
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecycled tyre rubber-thermoplastic composites through interface optimisation
Yonghui Zhouab and
Mizi Fan *ab
*ab
aCollege of Material Engineering, Fujian Agricultural and Forestry University, P. R. China
bDepartment of Civil Engineering, College of Engineering, Design and Physical Sciences, Brunel University London, UB8 3PH, UK. E-mail: fanfafu@gmail.com; Fax: +44(0) 1895 256392; Tel: +44(0) 7790390554
First published on 5th June 2017
Abstract
This paper presents the development of a rubber–polyethylene (PE) composite based on recycled materials with the aim of its interfacial optimisation by the use of maleated and silane coupling agents (maleated polyethylene (MAPE), bis(triethoxysilylpropyl)tetrasulfide (Si69) and vinyltrimethoxysilane (VTMS)). ATR-FTIR analysis revealed (1) the macromolecular entanglements between the grafted PE moiety in MAPE and the polymer chains of both rubber and PE in the composite, and (2) the chemical crosslinking between dissociated Si69 and rubber molecules followed by the entangling with PE polymer. These chemical interactions benefited the improvement of the constituent compatibility, rubber wettability, and interfacial adhesion of the corresponding composites, which were evident in SEM observations. The higher loss moduli, shift of glass transition peaks and inferior tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ of the treated composites indicated the segmental immobility of the macromolecules after the treatments, which was confirmed by the NMR analysis by showing comparatively broader resonance peaks. The optimised interface led to the increase of the mechanical properties of the composites including storage modulus, tensile stress and strain. VTMS treatment was not as effective as MAPE and Si69 treatments in terms of interface refinery and property strengthening.
δ of the treated composites indicated the segmental immobility of the macromolecules after the treatments, which was confirmed by the NMR analysis by showing comparatively broader resonance peaks. The optimised interface led to the increase of the mechanical properties of the composites including storage modulus, tensile stress and strain. VTMS treatment was not as effective as MAPE and Si69 treatments in terms of interface refinery and property strengthening.
1 Introduction
Used tyres are among the largest and most problematic sources of waste, due to the large volume produced and their durability. The environmental issues from the global disposal of these tyres have led to increasing interest in economic recycling of tyre rubber.1–5 The rubber in tyres is vulcanised and cannot be melted or dissolved, which makes the recycling challenging.6–9 As a result, a large number of used, worn out tyres are ground for the benefits of expanding their applications.10,11 The application related to the ground or powdered tyre rubber includes outdoor flooring and pavements, sports tracks, road construction, etc., which fall into the sectors with limited demanding and added value.Thermoplastic elastomers are a class of polymer blends or compounds which consist of materials with both thermoplastic character and elastomeric behaviour.12–15 It is widely accepted that the successful incorporation of even small amount of waste tyre rubber into thermoplastics would lead to a considerable increase of waste tyre consumption due to the massive market share of thermoplastic materials, which in the meantime transforms the waste tyre to value-added products.15,16 Nevertheless, the highly crosslinked characteristic of waste tyre rubber is not capable of entangling with the polymer molecules composing the thermoplastic matrices, leading to insufficient adhesion between two phases and poor mechanical properties of the resulted composites. To overcome this drawback, various modification strategies, such as the devulcanisation of tyre rubber, inclusion of compatibilisers, surface activations, have been attempted to improve the interfacial interaction between the tyre rubber and thermoplastic phases.17–20
Maleated olefins, such as maleated polypropylene (MAPP) or maleated polyethylene (MAPE), had been proven to improve the adhesion between tyre rubber and polyolefins due to the presence of the maleic anhydride functional group, resulting in smoother surface and better particle dispersion in a continuous matrix.21 The compatibilisation treatment between ground tyre rubber and high density polyethylene (HDPE) using peroxide suggested that the blends exhibited greater elongation at break and impact energy than uncompatibilised blend due to a co-crosslinking phenomenon which created interfacial adhesion between rubber and PE.22 Silane coupling agents were also found to be of the capability of enhancing the compatibility between recycled rubber and thermoplastics or thermosets by promoting rubber to interact with polymer matrix, which provides the compatibilisied composites with more desirable mechanical properties than the uncompatibilised counterpart.23–25 The effectiveness of these treatments was in general evaluated by the improvement in the physical and mechanical properties of the formulated composites. Nevertheless, the generation and variation of the chemical functionalities and structure during compatibilisation treatments, which undoubtedly concern the bonding scenario, microstructure and thus performance of the composites, were unfortunately not yet been thoroughly investigated.
In the present work, rubber–PE composites were developed by the use of recycled tyre rubber and HDPE aiming at improving the sustainability of the recycling process of ground tyre rubber and thermoplastics and reducing the environmental impact from waste disposal. In order to formulate a reasonable composite, three different coupling agents, i.e. maleated polyethylene (MAPE), bis(triethoxysilylpropyl)tetrasulfide (Si69) and vinyltrimethoxysilane (VTMS), were attempted to improve the compatibility, homogeneity and interfacial adhesion. The focus of this work was to reveal the chemical functionalities, structure and bonding of the formulated composites by carrying out attenuated total reflectance-Fourier Transform Infrared spectroscopy (ATR-FTIR) and solid state 13C Nuclear Magnetic Resonance spectroscopy (NMR) analyses, thus, to explore their correlation with the microstructure and bonding scenarios, and eventually the contribution to the mechanical properties of the composites.
2 Materials and methods
2.1 Materials
Recycled tyre rubber used in this work was supplied by J. Allcock & Sons Ltd (UK), with the particle size between 0.25 mm and 0.5 mm and bulk density of 0.36 kg m−3; recycled polyethylene pellet with the MFI of 0.6 g/10 min at 190 °C and bulk density of 0.96 kg m−3 was obtained from JFC Plastics Ltd (UK); lubricants Struktol TPW 709 (A unique proprietary blend of processing aids made by Struktol company) and 12-HSA (12-hydroxyoctadecanoic acid) were both purchased from Safic Alcan UK Ltd (Warrington, UK); coupling agents, MAPE (MFI of 1.9 g/10 min at 190 °C, 0.5 wt% of maleic anhydride), Si69 (>95% purity, 250 °C boiling point) and VTMS (>98% purity, 123 °C boiling point), were purchased from Sigma-Aldrich (Dorset, UK), and their chemical formulae were presented in Fig. 1. All the raw materials and additives were stored in a cool and dry place before uses.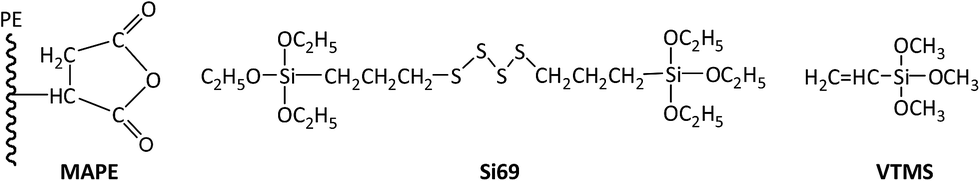 | ||
| Fig. 1 Chemical formulae of the coupling agents. | ||
2.2 Formulation of composites
The formulation of untreated and treated rubber–PE composites with specific ratio was summarised in Table 1. All the composites were carefully prepared under the same processing condition as follows: the required amount of PE for each batch was first placed in a Brabender Plastograph twin-screw mixer and allowed to melt at 100 rpm and 190 °C for 2 min, and subsequently mixed with rubber powder for 3 min. The lubricants and/or coupling agents were thus added into system and mixed for another 10 min. The resulted mixture was thus ground to pellets by using a Retsch cutting mill (SM 100, Germany). The ground blends were compression moulded on an electrically heated hydraulic press. Hot-press procedures involved 20 min preheating at 190 °C with no load applied followed by 10 min compressing at the same temperature under the pressure of 9.81 MPa, and subsequently air cooling under load until the mould reached 40 °C.| Sample | Rubber (%) | PE (%) | TPW 709 (%) | 12HSA (%) | MAPE (%) | Si69 (%) | VTMS (%) |
|---|---|---|---|---|---|---|---|
| Untreated | 50 | 43 | 3.5 | 3.5 | 0 | 0 | 0 |
| MAPE treated | 50 | 40 | 3.5 | 3.5 | 3 | 0 | 0 |
| Si69 treated | 50 | 40 | 3.5 | 3.5 | 0 | 3 | 0 |
| VTMS treated | 50 | 40 | 3.5 | 3.5 | 0 | 0 | 3 |
2.3 Solid state 13C NMR analysis
Solid state 13C NMR analysis was conducted on a Bruker spectrometer with a CP MAS probe operating at 100 MHz. The measurements were performed at ambient probe temperature with high power decoupling. Samples were packed in zirconium oxide rotors of 7 mm diameter fitted with Kel-F caps. Spectra were acquired at the spinning rate of 6 kHz, with 4096 scans per spectrum collecting in the region between −130 ppm and 270 ppm.2.4 ATR-FTIR analysis
The FTIR spectra of the composites were recorded on a PerkinElmer Spectrum one Spectrometer with the following measuring condition: 4000–650 cm−1 wave number range, 4 cm−1 resolution and 16 scans. The average of three measurements was used.2.5 Scanning electron microscope (SEM) analysis
All the composites were transversely cut by using a sliding microtome with the nominal thickness of around 25 microns for the morphological investigation of the cross sections. The observation was conducted on a Leo 1430VP SEM operating at 10 kV, all the samples were conductively plated with gold by sputtering for 45 s before imaging.2.6 Dynamic mechanical analysis (DMA)
Dynamic mechanical properties of the composites were measured by using a dynamic mechanical analyser (Q800, TA Instruments, New Castle, USA) under single cantilever strain-controlled mode. The temperature ranges from −100 °C to 120 °C with a heating rate of 3 °C min−1. The oscillation amplitude was 20 μm, the frequency was 1 Hz, and the specimen dimension was 17.5 mm × 10.8 mm × 1.4 mm.2.7 Tensile property analysis
Tensile properties of the composites were determined according to the standard BS EN ISO 527-2:2012 on an Instron 2580 testing machine with 30 kN load capacity. For each sample, the tensile property reported is the average of six measurements.3 Results and discussions
3.1 Chemical functionality and structure
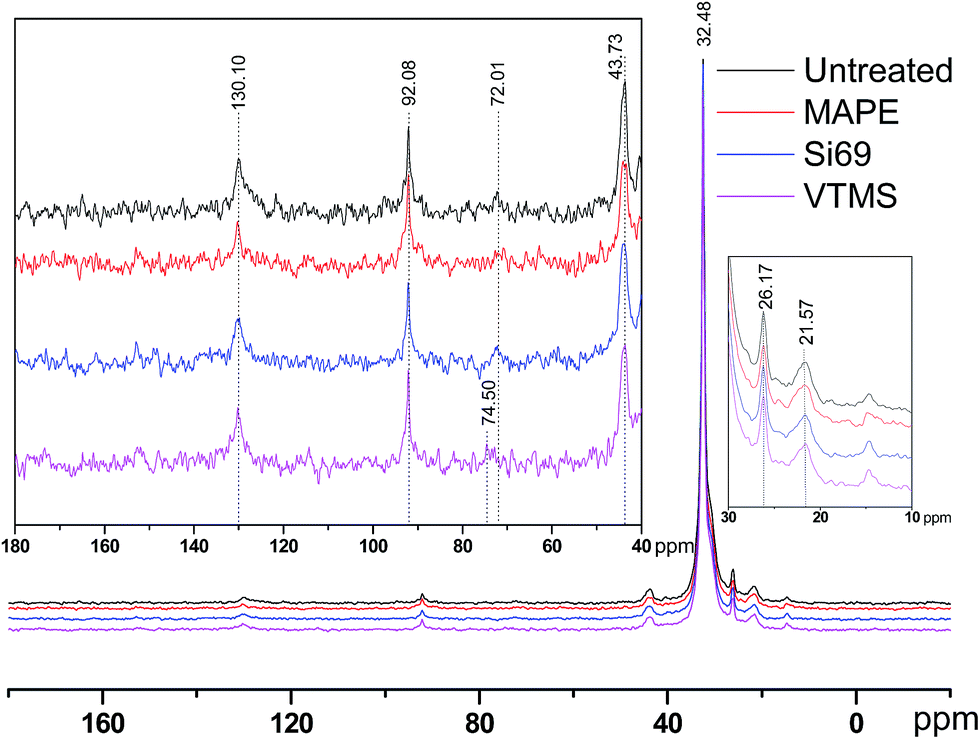 | ||
| Fig. 2 13C NMR spectra of untreated, MAPE, Si69 and VTMS treated rubber–PE composites. | ||
 | ||
| Fig. 3 Chemical structure of SBR unit and NR unit. | ||
It was noticed that apart from the chemical shifts of rubber and PE, there was an additional peak presented at 74.50 ppm in the spectra of VTMS treated composite, which might be resulted from the oxidation of C–C bonds in rubber molecules under high temperature and pressure with the incorporation of VTMS. The general region of carbons with a single bond to oxygen ranged from 73 ppm to 83 ppm.32,33 Epoxides were often anticipated to be the products of rubber oxidation, while the chemical shifts of which were rather around 55 ppm. Chemical shifts of secondary alcohols and in particular ethers could reach 73 ppm depending on the processing environment.33 Peroxides and hydroperoxides might be the most downfield signals in the region between 72 and 75 ppm.32,34
It can be seen that the peaks at 92.08 ppm and 43.73 ppm in the spectra of treated composites were slightly broader than those in the spectrum of the untreated counterpart. This was primarily due to the fact that the resonance line width was correlated to the segmental polymer motion. If the polymer molecules were free to move in any direction, then the spectral line would be narrow, and vice versa.34 In terms of the treatment processing, this might indicate the addition of coupling agents into the composites resulted in more solid-like polymers with restricted molecular mobility. With respect to the potential crosslinking reactions between the coupling agents and raw materials owing to the treatments, NMR determination results did not unambiguously suggest the formulation of relevant chemical bonds. Specifically, C–S bonds and C–O–Si bonds were expected to be observed in the region of 45–55 ppm and 55–65 ppm respectively.35–37 In this regard, the lack of these bands in the corresponding NMR spectra might be explained by a number of reasons, including: (1) insufficient concentration of these bonds to be detected; (2) T1 relaxation times of these units being much longer in comparison to the relaxation times of other molecular units in the polymers; and (3) T2 relaxation times of these units being short enough to cause the signals to be broadened beyond detection.38 Further scrutinising of the potential crosslinking between the coupling agents and constituents of the composites by other analytical technique would be of great significance for better understanding the impact of the treatments on the variations of chemical functionalities and structure of the composites, which was carried out in the next section.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC stretching vibrations of maleic anhydride moiety.39–41 With respect to the spectral variation of rubber constituent, the band at 1062 cm−1 referring to the C–S–C stretching in the C–S bonds was shifted to 1095 cm−1 and became sharper after the treatment.42,43 This might be an indication of the macromolecular entanglements between the rubber polymer chain and the grated polyethylene in MAPE.
O and CC stretching vibrations of maleic anhydride moiety.39–41 With respect to the spectral variation of rubber constituent, the band at 1062 cm−1 referring to the C–S–C stretching in the C–S bonds was shifted to 1095 cm−1 and became sharper after the treatment.42,43 This might be an indication of the macromolecular entanglements between the rubber polymer chain and the grated polyethylene in MAPE.
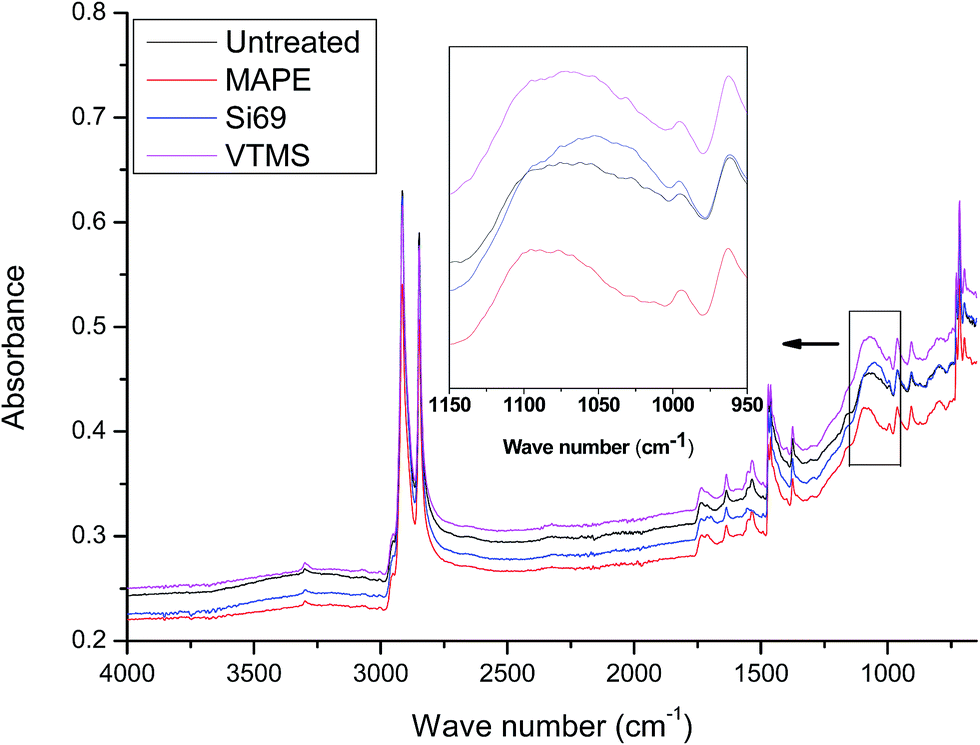 | ||
| Fig. 4 FTIR spectra of untreated, MAPE, Si69 and VTMS treated rubber–PE composites. | ||
Si69 has been proven to be an effective coupling agent for improving the interfacial and overall property of rubber based composites, such as lignocellulosic fibre–rubber, geopolymer–rubber and carbon black-rubber composites.44–49 The effect of the silane treatment on the chemical structure of the composite could be clearly seen from the FTIR result. The Si69 treated composite demonstrated a much more intense band at 1053 cm−1 attributing to C–S–C stretching vibration, while the counterpart from the untreated was observed at 1062 cm−1.42,43,50. The shift of wave number and increase of intensity may be attributed to the introduced Si–O–C bonds in Si69 and the C–S linkages formed between the silane and rubber polymers which may be proposed in Fig. 5, namely the coupling agent Si69 with a sulfidic linkage between triethoxysilylpropyl groups was dissociated to form radicals under high temperature and pressure, thus the sulfide groups crosslinked with both SBR and NR macromolecules in rubber.46 The crosslinking with silane of rubber may trigger its further entanglement and/or chemical coupling with PE matrix owing to the activated surface,15 which automatically reduced the chance to form sulfur-crosslinking within the sulfur-rich rubber molecules and simultaneously prevented the accumulation of rubber particles in the matrix to some extent.46,48 Thongsang et al.48 examined the effect of Si69 treatment on the properties of fly ash/NR composite, it was pointed out that with high loading of Si69 (4–8 wt%), the bulky triethoxysilylpropyl groups in Si69 might cause the steric hindrance to the linkage formation between the rubber molecules and the fly ash particles. In addition, a self-condensation of Si69 could occur at high Si69 contents, resulting in the formation of mono- and poly-layers of polysiloxane molecules on the surface of fly ash.
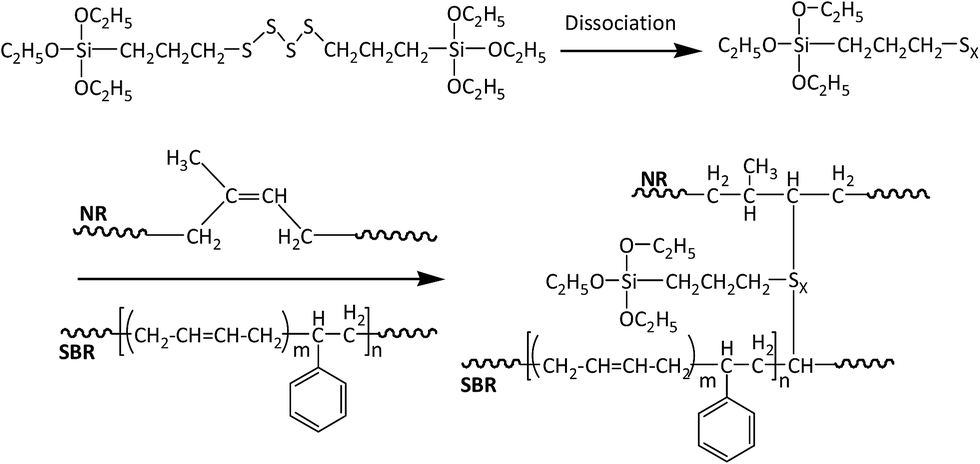 | ||
| Fig. 5 Proposed chemical reaction between Si69 and rubber polymers. | ||
It was worth noting that the diagnostic characteristic of Si69, i.e. Si–O–C stretching at approximately 1100 cm−1 and 1072 cm−1, was not found in the spectrum, which should have overlapped with the band of C–S–C stretching after being incorporated into the composite.51,52 These spectral characteristics unveiling the chemical interaction between the coupling agent and raw materials were unfortunately not detected in the above NMR analysis (Section 3.1.1) probably due to insufficient concentration or inappropriate relaxation time of the corresponding bonds (C–O and C–O–Si).
VTMS was another coupling agent applied for refining the interface of the composite. The FTIR spectra of the untreated and VTMS treated composites did not show considerable difference in terms of the band appearance and intensities, especially the bands corresponding to C–O–Si and C–S–C bonds (1020–1100 cm−1), which might be an indication of fairly limited crosslinking or entangling occurred between the coupling agent and raw materials. This result was consistent with its NMR analysis of this treatment.
3.2 Interface structure and bonding
The effect of the incorporation of the coupling agents on the interface structure and bonding scenario of rubber–PE composites was scrutinised by SEM, with results presenting in Fig. 6. Clear cracks and boundaries can be observed between the components of the untreated composite (Fig. 6a), suggesting the poor compatibility between the untreated raw materials. The incompatibility seemed not only to prevent the close interaction between rubber and PE, but also to impede the hydrodynamic flow of the polymer resin, which gave rise to the formation of a number of voids within the matrix as shown in Fig. 6a. These phenomena evidently indicated the inappropriate interfacial contact and adhesion of the untreated composite. The SEM image of MAPE treated composite (Fig. 6b) demonstrated an improvement of constituent compatibility and wettability of the rubber by the resin after the treatment, by showing a greater embedment of the rubber particles in the matrix with subtle cracks and voids. The scenario in the Si69 treated composite is completely different from the others, displaying well embedded rubber particles in the matrix along with firmly bonded interface (Fig. 6c). Moreover, the phase structure of the matrix was smoother and more compact than that of the other composites. These observations should be related to the enhanced chemical compatibility and interdiffusion through the intermolecular interactions between the MAPE and Si69 coupling agents and rubber and PE molecules, i.e. the macromolecular entanglements between the grafted PE moiety in MAPE and the polymer chains of both rubber and PE in the composite, and the chemical crosslinking between dissociated Si69 and rubber molecules followed by the entangling with PE polymer (Section 3.1.2). These chemical interactions gave rise to the creation of entangled and/or crosslinked rubber–PE network accompanied by the increase of rubber wettability by matrix, constituent compatibility and interfacial adhesion of the corresponding composites. It was presumed that the enhancement in the adhesion and bonding at the MAPE and Si69 treated rubber–PE interfaces would benefit the performance of the composites given the interface was recognised to play a predominant role in governing the global composite behaviour by controlling the stress transfer between the constituents of a composite. In comparison to the untreated, the VTMS treated composite did not demonstrate significantly distinct interface structure and bonding scenario (Fig. 6d), suggesting the inferior interface refinery by VTMS treatment. This was in a good agreement with the previously discerned comparatively limited chemical interactions between this coupling agent and the constituents of the composite (Section 3.1). | ||
| Fig. 6 Microstructures of cross section of untreated (a), MAPE treated (b), Si69 treated (c) and VTMS treated (d) composites. | ||
3.3 Mechanical properties
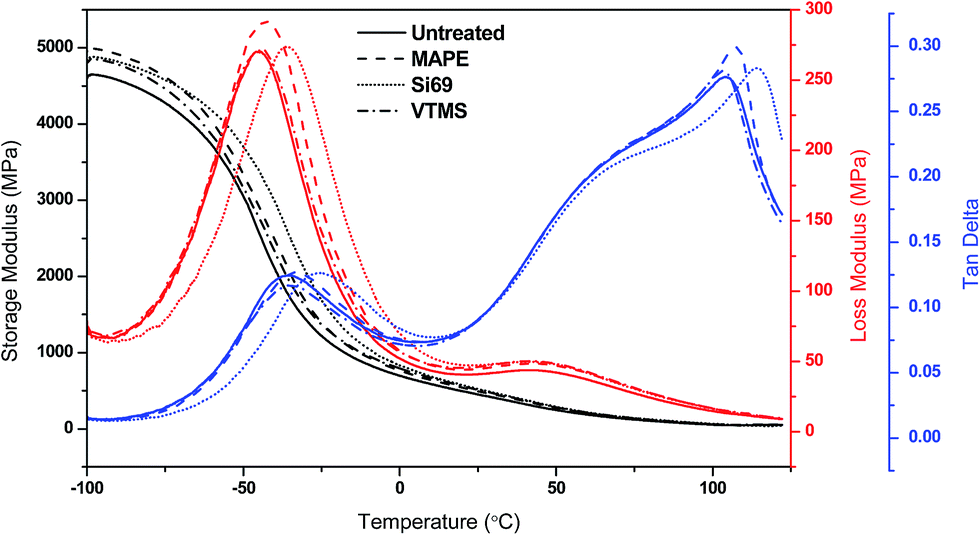 | ||
| Fig. 7 Storage modulus, loss modulus and tanδ of untreated and treated composites as a function of temperature. | ||
The variation of loss modulus as a function of temperature was also presented in Fig. 7 for the purpose of exploring the transition behaviour of the composites. Both the untreated and treated composites demonstrated two relaxation peaks in their curves, i.e. the peaks at around 45 °C were associated with the α transition of PE matrix, concerning the chain segment mobility in the crystalline phase due to the reorientation of defect area in the crystals,55 while the peaks at −45 °C to −35 °C were resulted from the molecular motion of rubber phase corresponding to its glass transition.56 It was observed that the treated composites especially the MAPE and Si69 treated possessed higher loss moduli than the untreated one, and their glass transition peaks had shifted towards higher temperature regions. These behaviours were associated with the generation of constraints on the segmental mobility of macromolecules at the relaxation temperatures due to the strengthened interfacial interaction and adhesion of the composites after the MAPE and Si69 treatments, which on the other hand accounted for the comparatively broader NMR resonance peaks as observed in Section 3.1.1.57,58 The larger the interface area and the stronger the interfacial interaction, the greater the molecular motion were restricted.59
The ratio of loss modulus to storage modulus tanδ was measured to further understand the damping behaviour and interface property of the composites (Fig. 7). In the glassy plateau, the MAPE and Si69 treated composites showed inferior tanδ amplitude than the untreated composite, and their glass transition temperatures (Tg) were determined to be 3.1 °C and 10.2 °C higher than that of the untreated (−36.0 °C) respectively. These findings substantiated the aforementioned enhanced interface bonding and immobility of molecular chains of the treated composites. When the composites were subjected to external stress, the external energy was dissipated by the friction between particle–particle and particle–matrix interaction through the interface.59 Therefore, the composites with comparatively poorer interface bonding (untreated and VTMS treated) were inclined to dissipate more energy due to the existence of particle–particle friction in weak agglomerates where particles touched each other and the particle-polymer friction at the interface where there was essentially no adhesion, leading to higher magnitude of the corresponding damping peaks.57,60,61 In addition, it was noted that the melting points of MAPE and Si69 treated composites were shifted to higher temperatures, namely 107.6 °C and 114.7 °C respectively, which should be ascribed to the crosslinking occurred between the coupling agents and the polymer molecules.
| Sample | Tensile stress at maximum load (MPa) | Tensile strain at maximum load (%) | Tensile modulus (MPa) |
|---|---|---|---|
| Untreated | 4.7 ± 0.1 | 7.7 ± 0.4 | 422.3 ± 17.4 |
| MAPE treated | 4.9 ± 0.3 | 11.3 ± 0.5 | 411.8 ± 20.4 |
| Si69 treated | 6.3 ± 0.2 | 13.1 ± 0.7 | 422.1 ± 22.1 |
| VTMS treated | 4.5 ± 0.3 | 8.3 ± 0.6 | 403.5 ± 18.6 |
4 Conclusions
The influence of the application of MAPE, Si69 and VTMS coupling agents on the chemical structure, microstructure and mechanical property of rubber–PE composites has been comprehensively investigated. FTIR results revealed the molecular entanglement and crosslinking between MAPE and Si69 coupling agents and the constituents of the composites, giving rise to the enhancement of chemical compatibility and interdiffusion, and thus the interfacial adhesion of the composites. SEM observations substantiated the improvement of the constituent compatibility, rubber wettability and embedment, and interfacial bonding after MAPE and Si69 treatments. VTMS treatment was not as effective as MAPE and Si69 treatments by showing comparatively limited crosslinking with the constituents and poorer interface within the composite. NMR analysis suggested the constraints on the segmental mobility of the polymers resulting from the treatments, which contributed to the shift of glass transition peaks and inferior tanδ amplitude as explored in DMA study. The mechanical properties including storage modulus, tensile strength and tensile strain of the composites were increased due to the better interfacial compatibility and adhesion as well as more efficient stress transfer from the matrix to rubber particles after the treatments.
Acknowledgements
The authors gratefully acknowledge the financial support from the European CIP-EIP-Eco-innovation-2012 (Project Number: 333083).References
- V. Torretta, E. C. Rada, M. Ragazzi, E. Trulli, I. A. Istrate and L. I. Cioca, Waste Manag., 2015, 45, 152–160, DOI:10.1016/j.wasman.2015.04.018.
- M. C. Zanetti, S. Fiore, B. Ruffino, E. Santagata, D. Dalmazzo and M. Lanotte, Waste Manag., 2015, 45, 161–170, DOI:10.1016/j.wasman.2015.05.003.
- A. K. Naskar, Z. Bi, Y. Li, S. K. Akato, D. Saha, M. Chi, C. A. Bridges and M. P. Paranthaman, RSC Adv., 2014, 4, 38213–38221, 10.1039/c4ra03888f.
- M. Echeverria, C. M. Abreu and C. A. Echeverria, RSC Adv., 2015, 5, 76057–76064, 10.1039/c5ra06453h.
- E. Lievana and J. Karger-Kocsis, Prog. Rubber, Plast. Recycl. Technol., 2004, 20, 1–10 CAS.
- I. Fuhrmann and J. Karger-Kocsis, Plast., Rubber Compos., 1999, 28, 500–504, DOI:10.1179/146580199101540088.
- R. Sonnier, E. Leroy, L. Clerc, A. Bergeret and J. M. Lopez-Cuesta, Polym. Degrad. Stab., 2006, 91, 2375–2379, DOI:10.1016/j.polymdegradstab.2006.04.001.
- D. De, A. Das, D. De, B. Dey, S. C. Debnath and B. C. Roy, Eur. Polym. J., 2006, 42, 917–927, DOI:10.1016/j.eurpolymj.2005.10.003.
- M. Sienkiewicz, J. Kucinska-Lipka, H. Janik and A. Balas, Waste Manag., 2012, 32, 1742–1751, DOI:10.1016/j.wasman.2012.05.010.
- C. R. Kumar, I. Fuhrmann and J. Karger-Kocsis, Polym. Degrad. Stab., 2002, 76, 137–144, DOI:10.1016/S0141-3910(02)00007-1.
- Z. Wang, Y. Zhang, F. Du and X. Wang, Mater. Chem. Phys., 2012, 136, 1124–1129, DOI:10.1016/j.matchemphys.2012.08.063.
- M. Mondal, U. Gohs, U. Wagenknecht and G. Heinrich, Mater. Chem. Phys., 2013, 143, 360–366, DOI:10.1016/j.matchemphys.2013.09.010.
- Z. Wang, X. Cheng and J. Zhao, Mater. Chem. Phys., 2011, 126, 272–277, DOI:10.1016/j.matchemphys.2010.11.027.
- H. Kang, X. Hu, M. Li, L. Zhang, Y. Wu, N. Ning and M. Tian, RSC Adv., 2015, 5, 23498–23507, 10.1039/c4ra17024e.
- J. Karger-Kocsis, L. Meszaros and T. Barany, J. Mater. Sci., 2013, 48, 1–36, DOI:10.1007/s10853-012-6564-2.
- A. Ramezani Kakroodi and D. Rodrigue, Polym. Degrad. Stab., 2013, 98, 2184–2192, DOI:10.1016/j.polymdegradstab.2013.08.017.
- L. Mészáros, T. Bárány and T. Czvikovszky, Radiat. Phys. Chem., 2012, 81, 1357–1360, DOI:10.1016/j.radphyschem.2011.11.058.
- L. Mészáros, M. Fejos and T. Bárány, J. Appl. Polym. Sci., 2012, 125, 512–519, DOI:10.1002/app.35675.
- P. S. Garcia, F. D. B. de Sousa, J. A. de Lima, S. A. Cruz and C. H. Scuracchio, eXPRESS Polym. Lett., 2015, 9, 1015–1026, DOI:10.3144/expresspolymlett.2015.91.
- S. Seghar, N. Ait Hocine, V. Mittal, S. Azem, F. Al-Zohbi, B. Schmaltz and N. Poirot, eXPRESS Polym. Lett, 2015, 9, 1076–1086, DOI:10.3144/expresspolymlett.2015.97.
- S. H. Lee, M. Balasubramanian and J. K. Kim, J. Appl. Polym. Sci., 2007, 106, 3209–3219, DOI:10.1002/app.26490.
- R. Sonnier, E. Leroy, L. Clerc, A. Bergeret, J. Lopez-Cuesta, A. Bretelle and P. Ienny, Polym. Test., 2008, 27, 901–907, DOI:10.1016/j.polymertesting.2008.07.003.
- C. Kaynak, E. Sipahi-Saglam and G. Akovali, Polymer, 2001, 42, 4393–4399, DOI:10.1016/S0032-3861(00)00734-5.
- C. Kaynak, C. Celikbilek and G. Akovali, Eur. Polym. J., 2003, 39, 1125–1132, DOI:10.1016/S0014-3057(02)00381-6.
- X. Colom, J. Cañavate, F. Carrillo, J. I. Velasco, P. Pagès, R. Mujal and F. Nogués, Eur. Polym. J., 2006, 42, 2369–2378, DOI:10.1016/j.eurpolymj.2006.06.005.
- N. Sombatsompop, K. Sungsanit and C. Thongpin, J. Appl. Polym. Sci., 2004, 92, 3167–3172, DOI:10.1002/app.20286.
- S. Renneckar, R. K. Johnson, A. Zink-Sharp, N. Sun and W. G. Glasser, Compos. Interfaces, 2005, 12, 559–580, DOI:10.1163/1568554054915219.
- T. M. Arantes, K. V. Leão, M. I. B. Tavares, A. G. Ferreira, E. Longo and E. R. Camargo, Polym. Test., 2009, 28, 490–494, DOI:10.1016/j.polymertesting.2009.03.011.
- J. Y. Buzaré, G. Silly, J. Emery, G. Boccaccio and E. Rouault, Eur. Polym. J., 2001, 37, 85–91, DOI:10.1016/S0014-3057(00)00081-1.
- N. M. Ricardo, M. Lahtinen, C. Price and F. Heatley, Polym. Int., 2002, 51, 627–634, DOI:10.1002/pi.936.
- J. T. Sakdapipanich, T. Kowitteerawut, S. Tuampoemsab and S. Kawahara, J. Appl. Polym. Sci., 2006, 100, 1875–1880, DOI:10.1002/app.23335.
- A. V. Aganov and V. L. Antonovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1982, 31, 247–250, DOI:10.1007/bf00948235.
- C. Kehlet, A. Catalano and J. Dittmer, Polym. Degrad. Stab., 2014, 107, 270–276, DOI:10.1016/j.polymdegradstab.2013.12.039.
- A. E. Somers, T. J. Bastow, M. I. Burgar, M. Forsyth and A. J. Hill, Polym. Degrad. Stab., 2000, 70, 31–37, DOI:10.1016/S0141-3910(00)00076-8.
- C. A. Fyfe and J. Niu, Macromolecules, 1995, 28, 3894–3897 CrossRef CAS.
- M. Brochier Salon, M. Abdelmouleh, S. Boufi, M. N. Belgacem and A. Gandini, J. Colloid Interface Sci., 2005, 289, 249–261, DOI:10.1016/j.jcis.2005.03.070.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle and D. L. Bryce, Spectrometric Identification of Organic Compounds, Petra Recter, USA, 2014 Search PubMed.
- M. Sitarz, C. Czosnek, P. Jeleń, M. Odziomek, Z. Olejniczak, M. Kozanecki and J. F. Janik, Spectrochim. Acta, Part A, 2013, 112, 440–445, DOI:10.1016/j.saa.2013.05.007.
- G. Martíínez-Barrera, H. López, V. M. Castaño and R. Rodrıíguez, Radiat. Phys. Chem., 2004, 69, 155–162, DOI:10.1016/S0969-806X(03)00452-3.
- J. Z. Lu, I. I. Negulescu and Q. Wu, Compos. Interfaces, 2005, 12, 125–140, DOI:10.1163/1568554053542133.
- J. Liu, C. Scott, S. Winroth, J. Maia and H. Ishida, RSC Adv., 2015, 5, 16785–16791, 10.1039/c4ra12086h.
- M. Akiba and A. S. Hashim, Prog. Polym. Sci., 1997, 22, 475–521, DOI:10.1016/S0079-6700(96)00015-9.
- A. K. Rai, R. Singh, K. N. Singh and V. B. Singh, Spectrochim. Acta, Part A, 2006, 63, 483–490, DOI:10.1016/j.saa.2005.05.034.
- H. Ismail, S. Shuhelmy and M. R. Edyham, Eur. Polym. J., 2002, 38, 39–47, DOI:10.1016/S0014-3057(01)00113-6.
- H. Ismail, M. N. Nasaruddin and U. S. Ishiaku, Polym. Test., 1999, 18, 287–298, DOI:10.1016/S0142-9418(98)00030-0).
- S. Choi, Polym. Test., 2002, 21, 201–208, DOI:10.1016/S0142-9418(01)00071-X.
- N. Z. Noriman and H. Ismail, J. Appl. Polym. Sci., 2012, 124, 19–27, DOI:10.1002/app.34961.
- S. Thongsang and N. Sombatsompop, Polym. Compos., 2006, 27, 30–40, DOI:10.1002/pc.20163.
- D. Salimi, S. N. Khorasani, M. R. Abadchi and S. J. Veshare, Adv. Polym. Technol., 2009, 28, 224–232, DOI:10.1002/adv.20169.
- S. Gunasekaran, R. K. Natarajan and A. Kala, Spectrochim. Acta, Part A, 2007, 68, 323–330, DOI:10.1016/j.saa.2006.11.039.
- M. Abdelmouleh, S. Boufi, M. N. Belgacem and A. Dufresne, Compos. Sci. Technol., 2007, 67, 1627–1639, DOI:10.1016/j.compscitech.2006.07.003.
- M. Bengtsson and K. Oksman, Composites, Part A, 2006, 37, 752–765, DOI:10.1016/j.compositesa.2005.06.014.
- S. Mohanty, S. K. Verma and S. K. Nayak, Compos. Sci. Technol., 2006, 66, 538–547, DOI:10.1016/j.compscitech.2005.06.014.
- S. Mohanty and S. K. Nayak, J. Appl. Polym. Sci., 2006, 102, 3306–3315, DOI:10.1002/app.24799.
- M. Bengtsson, P. Gatenholm and K. Oksman, Compos. Sci. Technol., 2005, 65, 1468–1479, DOI:10.1016/j.compscitech.2004.12.050.
- M. K. A. Wahaba, H. Ismaila and N. Othmana, Polym.-Plast. Technol. Eng., 2012, 51, 298–303 CrossRef.
- R. Ou, Y. Xie, M. P. Wolcott, S. Sui and Q. Wang, Mater. Des., 2014, 58, 339–345, DOI:10.1016/j.matdes.2014.02.018.
- M. A. López-Manchado, J. Biagitti and J. M. Kenny, Polym. Compos., 2002, 23, 779–789, DOI:10.1002/pc.10476.
- H. Noor Azlinaa, H. A. Sahrima and R. Rozaidia, Polym.-Plast. Technol. Eng., 2011, 50, 1383–1387 CrossRef.
- J. M. Felix and P. Gatenholm, J. Appl. Polym. Sci., 1991, 42, 609–620, DOI:10.1002/app.1991.070420307.
- M. Ashida, T. Noguchi and S. Mashimo, J. Appl. Polym. Sci., 1984, 29, 661–670, DOI:10.1002/app.1984.070290222.
| This journal is © The Royal Society of Chemistry 2017 |