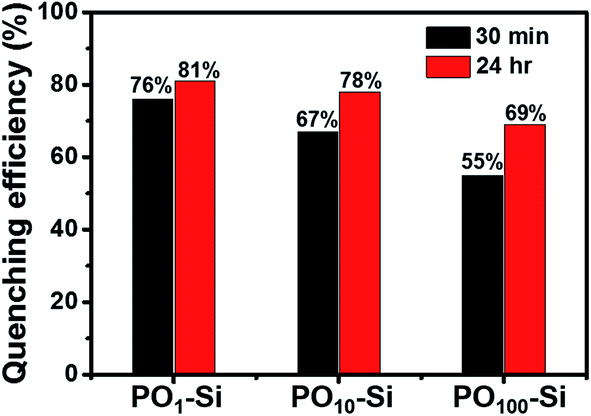 | ||
Fig. 10 Fluorescence quenching response of POx-Si films after exposure to vapors of aniline for known time interval ( ) 30 min, and ( ) 30 min, and ( ) 24 h. ) 24 h. | ||
3 Conclusions
A direct method for preparing functionalized optically anisotropic silica film at the liquid–liquid interface is reported. Structural changes and optical properties of the films were studied by varying the composition and concentration of the silane precursor molecules during silica film formation. The films formed were characterized using IR, SEM, XRD and TGA analyses. Wetting properties of the silica films were determined using contact angle measurements. O-Si film showed a water contact angle of ∼107° for alkyl chain side and ∼70° for the silica side of the film. The P-Si film showed a higher contact angle of ∼118°, which can be attributed to strong aggregation induced packing of perylene units on the surface of the film. To further explore the correlation between molecular organization, surface wetting and photophysical properties, films were synthesized using various combinations of perylenesilane and octadecylsilane (1:1, 1:10, 1:100 and 1:1000) molecules. An increase in concentration of octadecylsilane inside the film led to the formation of homogenous film, which resulted in changes in properties of the silica films. Also, organization of perylene molecules inside the silica films was examined using polarized absorption and emission anisotropy of the film, measured using a polarized spectrophotometer. PO100-Si film showed a high dichroic ratio (D) of 2.3 when compared to the values of P-Si film (D = 1.8) and drop casted film of perylenesilane (D = 1.08). Thus, PO100-Si films showed strong molecular alignment with an order of parameter, S = 0.3 and the least order of parameter was observed for drop casted film of perylenesilane (S = 0.02). Different films also showed interesting optical responses in presence of aniline vapors. In this study, we investigated the structure–property correlation of perylene incorporated anisotropic 2D silica films formed through hydrolysis and condensation of silane precursors at the interface of water and chloroform. Such ordered silica films may have potential applications in different areas and we are currently exploring them in our lab.
4 Experimental
4.1. Materials
Trichloro(octadecylsilane), octadecyltrimethoxysilane, polyethylenimine (PEI, 50%) were obtained from Sigma Aldrich. Ammonia solution (25%) was obtained from Merck and N-(2-ethylhexyl)-N′-(3-(triethoxysilyl)propyl)perylene-3,4,9,10-tetracarboxylic diimide were synthesized using a known literature report.45,48 Molecular structures of the starting materials are given in Fig. S1.† All chemicals and AR grade solvents were used as received. Deionised water was used to dissolve ammonia and PEI in the aqueous layer in all experiments.4.2. Setup for interface polymerization
The unimolecular thin silica films were prepared at the interface of two immiscible liquids (e.g. water and chloroform) using the condensation of relevant silica precursors dissolved in organic phase and catalyst dissolved in the water phase (Scheme 1). The film formation depends on the reactivity and concentration of the reagents. Clean glass cover slips were used to collect and fully characterize the films. Scooping the film from organic side to water phase will expose the hydrophilic surface to the air, whereas collecting the film from water side exposes the hydrophobic surface to the air. Films collected were dried at room temperature for a day for further characterization.4.3. Preparation of octadecylsilica (O-Si) film
In a typical procedure, chloroform (10 mL) and water (10 mL) were taken in a 25 mL transparent plastic container and the system was allowed to stabilize for a few minutes. Octadecyltrichlorosilane (0.031 mL) was slowly injected into chloroform (CHCl3) solution followed by addition of 2 wt% PEI solution into the aqueous layer on top of the CHCl3 solution. The system was kept at room temperature for film formation at the interface. A white film formed at the interface after 30 min was collected, washed thoroughly with water, dried and used for full characterization and property investigations.4.4. Preparation of perylenesilica (P-Si) film
The perylene silica film was prepared by placing chloroform (5 mL) and water (5 mL) in 25 mL beaker. N-(2-Ethylhexyl)-N′-(3-(triethoxysilyl)propyl)perylene-3,4,9,10-tetracarboxylic acid diimide (labelled as perylenesilane, 5 mL, 2 mM) and ammonia solution (5 mL, 2 M) were added into CHCl3 and water layers, respectively, and kept undisturbed at room temperature. The film formed at the interface after 24 h was collected, washed with water and dried at room temperature for further characterization.4.5. Preparation of hybrid perylene–octadecylsilica (POx-Si, x = 1, 10, 100, 1000) film
For the preparation of octadecyl - perylene silica film, a mixture of N-(2-ethylhexyl)-N′-(3-(triethoxysilyl)propyl)perylene-3,4,9,10-tetracarboxylic acid diimide (i.e. perylenesilane) and octadecyltrimethoxysilane (i.e. octadecylsilane) in molar ratios of 1:1, 1:10, 1:100 and 1:1000 were dissolved in CHCl3 (10 mL) in a beaker. Ammonia solution (10 mL) was added slowly on top of the CHCl3 solution without disturbing the system. The ammonia initiated the polymerization of monomers at the interface under ambient conditions.
4.6. Characterization
The IR spectra were recorded in the range of 4000–400 cm−1 using a Bruker ALPHA FT-IR Spectrophotometer with a resolution of 4.0 cm−1. The UV/Vis absorption studies were performed on a Shimadzu-1601 PC spectrophotometer. The steady state fluorescence studies were carried out on Agilent Cary Eclipse Fluorescence Spectrophotometer. Wide angle X-ray diffraction (WAXRD) were recorded using Bruker-AXS: D8 DISCOVER with GADDS powder X-ray diffractometer with Cu-Ka (λ = 1.5406 Å) at 40 kV and 40 mA over a range of 2θ angle from 5° to 80° and small angle X-ray diffraction from 2θ = 0° to 10°. The thermal analyses of the samples were performed on a TA instrument 2960 (DTA-TGA) analyzer within the temperature range of 30–800 °C. Surface morphologies of the silica films were determined using Field Emission Scanning Electron Microscope (FESEM on a JEOL JSM-6701F instrument) at 5 kV after coating the films with platinum. Transmission Electron Microscopy (TEM, JEOL JEM 2010) was used to establish the morphology of silica films collected on copper TEM grid. The contact angle measurements were carried out using the video-based fully automated Data Physics optical contact angle microlitre dosing system (OCA 40 Micro). Water drops (1 μL per drop) with known surface tension were dispensed using Teflon coated motor driven syringe. The contact angles were measured at ambient conditions and a video was recorded (72 frames per second) for every dispensed solvent droplet. Any dynamic changes to the droplet on the surface were precisely observed through this method. The contact angles were evaluated using the sessile drop technique.494.7. Absorption anisotropy measurement
For absorption anisotropy studies, a custom microscope attached with a polarizer and an analyzer was constructed. The light source used was a halogen and deuterium lamp (Ocean Optics D-2000 BAL) with a wavelength range from 250 to 2500 nm. The schematic diagram given in Fig. 11a shows the details of the absorption anisotropy setup. The input light comes through a fiber of about 600 μm core diameter and the light is collimated using a lens. After collimation, the light passes through a polarizer and a half-wave plate to control the incident light polarization. The light is focused on the sample with a 40× objective. The back-reflected light was passed into a polarization analyzer and finally collected into the optical fiber connected to the UV-Vis Spectrophotometer (Ocean Optics Q65000). Our measurement range was limited to the polarizer working range from 420 to 680 nm. The samples were mounted on a Teflon substrate and the background signal was measured using blank glass/quartz substrates.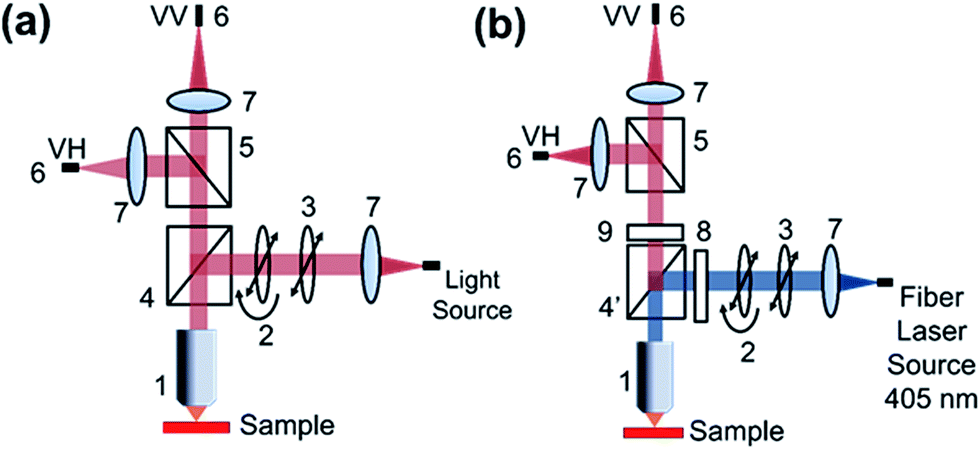 | ||
| Fig. 11 Schematic representation of solid state absorption anisotropy (a) and emission anisotropy setup (b). (1) Objective lens, (2) half wave plate, (3) polarizer, (4) beam splitter, (4′)dichroic beam splitter, (5) polarizing beam splitter, (6) fiber to spectrophotometer, (7) lens, (8) 405 notch filter, (9) 425 long pass filter. | ||
4.8. Emission anisotropy measurement
For photoluminescence measurements, a custom made spectrophotometer connected to a microscope with a polarizer and analyser was used (Fig. 11b). A fiber coupled with 405 nm laser was attached to the microscope and the polarization was controlled using a polarizer and a half-wave plate. After the dichroic and excitation long pass filter, the collected PL signal was analyzed with another polarizer. The spectrophotometer comprises of Andor Shamrock SR500 spectrograph that has a 600 line grating at a blaze of 500 nm typically used for photoluminescence measurements. The signal is recorded with a sensitive air-cooled Andor iVac camera. The measurements were conducted with an exposure time of 1 s and 10 accumulations to improve the SNR (signal-to-noise ratio).Acknowledgements
The authors acknowledge funding support from National University of Singapore, and technical support from Department of Chemistry, National University of Singapore. Meenakshi Annamalai, NUNNI-Nanocore (NUS) for Contact angle measurement. Deepa Sriramulu thanks Ministry of Education and National University of Singapore for a research scholarship towards her PhD program.References
- B.-H. Jeong, E. M. V. Hoek, Y. Yan, A. Subramani, X. Huang, G. Hurwitz, A. K. Ghosh and A. Jawor, J. Membr. Sci., 2007, 294, 1 CrossRef CAS.
- A. K. Ghosh, B.-H. Jeong, X. Huang and E. M. V. Hoek, J. Membr. Sci., 2008, 311, 34 CrossRef CAS.
- C. C. Wamser and M. I. Gilbert, Langmuir, 1992, 8, 1608 CrossRef CAS.
- G.-Y. Chai and W. B. Krantz, J. Membr. Sci., 1994, 93, 175 CrossRef CAS.
- S. J. Wagh, S. S. Dhumal and A. K. Suresh, J. Membr. Sci., 2009, 328, 246 CrossRef CAS.
- H. Ichiura, K.-i. Yamamoto and Y. Ohtani, Polym. Bull., 2015, 72, 2621 CrossRef CAS.
- R. Dsouza, D. Sriramulu and S. Valiyaveettil, RSC Adv., 2016, 6, 24508 RSC.
- G. N. B. Baroña, J. Lim, M. Choi and B. Jung, Desalination, 2013, 325, 138 CrossRef.
- F. MacRitchie, Trans. Faraday Soc., 1969, 65, 2503 RSC.
- S. S. Dhumal, S. J. Wagh and A. K. Suresh, J. Membr. Sci., 2008, 325, 758 CrossRef CAS.
- C. N. R. Rao, G. U. Kulkarni, V. V. Agrawal, U. K. Gautam, M. Ghosh and U. Tumkurkar, J. Colloid Interface Sci., 2005, 289, 305 CrossRef CAS PubMed.
- K. Biswas and C. N. R. Rao, J. Colloid Interface Sci., 2009, 333, 404 CrossRef CAS PubMed.
- C. Wang, Q. Hu, P. Fang and C. He, Wuhan Univ. J. Nat. Sci., 2013, 18, 213 CrossRef CAS.
- Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. Guo, H. Soyez, B. Dunn, M. H. Huang and J. I. Zink, Nature, 1997, 389, 364 CrossRef CAS.
- R. M. de Vos and H. Verweij, Science, 1998, 279, 1710 CrossRef CAS PubMed.
- S. Morishita, Y. Uchida and M. Matsumura, Jpn. J. Appl. Phys., Part 1, 1995, 34, 5738 CAS.
- A. Pfuch, A. Heft, R. Weidl and K. Lang, Surf. Coat. Technol., 2006, 201, 189 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- C. J. Brinker, A. J. Hurd, P. R. Schunk, G. C. Frye and C. S. Ashley, J. Non-Cryst. Solids, 1992, 147–148, 424 CrossRef CAS.
- N. J. Arfsten, A. Eberle, J. Otto and A. Reich, J. Sol-Gel Sci. Technol., 1997, 8, 1099 CAS.
- D. Meyerhofer, J. Appl. Phys., 1978, 49, 3993 CrossRef.
- Y.-Y. Huang and K.-S. Chou, Ceram. Int., 2003, 29, 485 CrossRef CAS.
- D. Ge, L. Yang, G. Wu and S. Yang, Chem. Commun., 2014, 50, 2469 RSC.
- M. M. Kulkarni, R. Bandyopadhyaya and A. Sharma, J. Mater. Chem., 2008, 18, 1021 RSC.
- L. Faget, A. Berman and O. Regev, Thin Solid Films, 2001, 386, 6 CrossRef CAS.
- M. Telepchak, T. August and G. Chaney, in Forensic and Clinical Applications of Solid Phase Extraction, Humana Press, 2004, p. 55 Search PubMed.
- R. Wang, G. Baran and S. L. Wunder, Langmuir, 2000, 16, 6298 CrossRef CAS.
- P. A. J. De Witte, J. Hernando, E. E. Neuteboom, E. M. H. P. van Dijk, S. C. J. Meskers, R. A. J. Janssen, N. F. van Hulst, R. J. M. Nolte, M. F. García-Parajó and A. E. Rowan, J. Phys. Chem. B, 2006, 110, 7803 CrossRef CAS PubMed.
- A. N. Parikh, M. A. Schivley, E. Koo, K. Seshadri, D. Aurentz, K. Mueller and D. L. Allara, J. Am. Chem. Soc., 1997, 119, 3135 CrossRef CAS.
- X. Zhang, Y. Wu, J. Li, F. Li and M. Li, Dyes Pigm., 2008, 76, 810 CrossRef CAS.
- W. S. Shin, H.-H. Jeong, M.-K. Kim, S.-H. Jin, M.-R. Kim, J.-K. Lee, J. W. Lee and Y.-S. Gal, J. Mater. Chem., 2006, 16, 384 RSC.
- C. Boehm, F. Leveiller, D. Jacquemain, H. Moehwald, K. Kjaer, J. Als-Nielsen, I. Weissbuch and L. Leiserowitz, Langmuir, 1994, 10, 830 CrossRef CAS.
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudhölter, J. Am. Chem. Soc., 2000, 122, 11057 CrossRef CAS.
- B. Fouzia, J. Ferguson, K. McKenna, L. E. McNamara, N. I. Hammer and H. Rathnayake, New J. Chem., 2015, 39, 2004 RSC.
- Y. Huang, W. Zhang, H. Zhai and C. Li, J. Mater. Chem. C, 2015, 3, 466 RSC.
- T. Ribeiro, C. Baleizão and J. P. S. Farinha, J. Phys. Chem. C, 2009, 113, 18082 CAS.
- R. Gómez, D. Veldman, R. Blanco, C. Seoane, J. L. Segura and R. A. J. Janssen, Macromolecules, 2007, 40, 2760 CrossRef.
- X. Li, L. E. Sinks, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 10810 CrossRef CAS PubMed.
- Y. Che, X. Yang, S. Loser and L. Zang, Nano Lett., 2008, 8, 2219 CrossRef CAS PubMed.
- E. E. Beauvilliers, M. R. Topka and P. H. Dinolfo, RSC Adv., 2014, 4, 32866 RSC.
- E. Dobruchowska, T. Marszalek and J. Ulanski, Thin Solid Films, 2014, 564, 361 CrossRef CAS.
- F. L. Arbeloa, V. Martínez, T. Arbeloa and I. L. Arbeloa, in Reviews in Fluorescence 2008, ed. C. D. Geddes, Springer New York, New York, NY, 2010, pp. 1–35 Search PubMed.
- Y. Zakrevskyy, C. F. J. Faul, Y. Guan and J. Stumpe, Adv. Funct. Mater., 2004, 14, 835 CrossRef CAS.
- M. T. Sims, L. C. Abbott, S. J. Cowling, J. W. Goodby and J. N. Moore, Chem.–Eur. J., 2015, 21, 10123 CrossRef CAS PubMed.
- D. Sriramulu and S. Valiyaveettil, Dyes Pigm., 2016, 134, 306 CrossRef CAS.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390 CrossRef CAS PubMed.
- J. Blechinger, R. Herrmann, D. Kiener, F. J. García-García, C. Scheu, A. Reller and C. Bräuchle, Small, 2010, 6, 2427 CrossRef CAS PubMed.
- M. Annamalai, K. Gopinadhan, S. A. Han, S. Saha, H. J. Park, E. B. Cho, B. Kumar, A. Patra, S.-W. Kim and T. Venkatesan, Nanoscale, 2016, 8, 5764 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular structure of precursors used for preparing silica films, SEM images of silica films, TGA-DTA analysis of O-Si, P-Si film and PO100-Si film, digital photograph of O-Si film surface collected on a glass substrate, optical images of film in the presence and absence of UV source. See DOI: 10.1039/c7ra05036d |
| This journal is © The Royal Society of Chemistry 2017 |