
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking the disulfide chemical bond using high energy photons: the dimethyl disulfide and methyl propyl disulfide molecules
L. R. Varas a,
L. H. Coutinhob,
R. B. Berninic,
A. M. Betancourtabd,
C. E. V. de Mourad,
A. B. Rochad and
G. G. B. de Souza*d
a,
L. H. Coutinhob,
R. B. Berninic,
A. M. Betancourtabd,
C. E. V. de Mourad,
A. B. Rochad and
G. G. B. de Souza*d
aEscuela de Ingeniería Química, Universidad de Costa Rica, Sede Central Rodrigo Facio, Facultad de Ingeniería, 5° piso, 11501-2060 San José, Costa Rica
bInstituto de Física, Universidade Federal do Rio de Janeiro, Cidade Universitária, Ilha do Fundão, 21949-900, Rio de Janeiro, RJ, Brazil
cInstituto Federal de Ciência e Tecnologia do Rio de Janeiro (IFRJ), Duque de Caxias, 25050-100, RJ, Brazil
dInstituto de Química, Universidade Federal do Rio de Janeiro, Cidade Universitária, Ilha do Fundão, 21949-900, Rio de Janeiro, RJ, Brazil. E-mail: gerson@iq.ufrj.br
First published on 24th July 2017
Abstract
In order to study the stability of the disulfide chemical bond in molecules subjected to a flux of high energy photons, the ionic fragmentation of two molecules containing a disulfide chemical bond, dimethyl disulfide (DMDS) and methyl propyl disulfide (MPDS), has been studied following excitation around the S 1s edge (∼2470 eV). Synchrotron radiation and electron-ion coincidence techniques were used. The core excited states of DMDS and MPDS have also been theoretically investigated at the multiconfigurational self-consistent field (MCSCF) level and multireference perturbation theory. Deep core excitation (S 1s) induces multiple ionization and intense fragmentation of the molecules, as clearly demonstrated by the observation of cations such as S+, S2+ and S3+. Splitting of the S+ peak is observed in the mass spectra of the two molecules when they are excited to a resonant state with antibonding (S–S) character. Although fragments associated with the breakage of the disulfide bond (S+ and CHnS+) play a dominant role in the dissociation of the molecules, a fragment containing the disulfide chemical bond, S2+, survives, nevertheless, at all photon energies.
1. Introduction
The formation and stability of chemical bonds is considered to basically involve interactions between valence-shell electrons, with core electrons assuming only a minor role in this endeavor. Nevertheless, in the last few decades a wealth of (mainly) experimental results has demonstrated that the excitation and ionization of core electrons may lead to an extensive breakage of chemical bonds and consequently to the dissociation of molecules. The dynamics of relaxation of highly excited molecules may actually involve a vast number of radiative and non-radiative processes. This subject has been the focus of several excellent reviews (for example, 1–3). For low Z atomic number elements (Z < 20), however, non-radiative processes predominate. For instance, following the excitation of a shallow core electron (here roughly associated with excitation energies below 1 keV photon energy) to a valence-excited electronic state, the excited state has a large probability of relaxing very rapidly through the emission of a valence electron, while another valence-shell electron fills the hole. In this resonant Auger process the final state will either be a valence single-hole state, if the excited electron participates in the relaxation process, or a valence-shell two hole/one particle state, if the excited electron remains as spectator. Above the ionization edge, ionization of a core electron will give rise to (normal) Auger processes: the core hole is filled by a valence electron while another valence electron is emitted. The final state in this case will be a doubly ionized state. In double Auger processes, emission of two valence-shell electrons will occur, giving rise to triply ionized states. Relaxation processes involving the emission of one or more valence electrons may of course have dire consequences in which concerns the stability of chemical bonds.Going a step further in excitation energy and examining the case in which the excitation and ionization process involves a deep core electron, yet another degree of complexity is introduced as relaxation of the core-excited or ionized state may also give rise to the formation of another core hole (for instance K shell relaxation may result in L-(M…) shell ionization). This cascade Auger process will result in multiply-ionized molecular species, usually of quite unstable and dissociative character.
In plain terms, excitation and ionization of core electrons may result in extensive depletion of the valence shell, leading to efficient breakage of chemical bonds and consequently to the dissociation of molecules. Excitation of core electrons is also element-specific, paving the way to the study of the dissociation of a molecule following excitation around one of its constituent atoms. In this paper, the breakage of a very important chemical bond – the disulfide chemical bond – is studied as a function of the photon energy tuned around the S 1s edge of two volatile molecules containing this bond.
Disulfide (–S–S–) chemical bonds (or disulfide bridges) play an important role in the stability and functioning of several peptides and proteins. Proteins such as insulin and immunoglobulins contain one or more residues of the amino acid cysteine and these may be involved in the formation of intra- or inter-molecular disulfide bonds.4,5 In the present study we address our attention to the stability of the disulfide chemical bond in the dimethyl disulfide molecule, CH3S–SCH3 (DMDS), excited around the S 1s edge, using synchrotron radiation. Time-of-Flight Mass Spectrometry (TOFMS) and Near Edge X-ray Absorption (NEXAFS) techniques are employed. The DMDS molecule can be considered as a simple prototype molecule containing a disulfide bond. It is related to the oceanic sulfur cycle and to the formation of acid rain.6,7 A brief review of previous studies concerning the valence-shell photoionization of DMDS may be found in our previous paper.8 The shallow core level (S 2p) excitation and fragmentation of the DMS (CH3SCH3) and the DMDS molecules was studied using synchrotron radiation and theoretical calculations in order to describe the associated excited electronic states. The S 1s photoabsorption spectrum of DMDS in the gas phase has been previously measured by Hitchcock et al.9 Baba studied the core (S 1s) photoexcitation and ionic desorption in DMDS in solid phase.10 More recently, Pickering and co-workers obtained the S 1s photoabsorption spectrum of DMDS along with similar spectra obtained for a range of organic disulfides.11
Excitation and fragmentation of a related molecule, methyl propyl disulfide, CH3S–SCH2CH2CH3 (MPDS), around the S 1s edge will also be discussed in the present paper. Substitution of a methyl by a propyl group breaks the symmetry around the disulfide bond and may give rise to additional excitation and fragmentation mechanisms.
Study of the soft X-ray induced fragmentation of molecules containing a disulfide chemical bond, such as DMDS and MPDS, may also bring crucial information towards the understanding of the radiation damage in sulfur-containing proteins. Weik et. al. discussed the effect of synchrotron radiation on proteins containing the disulfide bridges;12 Sutton et al. discussed the radiation damage associated with disulfide bonds in protein crystallography.13
2. Experimental method
The measurements were performed at the SXS (Soft X-ray absorption spectroscopy) beam line of the Brazilian National Synchrotron Radiation Laboratory, LNLS. This bending magnet beam line covers the energy range from 1 to 5 keV, with energy resolution of 10−4, and has been previously described in detail.14 The samples were studied in the gas phase. The base pressure inside the vacuum chamber was on 10−8 Torr, rising to 10−6 Torr during the measurements. The NEXAFS spectra were recorded in the total ion yield (TIY) detection mode for both molecules. The energy calibration of the beamline was accomplished by recording the NEXAFS spectrum of SF6 in the S 1s edge and comparing to published results.15 Multi-coincidence spectra were recorded at selected photon energies, using a time-of-flight mass spectrometer (TOF-MS) to identify the positive ions resulting from the photoionization process. Coincidences between one electron and one cation (PhotoElectron PhotoIon COincidence – PEPICO) were recorded and will be discussed. The TOF spectrometer used for these measurements has been described in detail before.16 The gas sample is inserted in the ionization region through a needle, and is intercepted by the photon beam. The spectrometer axis is collinear with respect to the electric vector of the polarized photons. It is able to detect electrons with initial kinetic energies up to 150 eV, and ions up to 30 eV. The electrons and ions are not energy selected before detection. The alignment and calibration of the TOF spectrometer was accomplished by recording spectra of argon and SF6.The samples of DMDS and MPDS were purchased from Sigma Aldrich with 95% purity and used without further purification, except for a series of freeze-pump-thaw cycles prior to their admission to the vacuum chamber. During the experiments conducted with SF6, DMDS and MPDS, the chamber was cleaned prior to a different sample being inserted. With this purpose, we flushed argon into the chamber through the needle to remove any material left in the admission system. In the mass spectra of DMDS and MPDS (mainly) taken with 2465.0 eV photons (below the S 1s edge), peaks related to argon can be identified (m/z 8, 10, 13.3 and 20).
3. Theory
Theoretical investigation of core excited states of DMDS and PMDS at S K-edge was initially done at multiconfigurational self-consistent field (MCSCF) level and then supplemented by multireference perturbation theory, as explained later. If some restrictions are imposed on electron occupation and on the mixing of orbitals during the SCF procedure, MCSCF can be applied to core states.17–19 These constraints are necessary to avoid the variational collapse to a low-lying state. In the present work, the active space is chosen by splitting the orbitals in groups, using Occupation Restricted Multiple Active Space wave function (ORMAS).20,21 The first group contains sulfur atoms 1s-like orbitals and the other groups contain valence orbitals related to each final state. The latter are composed by the bonding orbital and two anti-bonding orbitals. Then, the composition for S 1s → σ* (S–S) transitions for both molecules is ORMAS (3,2) (3,3), where the numbers in the brackets indicate the number of electrons and orbitals in each group respectively, and the brackets indicate different groups. To describe transitions to final states located at S–C bonds, orbitals of S–S bond were also included in the active space in order to improve convergence. Accordingly, in DMDS, for transitions of the type S 1s → σ* (S–CH3), one has ORMAS (3,2) (2,2) (5,6). The final group, (5,6), was chosen in a such a way to set all S–C orbitals to the same group. For MPDS molecule, two types of calculation were done, i.e., S 1s → σ* (S–CH3) and S 1s → σ* (S–CH2CH2CH3). In these cases, active space is ORMAS (3,2) (2,2) (3,3).So as to make results more quantitative, dynamic correlation was recovered using Multireference Möller–Plesset Perturbation Theory (MRMP), in the form of the General Multi Configuration Self-Consistent Field Quasi-Degenerate Perturbation Theory (GMC-QDPT), developed by Nakano et al.22 General forms of MCSCF function, like ORMAS, can be treated by this formalism and show good results with IS-MCSCF method with low additional computational cost.17,18
Transition moments were calculated at state-averaged CASSCF level. For both molecules, a CAS (4,5), considering the ground state and the first core excited states was used. Oscillator strengths were calculated from these transition moments. The transition energies were obtained as explained in the previous paragraph.
All calculations were done with Dunning's aug-cc-pCVDZ basis set.23,24 Scalar relativistic effects are described by Douglas–Kroll–Hess method to 3rd order.25–27 Multi configurational self-consistent calculations were done with MOLPRO.28,29 Perturbation theory calculations were done with GAMESS.30,31
4. Results and discussion
4.1 Excitation and fragmentation of the DMDS molecule
The NEXAFS (photoabsorption) spectrum of DMDS, measured around the S 1s edge is shown in Fig. 1. The energies for the main peaks and respective assignments are presented in Table 1. Our spectrum shows an overall agreement with previous gas phase results obtained by Hitchcock et al.9 as well as with the condensed phase measurements made by Baba.10 The first structure in the spectrum is composed by two main σ resonances, observed at 2470.9 eV and 2471.8 eV while a broad shape resonance is observed around 2480 eV. These energy values are in very good agreement with the recent values obtained by Pickering et al.11 Our theoretical results for the DMDS S 1s photoabsorption, obtained at IS-MRMP with aug-cc-pCVDZ/DKH3 show an excellent numerical agreement with the experimental data and confirms that the first peak in the experimental spectrum can be attributed to a transition to a σ* (S–S) orbital while the second peak corresponds to a transition to a σ* (S–CH3) orbital. More detailed results, as well ionization energies, are presented in Table 1. The antibonding character of the σ* (S–S) has been demonstrated by electron transmission spectroscopy (ETS) and dissociative electron attachment spectroscopy (DEAS).32 The accepted experimental S 1s ionization potential in DMDS is 2477.5 eV, as previously determined by Sodhi et al.33 and corrected by Carroll et al.34 This is also in very good agreement with our theoretical results.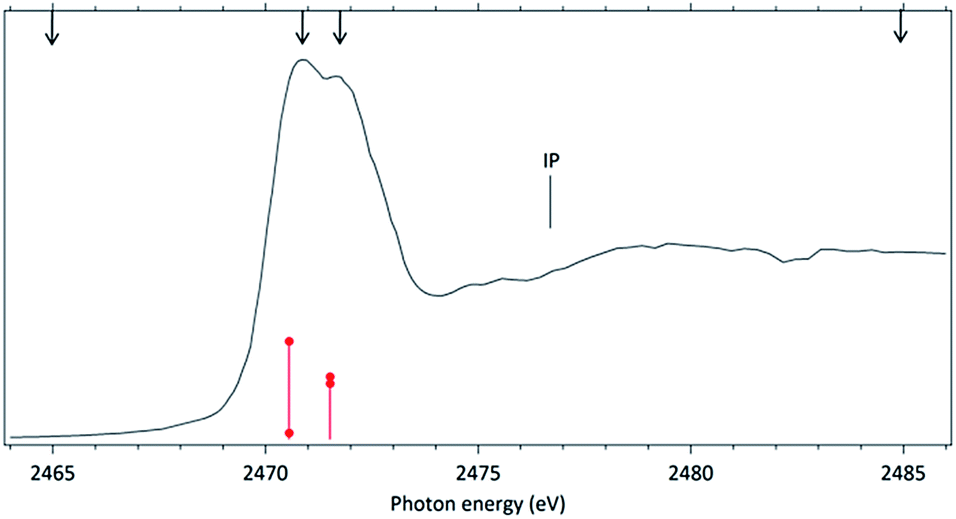 | ||
| Fig. 1 Photoabsorption spectrum (NEXAFS) of the DMDS molecule obtained around the S 1s edge. The ionization potential (IP) position was included. The arrows indicate the energy positions in which the TOF spectra were recorded. Calculated values are shown in red, with heights proportional do the oscillator strengths for each transition (red circles). | ||
In order to study the stability of the disulfide bond with respect to core electron excitation and ionization, ionic fragmentation spectra of the DMDS molecule have been obtained at four photon energies, around the S 1s edge: 2465.0 eV (below resonances), 2470.9 eV (on the first resonance, σ* (S–S character)), 2471.8 eV (second resonance, σ* (S–CH3 character)) and 2485.0 eV (above the S 1s ionization edge). The four mass spectra are shown in Fig. 2. The observed fragments and assignments are presented in Table 2. The branching ratios and their energy dependence will be discussed in a forthcoming paper.35 The subsequent discussion concentrates mostly in fragmentation processes directly involved with the survival or breakage of the disulfide chemical bond.
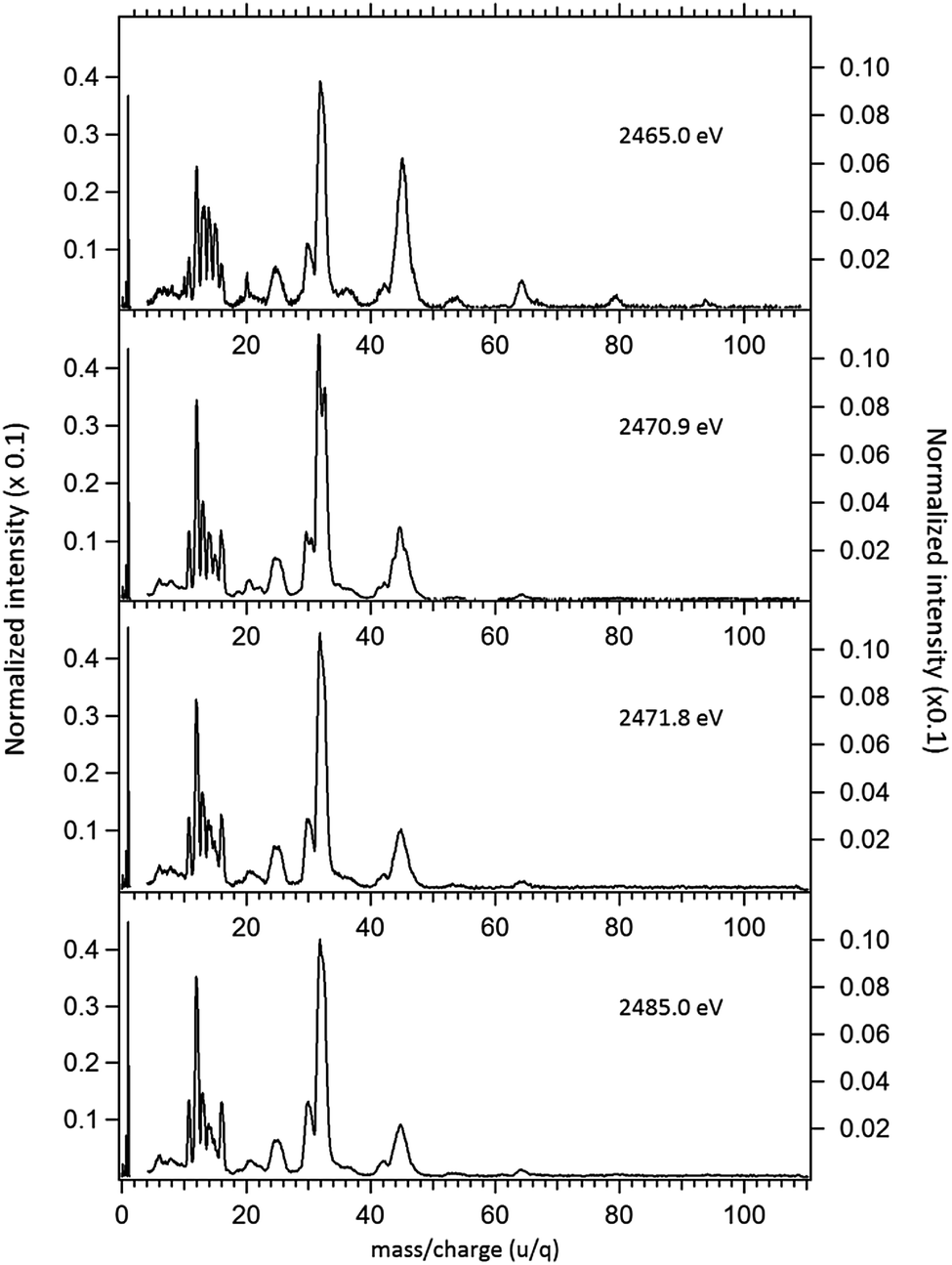 | ||
| Fig. 2 Normalized mass spectra of the DMDS molecule obtained below resonances (2465.0 eV), on the resonances (2470.9 eV and 2471.8 eV) and above the S 1s ionization potential (2485.0 eV). The left axis applies only to the intensity of the H+ ion, while the right axis applies to the intensities of the remaining fragments. | ||
| m/z (u/q) | Fragment DMDS | Fragment MPDS |
|---|---|---|
| 1 | [H]+ | [H]+ |
| 2 | [H2]+ | [H2]+ |
| 6 | [C]2+ | [C]2+ |
| 8 | [S]4+ | [S]4+ |
| 10.7 | [S]3+ | [S]3+ |
| 12 | [C]+ | [C]+ |
| 13 | [CH]+ | [CH]+ |
| 14–15 | [CH2]+, [CH3]+ | [CH2]+, [CH3]+ |
| 16 | [S]+2 | [S]+2 |
| 24–26 | [C2]+, [C2H]+, [C2H2]+ | [C2]+, [C2H]+, [C2H2]+ |
| 27 | [CH2CH]+ | |
| 29.5 | [CH3CS]2+ | |
| 30 | [CH3–CH–S]2+ or [CH3CH3]+ | [CH3–CH–S]2+ or [CH3CH3]+ |
| 32 | [S]+ | [S]+ |
| 36 | [C3]+ or [CH3CHC–S]2+ | |
| 37 | [C3H]+ | |
| 39 | [C3H3]+ | |
| 41 | [CH2–CH2–CH]+ | |
| 42 | [CH2–CH2–CH2]+ | |
| 43 | [CH3–CH2–CH2]+ | |
| 44–46 | [CS]+, [H–C–S]+, [H2–C–S]+ | [CS]+, [H–C–S]+, [H2–C–S]+ |
| 56 | [C2S]+ | |
| 64 | [S2]+ | [S2]+ |
| 79 | [CH2–S2–H]+ | [CH2–S2–H]+ |
| 94 | [DMDS]+ | |
| 123 | [MPDS]+ |
The mass spectra were obtained in the photoelectron-photoion coincidence (PEPICO) mode. For each spectrum, an average baseline was determined summing up the counts on a region where no ions were observed, and dividing by the number of time channels in this m/q interval. This baseline was subtracted from the whole spectrum, which was then normalized to the total number of counts.36
As shown in Fig. 2, photoabsorption around the S 1s edge induces strong overall dissociation of the molecule, with predominance of processes involving the breakage of the disulfide bond, as demonstrated by the important contribution to the spectra of fragments such as S+ (m/z = 32) and CHnS+ (m/z = 44–47). Breakage of the S–C bond is also relevant, as evidenced by the presence of CHn+ (m/z = 12–15) fragments. Interestingly, fragments such as S2+ (m/z = 64) and CH3S2+ (m/z = 79) in which the disulfide bond is preserved, are still observed, albeit with lower intensities, even when the molecule is subjected to photon energies as high as 2465.0 eV. The molecular ion, CH3S2CH3+ (m/z = 94), can also still be observed at this photon energy, with much reduced intensity. These fragments are also observed, with much higher relative intensities in the DMDS electron impact mass spectrum, obtained both at low impact energy (NIST MS Number 291515) and at high electron incident energy (800 eV).37 They are also observed in lower energy photon induced mass spectra, as described below.
The photon-induced breakage of the disulfide bond in DMDS at lower photon energies has been previously described. For instance at 20.63 eV (valence-shell ionization),38 although fragments with intact disulfide bonds (S2+, CH3S2+ and CH3S2CH3+), make important contributions to the mass spectrum, fragments such as CHnS+ (n = 1–6) which may be considered as a signature of the symmetric breaking of the disulfide bond, are also clearly observed.
At somewhat higher photon energies, the role of shallow core excitation in the fragmentation of DMDS has been studied by Bernini et al.8 who used synchrotron radiation to excite the molecule around the S 2p edge (160–180 eV). We here briefly highlight the main observations of that paper in which concerns the breakage of the disulfide bond. At 160 eV (below the S 2p resonances) ionization of the molecule can only occur via direct processes. Single, double and triple ionization of the molecule may contribute to the molecular dissociation. We roughly estimate the triple ionization potential of DMDS to be of the order of 50 eV, by comparison with a value of 53.1 eV obtained for CS2 (ref. 38) and approximately 50 eV for the OCS molecule.39 In addition to the fragments observed at 20.63 eV, a strong contribution from the S+ fragment appears in the 160 eV mass spectrum, an experimental evidence of the appearance of two (or more) bond breakage processes. Fragments such as H+, C+, along with S+ also pointed out to the possible atomization of the molecule at 160 eV photon energy. At the photon energies corresponding to the S 2p resonances (163.8 eV, 166.5 eV and 167.6 eV) and above the S 2p edge (168.3 eV (ref. 8)), CHnS+ and CHn+ become the most intense fragments, while the relative intensity of fragments with preservation of the disulfide bond, such as CH3S2+ and CH3S2CH3+, was seen to decrease.
Around the S 1s edge, the photon-induced fragmentation of the molecule increases significantly, with multiple Auger and cascade Auger processes playing important roles. The large degree of fragmentation can be exemplified by the observation that at this energy range the most intense cation in the mass spectra corresponds to S+. The overall fragmentation pattern around the S 1s edge is similar both below (2465.0 eV), on the first and second resonances (2470.9 eV and 2471.8 eV) and above the ionization edge (2485.0 eV). Previously unobserved doubly and triply charged species such as S2+ (m/z = 16) and, S3+ (m/z = 10.7) as well as rearrangement ions such as C2Hn+ (n = 0, 1, 2) and C2H6+ (m/z = 30) are clearly observed in our data. These rearrangement ions are probably associated with the dissociation of multiply-charged ionic species. Taking into consideration that S 1s excited states have lifetimes of the order of a femtosecond,40 these ions may be the result of extremely fast (femtosecond or attosecond regime) chemical reactions. From the three previously mentioned fragments containing an intact disulfide bonding, only S2+ survives, following excitation of the molecule to the excited state at 2470.9 eV, corroborating the strong antibonding character of this state. Above the S 1s ionization potential, again the only surviving fragment with a disulfide bond is S2+. Several ionic fragments previously observed in the spectra obtained around the S 2p edge, such as CH3+, S+, CHnS+, are also observed around the S 1s edge. An additional and interesting feature is the observed splitting of the peak corresponding to the S+ fragment in the mass spectrum obtained at the S 1s first resonance. In the enlargement of the mass spectra obtained at 2470.9 eV and 2471.8 eV and presented in Fig. 3 it can be clearly observed that the splitting of the S+ peak only occurs at the first resonance. A similar peak splitting has been previously observed in the fragmentation of the chloroform (CHCl3)36 and dichloromethane (CH2Cl2)41 molecules, following excitation of Cl 1s electrons to an antibonding unoccupied molecular orbital. This “Doppler effect” associated with the ultrafast dissociation of molecules, is related to the preservation of molecular alignment with respect to the polarization axis of the X-ray electromagnetic radiation. Within our knowledge, this is the first experimental observation of this kind of splitting in a sulfur containing molecule, excited around the S K-edge. The fact that this splitting has by now been observed at the fragmentation of both chlorine and sulfur containing molecules following Cl 1s and S 1s excitation to antibonding virtual states may indicate that this splitting can be viewed as a general feature associated with the ultrafast dissociation of deep-core electron excited molecules.
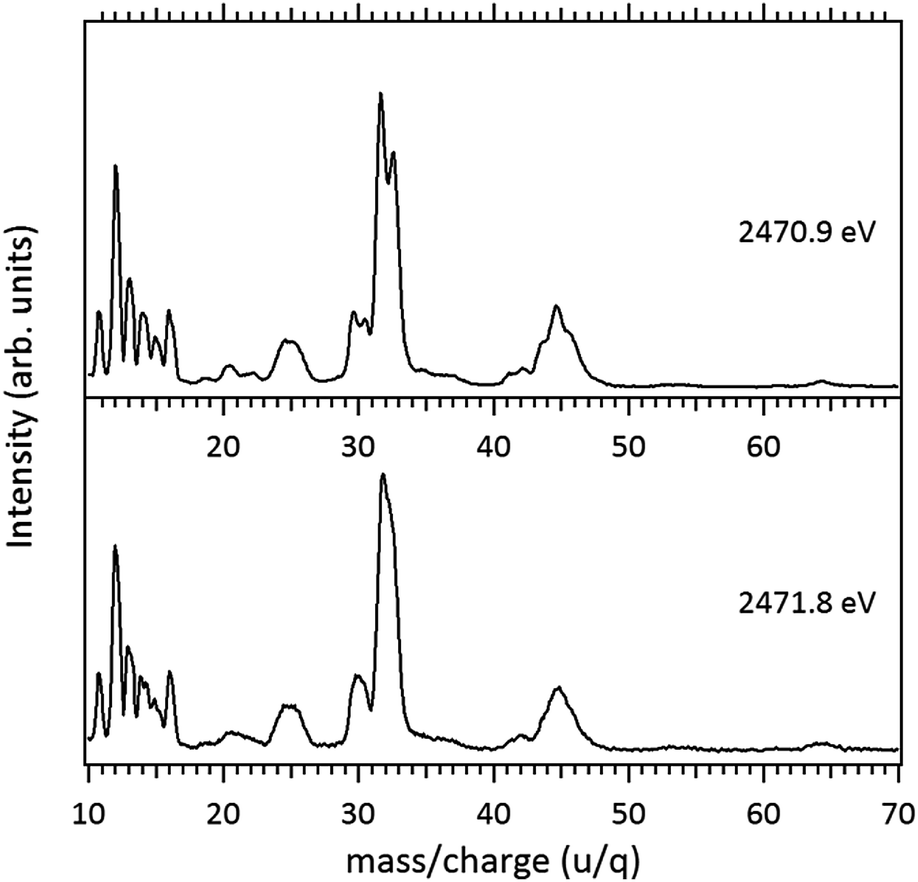 | ||
| Fig. 3 Enlargement of the mass (PEPICO) spectra of DMDS measured at 2470.9 eV and 2471.8 eV. | ||
4.2 Excitation and fragmentation of the MPDS molecule
In order to further explore the breakage of the disulfide chemical bond in molecules excited around the S 1s edge, we next investigate the excitation and fragmentation of methyl propyl disulfide (MPDS). This molecule may be seen as a “substituted” DMDS. The addition of methyl groups decreases the total molecular symmetry and in particular introduces an asymmetry around the S–S chemical bond.The NEXAFS (photoabsorption) spectrum of MPDS, measured around the S 1s edge (2460–2490 eV) is shown in Fig. 4. The energies for the main peaks and assignments are presented in Table 3. Within our knowledge the S 1s photoabsorption spectrum for the methyl propyl disulfide molecule (MPDS) is here presented and discussed for the first time.
 | ||
| Fig. 4 Photoabsorption spectrum (NEXAFS) of the MPDS molecule obtained around the S 1s edge. The ionization potential (IP) position was included. The arrows indicate the energy positions in which the TOF spectra were recorded. Calculated values are shown in red, with heights proportional do the oscillator strengths for each transition (red circles). | ||
| Peak | Experiment (eV) | Theory (eV) | Assignment | |
|---|---|---|---|---|
| S#1 | S#2 | |||
| 1 | 2470.7 | 2470.1 | 2470.3 | σ* (S–S) |
| 2 | 2472.0 | 2471.4 | 2471.7 | σ* (S–CH3) |
| 3 | 2471.3 | 2471.7 | σ* (S–CH2CH2CH3) | |
| 2476.2 | IP | |||
The MPDS photoabsorption spectrum resembles the NEXAFS spectrum of DMDS in the same photon energy range, but a richer fine structure may be observed between the two main resonances, pointing out to the existence of a more complex excitation manifold.
For the MPDS molecule, our theoretical results at IS-MRMP with aug-cc-pCVDZ/DKH3 show three peaks. The first peak in the photoabsorption spectrum of MPDS can be attributed to the transition to a σ* (S–S) orbital, the second and third corresponding to pairs of excitations to σ* (S–CH3) and σ* (S–CH2CH2CH3) orbitals. Transitions to σ* (S–CH2CH2CH3) orbitals have lower transition energy when compared to transitions to σ* (S–CH3) orbitals. More detailed results, as well as ionization energies, are presented in the Table 3.
Fragmentation of the MPDS molecule around the S 1s edge has been studied at photon energies corresponding to below, on and above the resonances. The resulting mass spectra are presented in Fig. 5. The observed fragments and assignments are presented in Table 2. In these spectra the same baseline subtraction and normalization procedures applied to the DMDS molecule were employed.
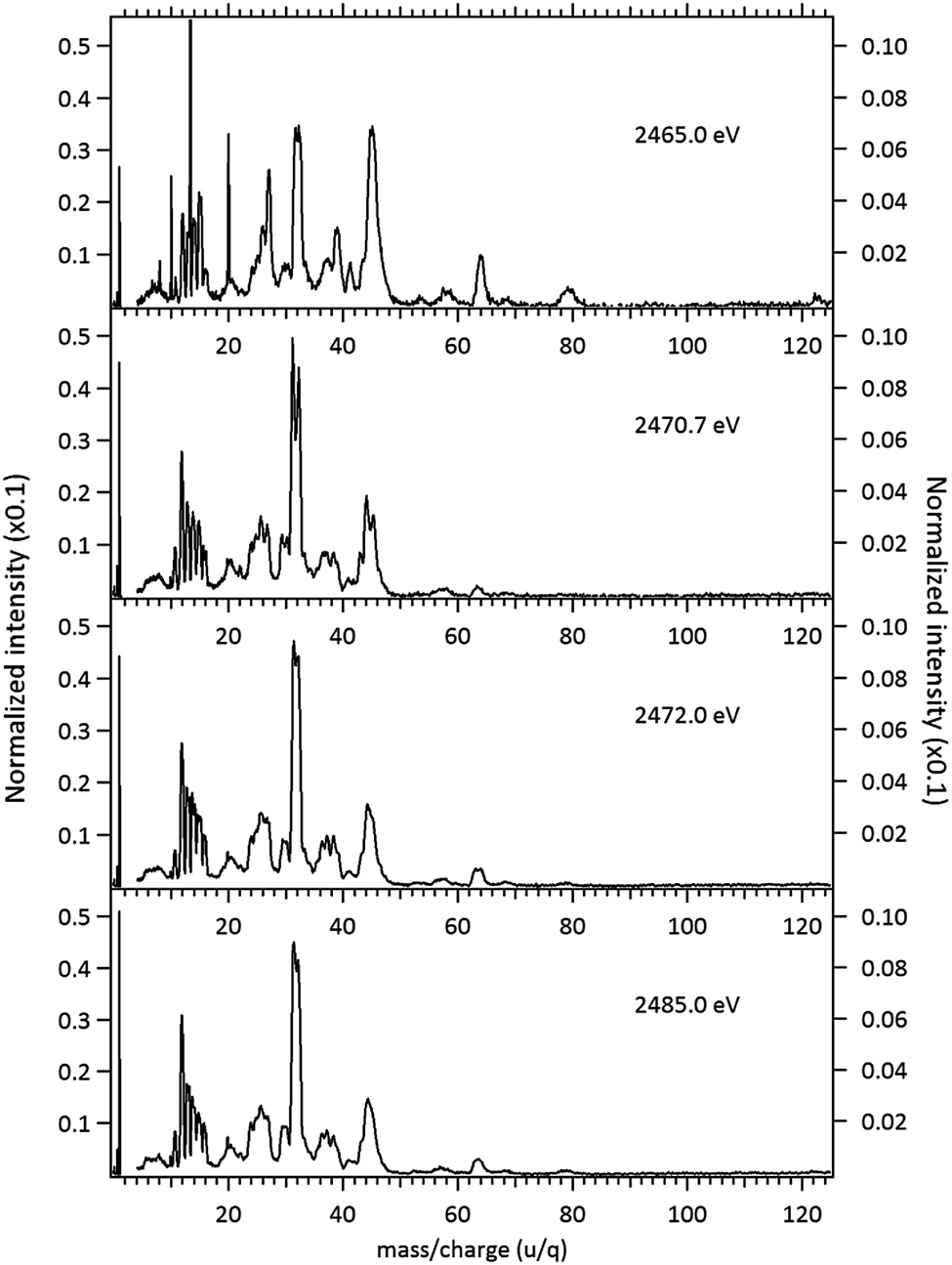 | ||
| Fig. 5 Normalized mass spectra of the MPDS molecule obtained below resonances (2465.0 eV), on the resonances (2470.7 eV and 2472.0 eV) and above the S 1s ionization potential (2485.0 eV). The left axis applies only to the intensity of the H+ ion, while the right axis applies to the intensities of the remaining fragments. The narrow peaks observed at the spectrum obtained below resonances (2465.0 eV) are due to argon contamination (m/z = 8; 10; 13.3; 20). | ||
The fragmentation pattern of MPDS presents an overall similarity with respect to the DMDS case. Singly-charged fragments such as H+, C+, CHn+, S+, CHnS+, S2+, CH3S2+ along with doubly and triply charged fragments: S2+ (m/z = 16), S3+ (m/z = 10.7), CH3SCH22+ (m/z = 30), are seen in both cases. As expected, additional fragments, related to the propyl group can now be observed: C2Hn+ (n = 0–3) (m/z = 24–27) and C3Hn+ (n = 1, 3, 5) (m/z = 37, 39, 41). Fragments with disulfide bond preservation, S2+ (m/z = 64) and CH3S2+ (m/z = 79), are still observed at 2466 eV photon energy. The molecular ion, CH3S2CH2CH2CH3+ (m/z = 122), can also be observed, with much reduced intensity. Above the S 1s ionization potential, again the only surviving fragment with a disulfide bond is S2+. In the mass spectrum obtained at the photon energy corresponding to the first resonance (2470.7 eV), a Doppler splitting of the S+ peak is observed. This splitting is also observed in other sulfur-containing fragments: CHnS+, S2+ and CH3SCH22+.
Splitting of sulfur-containing fragments in the photoionization of the PMDS molecule is emphasized in the enlargement of the mass (PEPICO) spectra obtained at the first (2470.7 eV) and second (2472.0 eV) resonances, as shown in Fig. 6.
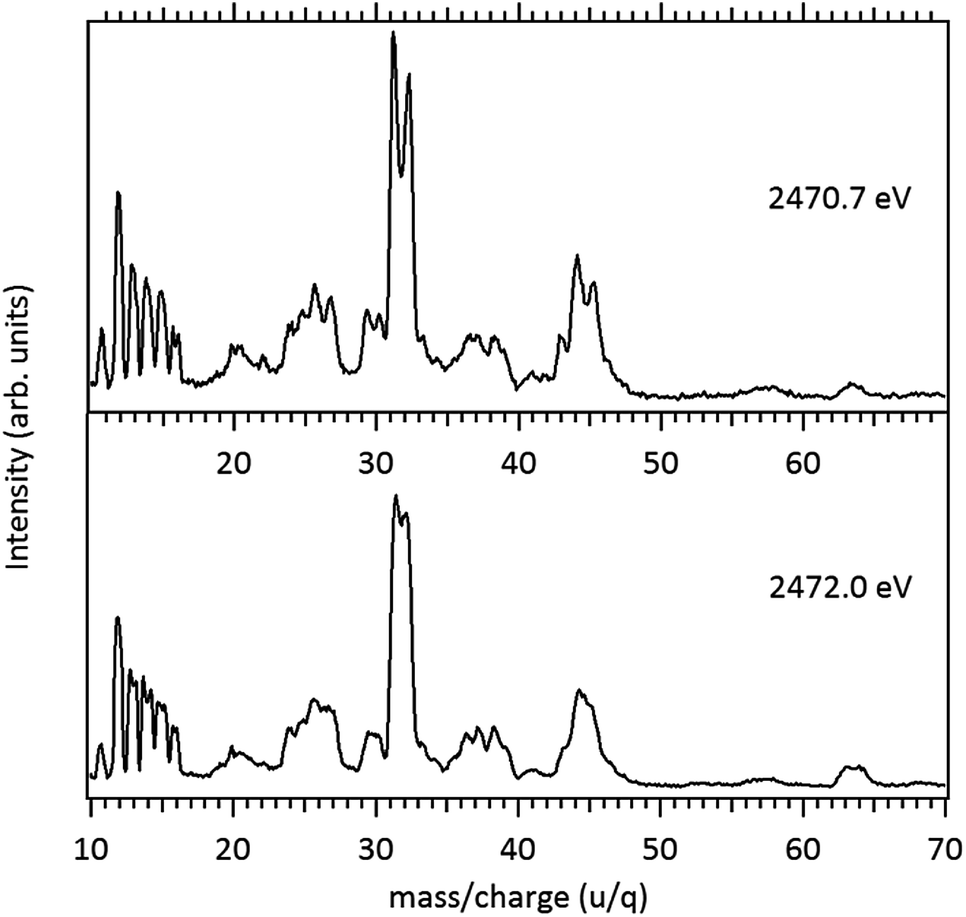 | ||
| Fig. 6 Enlargement of the Mass (PEPICO) spectra of PMDS measured at 2470.7 eV and 2472.0 eV. | ||
4.3 Photostability of the S2+ cation
A final but important remark consists in emphasizing the observation of the fragment S2+ in the mass spectra of both molecules, obtained below, on and above the S 1s resonances. This cation, whose existence is so obviously related to the formation and stability of the sulfur–sulfur chemical bond, has been the focus of several studies. Aitchison and Eland observed the formation of S2+ in association with the dissociative ionization of CS2.42 In a detailed theoretical study, Urban et al.43 discussed the electronic configuration of S2, S2+, S22+ and S23+ and the metastability of the doubly and triply charged cations. More recently, a theoretical investigation of the S2+ cation in gas phase was performed by Yu et al.44 Spectroscopic observation using the Hubble Space Telescope has evidenced that S2 is a common product of volcanic reactivity on Io, one of Jupiter's satellites.45 The present work demonstrates that following deep core excitation or deep core ionization of two sulfur containing molecules – DMDS and MPDS – the only disulfide bond-containing surviving fragment is S2+, attesting its chemical stability. At this point it seems in order to point out that the S2+ cation is also observed in the low photon energy regime37 as well as under electron induced dissociation of the DMDS molecule.36 Careful analysis shows that the S2+ fragment is not subjected to the Doppler splitting observed mainly in the S+ fragments, when the two molecules, DMDS and PMDS are excited to their first excited states (2470.9 eV and 2470.7 eV, respectively).5. Conclusions
In order to study the stability of the disulfide (–S–S–) chemical bond in molecules subjected to high energy photons, the ionic fragmentation of the dimethyl disulfide (DMDS) and methyl propyl disulfide (MPDS) molecules, has been studied around the S 1s edge, using synchrotron radiation and advanced TOF-MS techniques. A detailed theoretical investigation of the core excited states of DMDS and MPDS at S K-edge was done at multiconfigurational self-consistent field (MCSCF) level, supplemented by multireference perturbation theory. Intense fragmentation of the molecules is observed, with the S+ and CHnS+ cations being the most prominent features of the mass spectra for both molecules. Stable doubly and triply charged fragments, S2+, S3+ and CH3SCH22+, are observed for the first time and their existence is related to the existence of complex Auger processes (double and triple Auger, Cascade Auger). Splitting of the S+ peak is observed in the mass spectra of the two molecules when excited to the first resonance, associated with an antibonding state with S–S character. We also stress the stability of the S2+ cation, which appears in the mass spectra obtained at all photon energies. Further analysis of the fragmentation and multiple ionization of the DMDS molecule is under progress.Acknowledgements
We gratefully acknowledge the financial and technical assistance of the Brazilian Synchrotron Light Facility (LNLS). The authors are particularly indebted to Dr Flavio Vicentim for his help during the course of the experiments. Work partially supported by the Brazilian Agencies CNPq and FAPERJ.References
- I. Nenner and P. Morin in VUV and Soft X-Ray Photoionization, ed. U. Becker and D. A. Shirley, Plenum Press, New York, 1996 Search PubMed.
- R. Feifel and M. N. Piancastelli, J. Electron Spectrosc. Relat. Phenom., 2011, 183, 10 CrossRef CAS.
- C. Miron and P. Morin, in Handbook of High Resolution Spectroscopy, ed. M. Quack and F. Merkt, John Wiley & Sons, Ltd., 2011 Search PubMed.
- N. A. Patil, J. Tailhades, R. A. Hughes, F. Separovic, J. D. Wade and M. A. Hossain, Int. J. Mol. Sci., 2015, 16, 1791 CrossRef CAS PubMed.
- M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614 CrossRef CAS PubMed.
- S. M. Siviert, R. P. Kiene and H. N. Schulz-Vogt, Oceanography, 2007, 20, 117 CrossRef.
- A. J. Kettle and M. O. Andreae, J. Geophys. Res., 2000, 105, 26793 CrossRef CAS.
- R. B. Bernini, L. B. G. da Silva, F. N. Rodrigues, L. H. Coutinho, A. B. Rocha and G. G. B. de Souza, J. Chem. Phys., 2012, 136, 144307 CrossRef CAS PubMed.
- A. P. Hitchcock, S. Bodeur and M. Tronc, Phys. B, 1989, 158, 257 CrossRef CAS.
- Y. Baba, K. Yoshii and T. A. Sasaki, J. Chem. Phys., 1996, 105, 8858 CrossRef CAS.
- I. J. Pickering, M. Barney, J. J. H. Cotelesage, L. Vogt, M. J. Pushie, A. Nissan, R. C. Prince and G. N. George, J. Phys. Chem. A, 2016, 120, 7279 CrossRef CAS PubMed.
- M. Weik, R. B. Ravelli, G. Kryger, S. McSweeney, M. L. Raves, M. Harel, P. Gros, I. Silman, J. Kroon and J. L. Sussman, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 623 CrossRef CAS.
- K. A. Sutton, P. J. Black, K. R. Mercer, E. F. Garman, R. L. Owen, E. H. Snell and W. A. Bernhard, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 2381 CAS.
- M. Abbate, F. C. Vicentin, V. C. Cailhol, M. C. Rocha and H. Tolentino, J. Synchrotron Radiat., 1999, 6, 964 CrossRef CAS.
- E. Hudson, D. A. Shirley, M. Domke, G. Remmers, A. Puschmann, T. Mandel, C. Xue and G. Kaindl, Phys. Rev. A, 1993, 47, 361 CrossRef CAS.
- F. Burmeister, L. H. Coutinho, R. R. T. Marinho, M. G. P. Homem, M. A. A. de Morais, A. Mocellin, O. Björneholm, S. L. Sorensen, P. d. T. Fonseca, A. Lindgren and A. Naves de Brito, J. Electron Spectrosc. Relat. Phenom., 2010, 180, 6 CrossRef CAS.
- A. B. Rocha and C. E. V. de Moura, J. Chem. Phys., 2011, 135, 224112 CrossRef PubMed.
- C. E. V. de Moura, R. R. Oliveira and A. B. Rocha, J. Mol. Model., 2013, 19, 2027 CrossRef PubMed.
- A. B. Rocha, J. Chem. Phys., 2011, 134, 024107 CrossRef PubMed.
- J. Ivanic, J. Chem. Phys., 2003, 119, 9364 CrossRef CAS.
- J. Ivanic, J. Chem. Phys., 2003, 119, 9377 CrossRef CAS.
- H. Nakano, R. Uchiyama and K. Hirao, J. Comput. Chem., 2002, 23, 1166 CrossRef CAS PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1995, 103, 4572 CrossRef CAS.
- K. A. Peterson and T. H. Dunning, J. Chem. Phys., 2002, 117, 10548 CrossRef CAS.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 32, 756 CrossRef CAS.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89 CAS.
- B. A. Hess, Phys. Rev. A, 1986, 33, 3742 CrossRef CAS.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. S. Tz, P. Celani, W. G. Orffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. K. Oppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, D. P. ONeill, P. Palmieri, D. Peng, K. P. Uger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs, http://www.molpro.net/ Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 242 CrossRef.
- M. Gordon and M. W. Schmidt, Theory and Applications of Computational Chemistry, 2005, p. 1167 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- A. Modelli, D. Jones, G. Distefano and M. Tronc, Chem. Phys. Lett., 1991, 181, 361 CrossRef CAS.
- R. Sodhi and R. G. Cavell, J. Electron Spectrosc. Relat. Phenom., 1986, 41, 1 CrossRef CAS.
- T. X. Carroll, J. I. De, D. C. Maclaren, T. Darrah Thomas and L. J. Saethre, J. Electron Spectrosc. Relat. Phenom., 1987, 42, 281 CrossRef CAS.
- L. R. Varas, L. H. Coutinho, R. B. Bernini, A. M. Betancourt and G. G. B. de Souza, to be published.
- A. F. Lago, R. L. Cavasso-Filho, G. G. B. de Souza, A. C. F. Santos, W. C. Stolte and A. S. Schlachter, Chem. Phys. Lett., 2012, 543, 34 CrossRef CAS.
- L. R. Varas, F. C. Pontes, A. C. F. Santos and G. G. B. de Souza, Rapid Commun. Mass Spectrom., 2015, 29, 1571 CrossRef CAS PubMed.
- J. H. D. Eland, C. F. Rigby, E. Andersson, J. Palaudoux, L. Andric, F. Penent, P. Linusson, L. Hedin, L. Karlsson, J.-E. Rubensson, Y. Hikosaka, K. Ito, P. Lablanquie and R. Feifel, J. Chem. Phys., 2010, 132, 104311 CrossRef CAS PubMed.
- T. Masuoka and H. Doi, Phys. Rev. A: At., Mol., Opt. Phys., 1993, 47, 278 CrossRef CAS.
- T. Marchenko, S. Carniato, L. Journel, R. Guillemin, E. Kawerk, M. Zitnik, M. Kavcic, K. Bucar, R. Bohinc, M. Petric, V. Vas da Cruz, F. Gel'mukhanov and M. Simon, Phys. Rev. X, 2015, 5, 031021 Search PubMed.
- A. F. Lago, J. Z. Dávalos, U. Kerdpin and A. S. Schlachter, J. Phys. Chem. A, 2006, 110, 13717 CrossRef CAS PubMed.
- D. Aitchison and J. H. D. Eland, Chem. Phys., 2001, 263, 449 CrossRef CAS.
- M. Urban, G. H. F. Diercksen and M. Jurek, Mol. Phys., 1998, 94, 199 CrossRef CAS.
- W. Yu, Z. Zhu, C. Cheng and D. Shi, Can. J. Chem., 2014, 92, 1041 CrossRef CAS.
- J. R. Spencer, K. L. Jessup, M. A. McGrath, G. E. Ballester and R. Yelle, Science, 2000, 288, 1208 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |