
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(±)-Applanatumines B–D: novel dimeric meroterpenoids from Ganoderma applanatum as inhibitors of JAK3†
Qi Luoab,
Zhen Wangc,
Jin-Feng Luoc,
Zheng-Chao Tu*c and
Yong-Xian Cheng *ad
*ad
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Kunming 650204, People's Republic of China. E-mail: yxcheng@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Yuquan Road 19, Beijing 100049, People's Republic of China
cDrug Discovery Pipeline, Guangdong Provincial Key Laboratory of Biocomputing, Guangzhou Institutes of Biomedicine and Health, Guangzhou 510530, People's Republic of China. E-mail: tu_zhengchao@gibh.ac.cn
dSchool of Pharmaceutical Sciences, Shenzhen University Health Sciences Center, Shenzhen 518060, People's Republic of China
First published on 2nd August 2017
Abstract
Applanatumines B–D (1–3), three pairs of dimeric meroterpenoid enantiomers featuring the presence of a 6-oxo-4,4a,5,5a,6,8,8a,8b-octahydrofuro[3′,4′:4,5]cyclopenta[1,2-b]pyran-3-carbaldehyde structure core, were isolated from the fruiting bodies of Ganoderma applanatum. Their structures and absolute configurations were assigned by using spectroscopic methods and ECD calculations. Biological evaluation found that all the compounds are JAK3 inhibitors. In addition, the enantiomers of 1 are active towards DDR1 with IC50 values of 8.2 ± 0.8 μM and 6.9 ± 0.8 μM. Finally, a plausible biogenic pathway for compounds 1–3 was proposed.
1. Introduction
Ganoderma (Ganodermataceae) are mainly spread over tropical and subtropical regions.1 Of hundreds of fungal species in this genus, many are of great medicinal values and are even called cure-all medicine, implying their wide range of therapeutic applications.2 Despite the medicinal importance of Ganoderma, chemical investigation on this genus has mainly focused on triterpenoids and polysaccharides,3,4 which could only in part unveil the scientific connotation of Ganoderma-based traditional medicine; the chemical and biological profiles of Ganoderma fungi need further exploration. Oxidative stress and inflammation are implicated in multiple diseases; with this context, we have characterized a series of structurally novel meroterpenoids from Ganoderma with antioxidant or anti-inflammatory properties; a subsequent investigation revealed that they are either Nrf2 activators or TGF-β/Smad3 inhibitors,5–7 showing that meroterpenoids in Gnoderma are promising structure templates. Indeed, lingzhiols have attracted great attention since they were reported in 2013.8–14Janus kinase 3 (JAK3), discoidin domain receptor 1 (DDR1), histone deacetylase 1 (HDAC1) and bromodomain-containing protein 4 (BRD4) have emerged as promising drug targets for a number of diseases such as cancer, fibrosis, and rheumatoid arthritis.15–17 In consideration of wide applications of Ganoderma species, it is therefore necessary to examine whether substances in this genus possess inhibitory activities towards the above established targets. With this motivation, we have conducted a chemical investigation focusing meroterpenoids in Ganoderma applanatum and reported structurally novel applanatumine A.6,7,18 In a follow up study on this fungus, applanatumines B–D (1–3) (Fig. 1), three pairs of novel dimeric meroterpenoid enantiomers, were characterized and their biological activities against JAK3, DDR1, HDAC1, and BRD4 were evaluated.
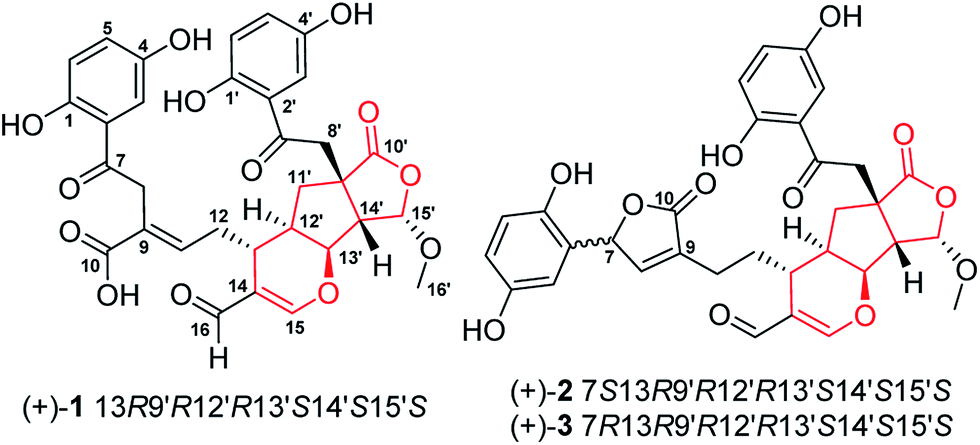 | ||
| Fig. 1 The structures of novel dimeric meroterpenoids 1–3. | ||
2. Results and discussion
2.1. Structure elucidation
The EtOH extract of G. applanatum was suspended in H2O and partitioned with EtOAc. A combination of chromatography of an EtOAc soluble fraction afforded 3 racemic meroterpenoid dimers.(±)-Compound 1 was obtained as yellow gum with the molecular formula of C32H30O13 determined by analysis of its HRESIMS (m/z 621.1605 [M − H]−, calcd for C32H29O13, 621.1614), 13C NMR and DEPT data, indicating 18 degrees of unsaturation. The 1H NMR spectrum (Table 1) contains two typical ABX proton coupling systems [δH 7.36 (1H, d, J = 2.8 Hz, H-3), 7.01 (1H, dd, J = 8.8, 2.8 Hz, H-5) and 6.79 (1H, d, J = 8.8 Hz, H-6)] and [δH 7.18 (1H, d, J = 2.8 Hz, H-3′), 7.01 (1H, dd, J = 8.8, 2.8 Hz, H-5′) and 6.81 (1H, d, J = 8.8 Hz, H-6′)], suggesting the presence of two 1,2,4-trisubstituted benzene substructures.
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a Overlapped signals by measured solvent.b Signals were assigned by HMBC experiment. | ||||||
| 1 | 156.4 | 149.0 | 149.1 | |||
| 2 | 120.8 | 123.4 | 123.3 | |||
| 3 | 7.36 d (2.8) | 115.8 | 6.49 d (2.6) | 113.4 | 6.50 d (2.8) | 113.6 |
| 4 | 150.7 | 151.5 | 151.4 | |||
| 5 | 7.01 dd (8.8, 2.8) | 125.7 | 6.61 dd (8.6, 2.6) | 117.3 | 6.62 dd (8.6, 2.8) | 117.3 |
| 6 | 6.79 d (8.8) | 119.6 | 6.68 d (8.6) | 117.3 | 6.69 d (8.6) | 117.4 |
| 7 | 204.7 | 6.26 br s | 79.9 | 6.26 br s | 80.0 | |
| 8 | 4.05 s | 37.3b | 7.47 br s | 151.1 | 7.47 br s | 151.0 |
| 9 | 128.4b | 133.2 | 133.3 | |||
| 10 | 172.4b | 176.6 | 176.7 | |||
| 11 | 7.03 t (4.0) | 142.2b | 2.37 m | 23.6 | 2.38 m | 23.5 |
| 12 | Ha 2.53 m | 35.0 | Ha 1.86 m | 34.5 | Ha 1.86 m | 34.8 |
| Hb 2.30 m | Hb 1.64 m | Hb 1.63 m | ||||
| 13 | 2.65 dd (8.5, 4.7) | 30.0 | 2.52 dd (8.5, 4.7) | 29.3 | 2.53 dd (8.5, 4.7) | 29.2 |
| 14 | 120.5 | 121.4 | 121.3 | |||
| 15 | 7.51 s | 166.5 | 7.49 s | 166.3 | 7.48 s | 166.4 |
| 16 | 9.22 s | 193.3 | 9.22 s | 193.4 | 9.23 s | 193.4 |
| 1′ | 156.4 | 156.4 | 156.4 | |||
| 2′ | 120.2 | 120.1 | 120.1 | |||
| 3′ | 7.18 d (2.8) | 115.3 | 7.18 d (2.6) | 115.3 | 7.18 d (2.6) | 115.3 |
| 4′ | 150.9 | 150.8 | 150.8 | |||
| 5′ | 7.01 dd (8.8, 2.8) | 126.5 | 7.03 dd (8.8, 2.6) | 126.5 | 7.02 dd (8.8, 2.6) | 126.5 |
| 6′ | 6.81 d (8.8) | 119.9 | 6.80 d (8.8) | 119.9 | 6.80 d (8.8) | 119.9 |
| 7′ | 204.7 | 204.7 | 204.7 | |||
| 8′ | Ha 3.79 d (19.0) | 48.3 | Ha 3.78 d (19.0) | 48.5 | Ha 3.79 d (19.0) | 48.5 |
| Hb 3.43 d (19.0) | Hb 3.42 d (19.0) | Hb 3.42 d (19.0) | ||||
| 9′ | 53.0 | 53.0 | 53.0 | |||
| 10′ | 181.8 | 181.9 | 181.9 | |||
| 11′ | Ha 2.24 dd (13.5, 7.2) | 41.3 | Ha 2.20 dd (13.4, 7.3) | 41.0 | Ha 2.22 dd (13.4, 7.3) | 41.0 |
| Hb 1.63 t-like (13.5) | Hb 1.61 t (13.4) | Hb 1.61 t (13.4) | ||||
| 12′ | 2.37 m | 40.1 | 2.46 m | 39.9 | 2.46 m | 39.9 |
| 13′ | 4.85a | 81.7 | 4.85a | 81.6 | 4.85a | 81.6 |
| 14′ | 3.21 d (7.6) | 55.1 | 3.21 d (7.5) | 54.9 | 3.21 d (7.6) | 54.9 |
| 15′ | 5.94 d (7.6) | 105.7 | 5.94 d (7.5) | 105.6 | 5.94 d (7.6) | 105.6 |
| 16′ | 3.63 s | 59.0 | 3.64 s | 58.8 | 3.64 s | 58.9 |
The 13C NMR and DEPT spectra of 1 (Table 1) contain 32 resonances attributable to a methyl (oxygenated), four methylenes, fourteen methines (eight olefinics, an aldehyde group), thirteen quaternary carbons (eight sp2, two ketones, two carboxylic carbonyls, and an aliphatic carbon). The partial signals occur as pairs, suggesting that 1 might be a meroterpenoid dimer consisting of parts A (black drawing) and B (blue drawing) (Fig. 2). The ABX coupling pattern, the chemical shifts of C-1 (δC 156.4) and C-4 (δC 150.7), and HMBC correlations of H-3/C-7, H-11/C-8 (δC 37.3), and ROESY correlation of H-3/H-8 indicate the substructure of C-1–C-10. The HMBC correlations of H-15 (δH 7.51)/C-13, C-14, C-16 (δC 193.3), C-13′ (δC 81.7); H-13/C-13′; and H-16 (δH 9.22)/C-13, C-14, C-15 (δC 166.5), in combination with a 1H–1H COSY correlation of H-13/H-12′ (Fig. 2) indicate the existence of ring A with an aldehyde group attached to C-14. Further, the HMBC correlations of H-11/C-8, C-10, and COSY correlation of H-12/H-13 were observed, which reveals the presence of part A as shown. In addition to the signals for part A, the residual signals resemble those of lingzhilactone C, a meroterpenoid previously isolated from G. lingzhi.5 This conclusion gains supports from 2D NMR data, which show important interactions such as HMBC correlations of H-13′/C-9′, C-11′; H-14′/C-9′, C-10′, C-12′, C-13′, C-15′; H-15′/C-10′ and COSY correlations of H-9′/H-11′/H-12′ and H-14′/H-15′, indicating the ring systems in part B. Finally, parts A and B are connected via C-12′–C-13′ supported by the observed 1H–1H COSY correlations of H-13/H-12′/H-11′ as well as above mentioned H-15/C-13′. As a result, the planar structure of 1 was determined.
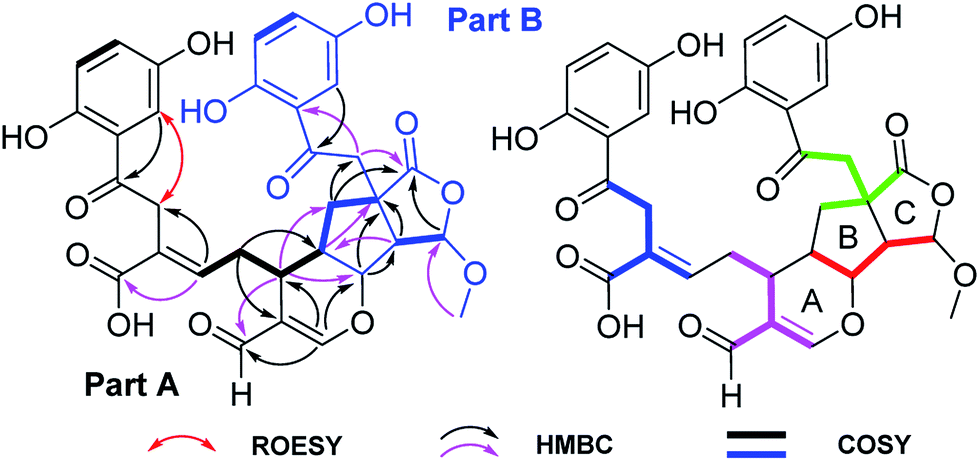 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of 1, and ROESY correlation of H-3/H-8; blue, pink, red, and green in 1 represent four independent isoprenyl moieties. | ||
The relative configuration of 1 was assigned by a ROESY spectrum (Fig. 3), which shows correlations of H-12/H-12′, H-13′, indicating the relative configuration of ring A. The ROESY correlation of H-8′/H-14′ suggests that these two protons are at the same side of ring B. Further, the observed ROESY correlations between H-13/Ha-11′, Hb-11′ and H-8′, H-14′/Hb-11′ indicate the stereo relationship between rings A and B. As for the relative configuration at C-15′, the observed ROESY correlation of 15′-OMe/H-12′ and weak correlation H-15′/Ha-8′ evidently suggests that the orientation of H-15′. Therefore, the relative configurations at chiral centers of rings B and C were determined. Besides, there is one double bond Δ9(11) at the side chain of part A, whose geometry was assigned as E-form based on the observed ROESY correlation of H-8/Hb-12.
 | ||
| Fig. 3 Key ROESY correlations of 1 and 2. | ||
(±)-1 was isolated as a racemate (Fig. S25, ESI†), which further was separated by chiral-phase HPLC. In order to determine the absolute configuration of (+)-1 and (−)-1, the electronic circular dichroism (ECD) calculations were carried out. It was found that the ECD spectrum of (13R,9′R,12′R,13′S,14′S,15′S)-1 agrees well with the experimental one of (+)-1, leading to the assignment of the absolute configuration at the stereogenic centers (Fig. 4). Consequently, the structures of (+)-1 and (−)-1 were finally assigned and named (±)-applanatumine B.
 | ||
| Fig. 4 Experimental and calculated ECD spectra of 1 at the B3LYP/6-311G(d,p) level in MeOH, σ = 0.3 eV. | ||
Simultaneously, a portion was purified by a semipreparative HPLC (Fig. S24, ESI†) to yield 2 (Rt = 17.5 min) and 3 (Rt = 19.8 min), and they are both racemic mixtures (Fig. S26 and S27, ESI†). Compounds 2 and 3 possess the same planar structure by analysis of their HRESIMS, 1D and 2D NMR data. Furthermore, a detailed interpretation of their 1D and 2D data (1–3), the only difference is that the existence of a five-membered lactone in 2 or 3, which was confirmed by the HMBC correlations of H-7/C-8 (δC 151.1), C-9 (δC 133.2), C-10 (δC 176.6), and H-8/C-7 (δC 79.9), C-9, C-10 (Fig. 5). The relative configuration of 2 was assigned by a ROESY spectrum which shows correlations of H-12′, H-13′/Hb-12, suggesting the relative configurations at C-13, C-12′, and C-13′. Likewise, the correlations of H-14′/Hb-8′, H-15′/Ha-8′, and 15′-OMe/H-12′, H-13′ (Fig. 3) reveal the relative configurations at C-9′, C-14′, and C-15′. As for the stereochemistry at C-7, it makes sense only to determine its absolute configuration.
 | ||
| Fig. 5 Key 1H–1H COSY and HMBC correlations of 2 or 3. | ||
Careful analysis of ROESY data of 3 reveals that compounds 3 and 2 bear the same relative configuration at C-13, C-9′, C-12′, C-13′, C-14′, and C-15′. Likewise, the assignment of stereochemistry at C-7 of 3 is challengeable. To clarify the absolute configuration at C-7, racemic 2 or 3 was submitted to chiral-phase HPLC to afford their respective enantiomers. By comparing the experimental spectra of 2 and 3, it was fortunately found that their experimental CD spectra have obvious difference at 197 nm and 207 nm [Δε197 −16.50, Δε207 +3.91 for (+)-2, no cotton effective for (+)-3], which makes it possible to use computational methods to solve the configuration at C-7. An effort of computation found that the calculated weighted ECD spectra of (7S,13R,9′R,12′R,13′S,14′S,15′S)-2 agree well with that of (+)-2 (Fig. 6 and 7). Hence, the absolute configurations of (+)-2 and (−)-2 were unambiguously assigned. In the same manner as that of 2, the absolute configurations of (+)-3 and (−)-3 were respectively assigned as 7R,13R,9′R,12′R,13′S,14′S,15′S and 7S,13S,9′S,12′S,13′R,14′R,15′R (Fig. 6 and 7). Obviously, (+)-3 is a 7-epimer of (+)-2.
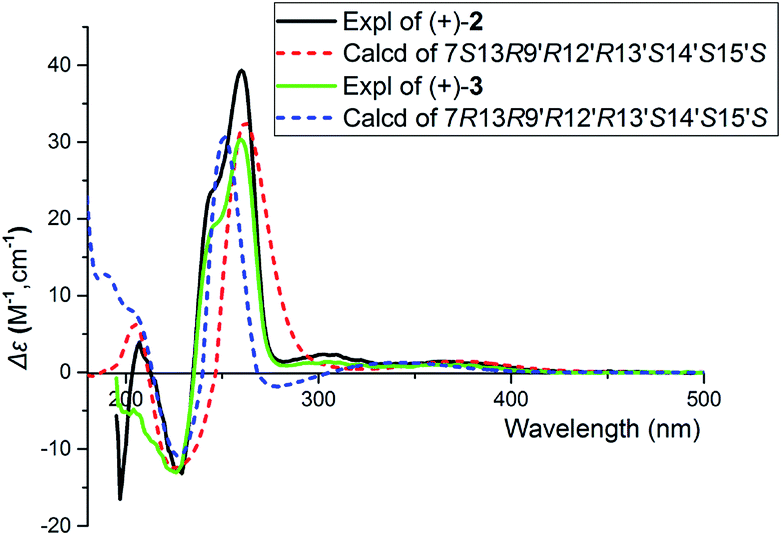 | ||
| Fig. 6 The calculated ECD spectra and experimental spectra of (+)-2 and (+)-3 at the B3LYP/6-311G(d,p) level in MeOH, σ = 0.3 eV. | ||
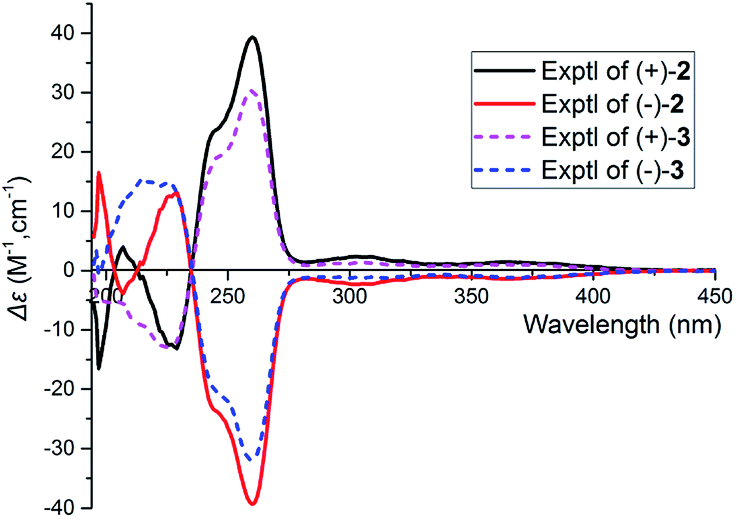 | ||
| Fig. 7 The experimental spectra of (+)-2, (−)-2, and (+)-3, (−)-3 in MeOH. | ||
Compounds 1–3 are novel meroterpenoid dimers. The formation of rings A–C leads to the presence of six continuous chiral centers in the structure, providing great challenge for synthetic chemistry. To aid total synthesis of this class of meroterpenoids, a plausible biosynthetic pathway was proposed. Compounds 1–3 are derived from the hybridization of shikimic acid pathway and mevalonic acid pathway18 (Scheme 1). First, two key intermediates A and B could be synthesized from 4-hydroxybenzonic acid (4HB)18 and isoprenyl moieties under geranyltransferase19 and VibMO1 (a monooxygenase).20 The intermediate A is further oxidized to give B. The monomer B could be cyclized via nucleophilic addition to generate C and F. In addition, the monomer E is formed by oxidation reaction and decarboxylization of C. Hence, 2 and 3 are further formed by a Diels–Alder reaction between E and F. Finally, the monomer G is formed by oxidation reaction and 1,3-hydrogen migration of B, and the ring A in 1 is formed via a Diels–Alder reaction between E and G.
 | ||
| Scheme 1 Proposed biogenetic pathway for compounds 1–3. | ||
2.2. Biological evaluation
To explore the biological activities of meroterpenoids in the genus Ganoderma, all the isolates were tested for their inhibitory activities towards JAK3, DDR1, HDAC1, and BRD4 (Table 2). It was found that almost all the compounds are active in these assays. Of particular, JAK3 kinase is sensitive for compounds 1–3 with IC50 values less than 10 μM. In addition, the enantiomers of 1 are active towards DDR1 with IC50 values of 8.2 ± 0.8 μM and 6.9 ± 0.8 μM, respectively. It is worthwhile that DDR1, HDAC1, and BRD4 are all promising epigenetic drug targets for a number of diseases. Despite the less potency of these compounds than synthetic agents, the present findings suggest that these natural meroterpenoids could be considered as a new scaffold for the development of more potent epigenetic agents.| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| JAK3 | DDR1 | HDAC1 | BRD4 | |
| a NA: no activity. | ||||
| (+)-1 | 1.8 ± 0.9 | 8.2 ± 0.8 | 212.0 ± 53.2 | 38.9 ± 5.3 |
| (−)-1 | 2.0 ± 0.9 | 6.9 ± 0.8 | 268.0 ± 0.2 | 46.5 ± 16.1 |
| (+)-2 | 0.8 ± 0.7 | ∼10.0 | 19.1 ± 10.4 | 39.5 ± 5.9 |
| (−)-2 | 6.1 ± 1.0 | NA | 45.4 ± 9.2 | 68.5 ± 19.4 |
| (+)-3 | 3.8 ± 1.0 | NA | 46.6 ± 20.1 | 19.9 ± 8.0 |
| (−)-3 | 3.8 ± 0.3 | NA | 36.1 ± 1.5 | 27.8 ± 1.5 |
| Staurosporine | (0.3 ± 0.01) × 10−3 | |||
| Dasatinib | (13.2 ± 0.1) × 10−3 | |||
| JQ-1 | (2.4 ± 0.6) × 10−3 | 0.1 ± 0.06 | ||
3. Experimental section
3.1. General procedure
Optical rotations were recorded on a Jasco P-1020. UV spectra were collected on a Shimadzu UV2401PC. CD spectra were measured on an Agilent applied photophysics. NMR spectra were determined on a Brucker AV 600 MHz. ESIMS and HRESIMS were measured on an Agilent UPLC/Q-TOF. RP-18 (40–60 μm, Daiso Co., Japan), MCI gel CHP 20P (75–150 μm, Tokyo, Japan), and Sephadex LH-20 (Amersham Biosciences, Sweden). Semipreparative HPLC was carried out using an Agilent 1200 liquid chromatograph and a LC-3000 high liquid chromatograph that made in China, the columns used were a 250 mm × 9.4 mm i.d., 5 μm, Zorbax SB-C18 and a 250 mm × 10 mm, i.d., 5 μm, Daicel Chiralpak (IC), flow rate: 2.5 mL min−1.3.2. Fungal material
The fruiting bodies of G. applanatum were purchased from Tongkang Pharmaceutical Co. Ltd. in Guangzhou Province, People's Republic of China, in September 2013. The material was identified by Prof. Zhu-Liang Yang at Kunming Institute of Botany, Chinese Academy of Sciences, and a voucher specimen (CHYX-0590) was deposited at the State Key Laboratory of Photochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, People's Republic of China.3.3. Extraction and isolation
Powdered fruiting bodies of G. applanatum (30 kg) were extracted by refluxing with 80% EtOH (3 × 120 L × 2 h) and concentrated under reduced pressure to give a crude extract, which was suspended in water followed by extraction with EtOAc to afford an EtOAc soluble extract. The EtOAc extract (0.9 kg) was divided into seven parts (Fr.1–Fr.7), by using a MCI gel CHP 20P column eluted with aqueous MeOH (20–100%). Fr.5 (32.0 g) was separated by MCI gel CHP 20P eluted with gradient aqueous MeOH (40–80%) to yield six fractions (Fr.5.1–Fr.5.6). Of which, Fr.5.5 (7.3 g) was further divided into six portions (Fr.5.5.1–Fr.5.5.6) by RP-18 with aqueous MeOH (60–80%). Fr.5.5.6 (1.1 g) was separated by Sephadex LH-20 (MeOH) followed by semipreparative HPLC (MeOH–H2O, 59%) to yield 1 (1.2 mg, Rt = 21.3 min), 2 (9.0 mg, Rt = 17.5 min), and 3 (12.0 mg, Rt = 19.8 min). Compounds 1–3 were isolated as racemate, which were further purified by chiral-phase HPLC on Daicel Chiralpak IC (flow rate: 2.5 mL min−1) to afford enantiomers [(−)-1 (0.5 mg, Rt = 17.5 min), (+)-1 (0.4 mg, Rt = 19.7 min), n-hexane–EtOH–HCOOH, 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:0.01%; (+)-2 (4.2 mg, Rt = 21.8 min), (−)-2 (4.3 mg, Rt = 25.9 min), n-hexane–EtOH, 80:20; and [(+)-3 (5.6 mg, Rt = 20.4 min), (−)-3 (5.7 mg, Rt = 23.0 min), n-hexane–EtOH, 80:20].
:20:0.01%; (+)-2 (4.2 mg, Rt = 21.8 min), (−)-2 (4.3 mg, Rt = 25.9 min), n-hexane–EtOH, 80:20; and [(+)-3 (5.6 mg, Rt = 20.4 min), (−)-3 (5.7 mg, Rt = 23.0 min), n-hexane–EtOH, 80:20].
3.4. Spectral data of 1–3
Applanatumin B (1): yellow gum, UV (MeOH) λmax (logε): 373 (3.51), 253 (3.91), 226 (4.00) nm; {[α]24D +163.8 (c 0.07, MeOH), CD (MeOH) Δε225 −17.08, Δε260 +38.26, for (+)-1; [α]24D −166.0 (c 0.05, MeOH), CD (MeOH) Δε224 +14.32, Δε260 −36.36, for (−)-1}; ESIMS: m/z 621 [M − H]−; HRESIMS: m/z 621.1605 [M − H]− (calcd for C32H29O13, 621.1614); 1H and 13C NMR data, see Table 1.
Applanatumin C (2): yellow gum, UV (MeOH) λmax (logε): 370 (3.53), 250 (4.17), 221 (4.25) nm; {[α]24D +159.3 (c 0.18, MeOH), CD (MeOH) Δε197 −16.50, Δε207 +3.91, Δε229 −13.10, Δε260 +39.30, for (+)-2; [α]24D −199.2 (c 0.16, MeOH), CD (MeOH) Δε197 +16.50, Δε208 −3.91, Δε229 +11.28, Δε260 −33.67, for (−)-2}; ESIMS: m/z 605 [M − H]−; HRESIMS: m/z 719.1590 [M + CF3COO]− (calcd for C34H30O14F3, 719.1593); 1H and 13C NMR data, see Table 1.
Applanatumin D (3): yellow gum, UV (MeOH) λmax (logε): 368 (3.62), 251 (4.24), 227 (4.30) nm; {[α]24D +208.9 (c 0.30, MeOH), CD (MeOH) Δε226 −13.03, Δε259 +30.31, for (+)-3; [α]24D −229.6 (c 0.33, MeOH), CD (MeOH) Δε225 +14.97, Δε260 −31.95, for (−)-3}; ESIMS: m/z 605 [M − H]−; HRESIMS: m/z 719.1587 [M + CF3COO]− (calcd for C34H30O14F3, 719.1593); 1H and 13C NMR data, see Table 1.
3.5. ECD calculation
The CONFLEX 7 searches based on molecular mechanics with MMFF94S force fields were performed for model compounds of 1–3, respectively. The predominant conformers were optimized and calculated by DFT calculation at B3LYP/6-311G(d,p) level with the PCM in MeOH. Under the circumstances, all the above calculations were carried out with the Gaussian 09 package of programs.21 For comparison of the calculated curves and experimental CD spectra, the program SpecDis22 was used.3.6. In vitro JAK3 kinase inhibitory activity assay
The inhibitory activity of compounds 1–3 against JAK3 kinase was performed using the FRET-based Z'-Lyte assay system in line with the manufacturer's instructions (Invitrogen, Carlsbad, USA) and previously described methods.23 In this study, staurosporine was used as a positive control. The data were analyzed using Graphpad Prism5 (Graphpad Software, Inc).3.7. In vitro DDR1 kinase inhibitory activity assay
Compounds 1–3 were evaluated for their inhibitory activity towards DDR1 kinase using a LanthaScreen Eu kinase activity assay technology (Invitrogen, USA) as previously described.23 In this study, dasatinib was used as a positive control.3.8. In vitro HDAC1 inhibition assay
The purified recombinant human HDAC1 and their corresponding substrates were purchased from BPS Bioscience (BPS Bioscience Inc., USA). The assays were carried out in 384-well format using the BPS fluorescent-based HDAC activity assay according to the manufacturer's protocol (BPS Bioscience Inc., USA). The HDAC reaction mixture was composed of HDAC assay buffer (BPS Bioscience Inc., USA), BSA, serial diluted test compounds, appropriate concentration of HDACs, and 20 μM fluorogenic substrate. The mixture was incubated at 37 °C for 60 min and then stopped by addition of developer containing trypsin and TSA. After 20 min of incubation, fluorescence was detected at the excitation wavelength of 360 nm and the emission wavelength of 460 nm using EnVision multilabel reader (PerkinElmer Inc., USA). JQ-1 was used as the positive control. The analytical software, GraphPad Prism 5.0 (GraphPad Software, Inc., USA) was used to generate IC50 value via nonlinear regression analysis.3.9. In vitro BRD4 inhibition assay
In vitro inhibitory activity of compounds on BRD4 was carried out in 384-well plates by TR-FRET assay using BRD4 (BD1) TR-FRET Assay Kit (BPS BioScience, San Diego, CA, USA) according to the manufacturer's instructions. In brief, 2.5 μL of BRD assay buffer was added 2.5 μL of BET bromodomain ligand and 2.5 μL of BRD4 (BD1) and appropriate serials of diluted compounds, subsequently mixed with GSH Acceptor beads (PerkinElmer #AL109C) and Streptavidin-conjugated donor beads (PE #6760002S). After 1 h room temperature incubation, the plates were measured in EnVision Multilabel Reader (Perkin Elmer, Inc.) using TR-FRET module with excitation 340 nm and emission 665 nm). JQ-1 was used as the positive control.4. Conclusions
In sum, the present study afforded three unprecedented meroterpenoid dimers from the fruiting bodies of G. applanatum. These metabolites were all isolated as racemic mixtures. ECD calculations were successfully used to clarify the absolute configurations of the enantiomers. Meroterpenoids 1–3 were found to be JAK3, DDR1, HDAC1, and BRD4 inhibitors with an unprecedented structure type. Of note, their inhibitory activities towards JAK3 and DDR1 are significant, we envision that compounds 1–3 might serve as a useful scaffold for the development of more potent therapeutic agents (including epigenetic agents) against these targets associated diseases such as cancer, fibrosis, and rheumatoid arthritis.Acknowledgements
This work was financially supported by the National Science Fund for Distinguished Young Scholars (81525026), National Natural Science Foundation of China (21472199), NSFC-Joint Foundation of Yunnan Province (U1202222), Guangdong Frontier and Key Technology Innovation Special Grant (2016B030229006), Guangzhou Science &Technology Project (2011Y2-00026 and 201508020131).Notes and references
- C. Richter, K. Wittstein, P. M. Kirk and M. Stadler, Fungal Divers., 2015, 71, 1 CrossRef.
- R. R. M. Paterson, Phytochemistry, 2006, 67, 1985 CrossRef CAS PubMed.
- K. Wang, L. Bao, W. P. Xiong, K. Ma, J. J. Han, W. Z. Wang, W. B. Yin and H. W. Liu, J. Nat. Prod., 2015, 78, 1977 CrossRef CAS PubMed.
- I. C. F. R. Ferreira, S. A. Heleno, F. S. Reis, D. J. Stojkovic, M. J. R. P. Queiroz, M. H. Vasconcelos and M. Sokovic, Phytochemistry, 2015, 114, 38 CrossRef CAS PubMed.
- (a) Y. M. Yan, J. Ai, Y. N. L. L. Zhou, A. C. K. Chung, R. Li, J. Nie, P. Fang, X. L. Wang, J. Luo, Q. Hu, F. F. Hou and Y. X. Cheng, Org. Lett., 2013, 15, 5488 CrossRef CAS PubMed; (b) F. J. Zhou, Y. Nian, Y. M. Yan, Y. Gong, Q. Luo, Y. Zhang, B. Hou, Z. L. Zuo, S. M. Wang, H. H. Jiang, J. Yang and Y. X. Cheng, Org. Lett., 2015, 17, 3082 CrossRef CAS PubMed; (c) Q. Luo, L. Tian, L. Di, Y. M. Yan, X. Y. Wei, X. F. Wang and Y. X. Cheng, Org. Lett., 2015, 17, 1565 CrossRef CAS PubMed; (d) Q. Luo, X. L. Wang, L. Di, Y. M. Yan, Q. Lu, X. H. Yang, D. B. Hu and Y. X. Cheng, Tetrahedron, 2015, 71, 840 CrossRef CAS; (e) Y. M. Yan, X. L. Wang, L. L. Zhou, F. J. Zhou, R. Li, Y. Tian, Z. L. Zuo, P. Fang, A. C. K. Chung, F. F. Hou and Y. X. Cheng, J. Ethnopharmacol., 2015, 176, 385 CrossRef CAS PubMed; (f) Q. Luo, Z. L. Yang, Y. M. Yan and Y. X. Cheng, Org. Lett., 2017, 19, 718 CrossRef CAS PubMed.
- (a) Q. Luo, X. H. Yang, Z. L. Yang, Z. C. Tu and Y. X. Cheng, Tetrahedron, 2016, 72, 4564 CrossRef CAS; (b) Q. Luo, L. Di, X. H. Yang and Y. X. Cheng, RSC Adv., 2016, 6, 45963 RSC; (c) Q. Luo, Z. C. Tu and Y. X. Cheng, RSC Adv., 2017, 7, 3413 RSC.
- Q. Luo, X. Y. Wei, J. Yang, J. F. Luo, R. Liang, Z. C. Tu and Y. X. Cheng, J. Nat. Prod., 2017, 80, 61 CrossRef CAS PubMed.
- R. Long, J. Huang, W. B. Shao, S. Liu, Y. Lan, J. X. Gong and Z. Yang, Nat. Commun., 2014, 5, 5707 CrossRef CAS PubMed.
- X. Y. Li, X. Y. Liu, X. Z. Jiao, H. G. Yang, Y. Y. Yao and P. Xie, Org. Lett., 2016, 18, 1944–1946 CrossRef CAS PubMed.
- K. S. Gautam and V. B. Birman, Org. Lett., 2016, 18, 1499–1501 CrossRef PubMed.
- D. Chen, H. M. Liu, M. M. Li, Y. M. Yan, W. D. Xu, X. N. Li, Y. X. Cheng and H. B. Qin, Chem. Commun., 2015, 51, 14594–14596 RSC.
- R. Rengarasu and M. E. Maier, Asian J. Org. Chem., 2017, 6, 108–117 CrossRef CAS.
- R. A. Hill and A. Sutherland, Nat. Prod. Rep., 2014, 31, 148–153 RSC.
- Y. Liu, C. J. Zhou, Q. J. Li and H. G. Wang, Org. Biomol. Chem., 2016, 14, 10362 CAS.
- J. Y. Song, X. Chen, J. Bai, Q. H. Liu, H. Li, J. W. Xie, H. Jing and J. N. Zheng, Tumor Biol., 2016, 37, 11509 CrossRef CAS PubMed.
- J. Y. Yan, Z. M. Zhang, J. Yang, W. Mitch and Y. L. Wang, J. Am. Soc. Nephrol., 2015, 26, 3060 CrossRef CAS PubMed.
- N. Ding, N. Hah, R. T. Yu, M. H. Sherman, C. Benner, M. Leblanc, M. X. He, C. Liddle, M. Dowens and R. M. Evans, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15713 CAS.
- Q. Luo, L. Di, W. F. Dai, Q. Lu, Y. M. Yan, Z. L. Yang, R. T. Li and Y. X. Cheng, Org. Lett., 2015, 17, 1110 CrossRef CAS PubMed.
- R. Boehm, S. Sommer, S. M. Li and L. Heide, Plant Cell Physiol., 2000, 8, 911 CrossRef.
- Y. L. Yang, H. Zhou, G. Du, K. N. Feng, T. Feng, X. L. Fu, J. K. Liu and Y. Zeng, Angew. Chem., Int. Ed., 2016, 55, 5463 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Y. Nakajima, O. Honda, H. Kitao, T. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243 CrossRef CAS PubMed.
- H. J. Zhu, Y. M. Yan, Z. C. Tu, J. F. Luo, R. Liang, T. H. Yang, Y. X. Cheng and S. M. Wang, Fitoterapia, 2016, 114, 163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ECD calculation methods, 1D, 2D, HRESIMS spectra, and HPLC chromatograms. See DOI: 10.1039/c7ra04862a |
| This journal is © The Royal Society of Chemistry 2017 |