
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metabolite identification and pharmacokinetic study of platycodi radix (Jiegeng) in vivo†
Zhongyao Tanga,
Yuanyuan Hou *a,
Xueyan Hua,
Aina Liua,
Leefong Yaub,
Tiantian Tongb,
Zhihong Jiangb and
Gang Baia
*a,
Xueyan Hua,
Aina Liua,
Leefong Yaub,
Tiantian Tongb,
Zhihong Jiangb and
Gang Baia
aState Key Laboratory of Medicinal Chemical Biology, College of Pharmacy, Tianjin Key Laboratory of Molecular Drug Research, Nankai University, Tianjin 300353, People's Republic of China. E-mail: houyy@nankai.edu.cn
bState Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Taipa, Macau, People's Republic of China
First published on 28th July 2017
Abstract
Platycodi radix (Jiegeng) is commonly used as a channel ushering drug for assistance in treating respiratory diseases, because of the activities of saponins that are called platycosides. In this study, using ultra high pressure liquid chromatography with quadrupole time of flight mass spectrometry (UPLC-Q/TOF-MS) and a strategy based on fragmentation behaviors, a total of thirteen platycosides were identified in the total saponins, and seven of these were found in rat plasma following a single oral dose of total saponins. The secondary platycosides were proved to be the main absorbed saponins in vivo. The major absorbed platycosides, 3-O-β-D-glucopyranosylplatyconic acid (GPA) and 3-O-β-D-glucopyranosylplatycodigenin (GP), were isolated from total saponins and the pharmacokinetics were evaluated in mice by liquid chromatography with triple quadrupole mass spectrometry (LC-QQQ). The results of the pharmacokinetic analysis indicated that GPA and GP were quickly absorbed with short Tmax (<45 min). Cmax (μg L−1), AUC0–t (μg h L−1) and MRT0–t (h) of GPA were 1147.75 ± 307.23, 2119.16 ± 530.84 and 4.10 ± 0.33 at a dose of 105 mg kg−1, and 3673.10 ± 1250.44, 8734.07 ± 2362.48 and 3.95 ± 0.82 at a dose of 350 mg kg−1. The above pharmacokinetic parameters were higher than those of GP at the same dosage, which showed that GPA was likely to be the main absorbed saponin in vivo. These findings provide useful information that will support the studies on identification of bioactive platycosides in vivo, and will lay the foundation for the development of Jiegeng.
1. Introduction
Platycodi radix (Jiegeng), the dried root of Platycodon grandiflorum A. DC. (Campanulaceae), has been widely used as an anti-tussive or anti-inflammatory medicine in China and East Asia.1–3 In addition, Jiegeng has been reported to have many other pharmacological activities, such as anti-oxidant,4,5 anti-tumor,6–9 anti-diabetic,10 anti-obesity,11,12 hepato-protective13,14 and cardiovascular system activities.15,16 All the above activities are due to the chemical constituents in Jiegeng, including polysaccharides, saponins, flavonoids, polyphenols, polyacetylenes and sterols.1 The saponins in Jiegeng are also called platycosides, which have been proved to be the major bioactive compounds.17 Currently, all known platycosides are oleanane-type pentacyclic triterpenoid saponins, which are mostly primary saponins.18 These platycosides primarily have two saccharide chains; one or two glucoses linked through linear glycosidic bonds at the C-3, and arabinose, rhamnose, xylose and apiose sequentially attached at C-28 by ester linkages.19 According to the different aglycones, they are classified into four types, including paltcodic acid, polygalic acid, platcogenic acid and its derivatives.1Although there are a large number of studies about the activities of platycosides at present, there are only a few studies on the pharmacokinetics of platycosides. The pharmacokinetic studies of platycodin D, platycodin D3 and deapioplatycodin D have shown that these platycosides have low oral bioavailability, which due to the poor permeability and degradation in the gastrointestinal tract.20–25 In that case, we consider that activities of Jiegeng could be caused by the metabolites of platycosides or other absorbed platycosides.
In this study, the total saponins were isolated from the platycodi radix extract, and the main platycosides were identified by the ultra high pressure liquid chromatography along with quadrupole time of flight mass spectrometry (UPLC-Q/TOF-MS). According to the result of identification based on the fragmentation behaviors of platycosides, the absorbed saponins were qualitatively analyzed by principal components analysis (PCA). Then, the secondary platycosides, 3-O-β-D-glucopyranosylplatycodigenin (GP) and 3-O-β-D-glucopyranosylplatyconic acid (GPA), with higher relative contents were isolated and identified. And their pharmacokinetic parameters were evaluated using the liquid chromatography along with triple quadrupole mass spectrometry (LC-QQQ). By this way, we hoped to utilize the information about metabolites of platycosides to discover the bioactive constituents in vivo, and provide pharmacokinetic data support for subsequent pharmacological experiments.
2. Materials and methods
2.1. Chemicals, reagents and animals
Platycodi radix extract (bath number TR20160103) was purchased from Tianrui Bio-Tech Co., Ltd (Xi'an, China). Acetonitrile (HPLC grade) and methanol (HPLC grade) were purchased from Merck (Darmstadt, Germany). Ultrapure water was prepared with a Milli-Q system (Millipore, Bedford, MA, USA). All other reagents were analytical grade and purchased from Concord Technology (Tianjin, China). Ginsenoside Rh1 (98% purity) was purchased from the Shilan Technology (Tianjin, China).Experimental animals were purchased from the Laboratory Animal Center of the Academy of Military Medical Sciences (SCXK 2012-0004, Beijing, China). The animals were housed under standard laboratory conditions (25 ± 1 °C, 50 ± 10% humidity, and 12 h photoperiod) with free access to food and water. The animal experiments were approved by the Animal Experiments Ethical Committee of Nankai University and complied with the Guide for the Care and Use of Laboratory Animals. The animals were fasted 12 h and had free access to water throughout the experimental period.
2.2. Instrumentation and conditions
The UPLC-Q/TOF-MS system consisted of a Waters Acquity UPLC System (Waters Co., Milford, MA, USA) and a Waters Q/TOF Premier instrument with an electrospray ionization system (Waters MS Technologies, Manchester, UK). An Acquity BEH C18 column (2.1 mm × 100 mm, 1.7 μm; Waters Co.) was used with a mobile phase of 0.1% formic acid (A)–acetonitrile (B). The elution gradient was as follows: 0 min, 5% B; 2 min, 15% B; 18 min, 35% B. 23 min, 55% B; 27 min, 100% B; 29 min 100% B. The flow rate was 0.40 mL min−1 and the column temperature was maintained at 30 °C. The injection volume of the test sample was 5 μL. Data were acquired using MassLynx V4.1 software (Waters Co.). The capillary voltage was set to 2.0 kV for the negative mode and 3.0 kV for the positive mode, and the sample cone voltage was set to 40 V. The desolvation gas was set to 800 L h−1 at a temperature of 450 °C, the cone gas was set to 50 L h−1, and the source temperature was 120 °C. The mass spectrometric data were collected in full scan mode, and the m/z ranged from 50 to 1900 Da in the positive and negative ion modes.The LC-QQQ platform was comprised of an Agilent 1290 infinity ultra-high performance liquid chromatography (UHPLC) system (Agilent Technologies, Santa Clara, USA) interfaced to an Agilent 6490 Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, USA), using a standard-flow ESI (Jet Stream) source. The LC system specifically consists of a binary pump, a thermostatted column compartment, and a thermostatted micro well-plate autosampler. The column was an Acquity BEH C18 column (2.1 mm × 100 mm, 1.7 μm; Waters Co.), and the mobile phase consisted of 0.1% formic acid (A) and acetonitrile (B) using a gradient elution program as follows: 0 min, 30% B; 8 min, 35% B; 10 min, 85% B. 10.1 min, 100% B; 12 min, 100% B. The column temperature was set to 25 °C, the flow rate was maintained at 0.35 mL min−1, and the injection volume was set at 2 μL. All data are processed with the Agilent MassHunter Quantitative Analysis software (version B.06.00, Agilent Technologies). The capillary voltage was set to 4.0 kV for the positive mode, and the fragmentor voltage was 250 V. The sheath gas was set to 16 L min−1 at a temperature of 250 °C. The nebulizer gas pressure was 35 psi.
2.3. Metabolite identification of the total saponins
The thawed plasma samples (500 μL) were precipitated protein with methanol (1.5 mL). After vortexing for 2 min and centrifuging at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C, the supernatants were transferred to another tube and evaporated to dryness by N2. The residues were dissolved in methanol (150 μL) and then centrifuged again at the same manner before the UPLC-Q/TOF-MS analysis. The metabolism data were analyzed by principal components analysis (PCA) using SIMCA-P software (version 13.0, UmetricsAB, Sweden).
000 rpm for 10 min at 4 °C, the supernatants were transferred to another tube and evaporated to dryness by N2. The residues were dissolved in methanol (150 μL) and then centrifuged again at the same manner before the UPLC-Q/TOF-MS analysis. The metabolism data were analyzed by principal components analysis (PCA) using SIMCA-P software (version 13.0, UmetricsAB, Sweden).
2.4. Pharmacokinetic study of GP and GPA
:MeOH = 9:1–1:1) to give five fractions (fr. A–E). Fraction B and C were collected and further separated by semipreparative HPLC (ACN:H2O = 35:65) to get GP. GPA, the new compound, was obtained by the same method from fraction D.Calibration standards and quality control (QC) samples were prepared as the following: working solution (25 μL) was evaporated to dryness by N2, and then blank plasma (100 μL) was added. The samples were prepared prior to the validation and pharmacokinetic study. The standards and QC samples were extracted on each analysis day with the same procedures for plasma samples as described below.
000 rpm for 10 min at 4 °C, the supernatants were transferred to another tube and evaporated to dryness by N2. The residues were redissolved in methanol (100 μL) and centrifuged again at the same manner. Subsequently, the supernatants were transferred into vials for injection.3. Results
3.1. Qualitative analysis of the components of total saponins
The optimal UPLC-Q/TOF-MS conditions were applied for the analysis of the total saponins. More ion information of platycosides was obtained from positive ion mode, so the positive ion mode was selected in the following investigations (Fig. 1).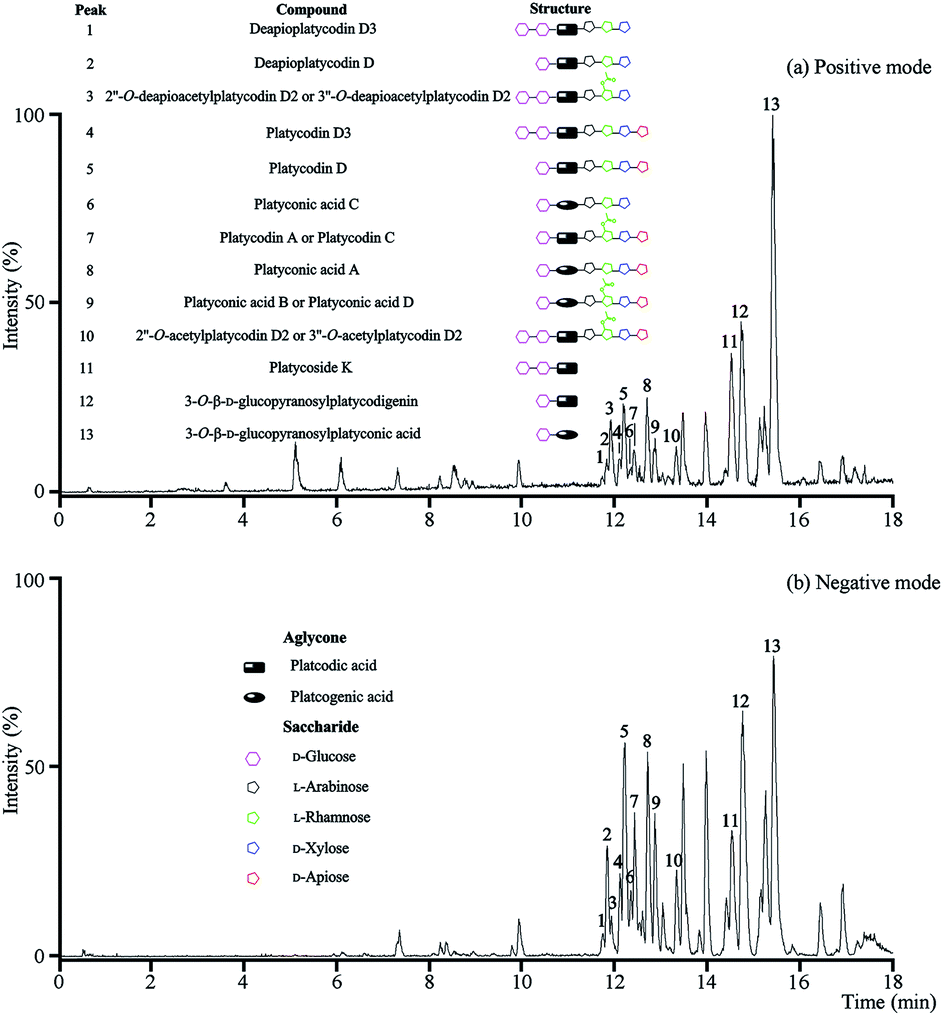 | ||
| Fig. 1 UPLC-Q/TOF-MS analysis of the total saponins. (a) The BPI chromatogram in positive mode; (b) the BPI chromatogram in negative mode. | ||
In positive mode, prominent peaks were mostly contained protonated platycoside molecules and their corresponding secondary saponin molecules. Taking the identify process of peak 5 as an example, it gave [M + H]+ ion at m/z 1225 in MS that presumed as platycodin D. Meanwhile it generated the prominent ions at m/z 1093, 961, 683 and 521 as base peaks in the MS/MS by sequential lost apiosyl, xylosyl, rhamosyl, and arabinosyl residues linked at C-28, and loss of glucosyl residues linked at C-3 of the triterpene, respectively. The above rule and structure were consistent with literature of platycodin D,26 so the peak 5 was identified as platycodin D. According to this process, peak 1, 2, 4, 5 and 7–12 were identified.
The above method was suitable for the identification of known platycosides, meanwhile the unknown compounds using reverse ideas for identifying. Taking peak 13 as the example, the prominent ion at m/z 535 was found in the positive MS/MS, and the m/z of [M + H]+ ion in MS was 697, which proved this compound was platcogenic acid-type platycoside with the exact molecular weight at 696.3615. According to the fragmentations and structural characteristics, this compound was presumed as a monodesmosidic platycoside with a glucosyl at C-3 named 3-O-β-D-glucopyranosylplatyconic acid. The identification of peak 3 was similar. These two platycosides were reported for the first time, which would be separated and further confirmed in the subsequent studies if necessary.
These methods were applicable for the fast and accurate identification of known and unknown platycosides. However, isomeric platycosides and constituents present at low levels were unidentifiable.
As a result, thirteen platycosides that respectively belonged to paltcodic acid- and platcogenic acid-type were identified accurately and effectively (Table S1†). The structures of these platycosides were described in Fig. S1.†
3.2. Qualitative analysis of total saponins constituents in vivo
The PCA score plots (Fig. 2a) presented the distribution among experimental and control group based on the metabolic profiles. Loading plots (Fig. 2b) indicated that markers made greater contribution to separation with farther away from original point, and the markers with the same retention time might be from the same compound.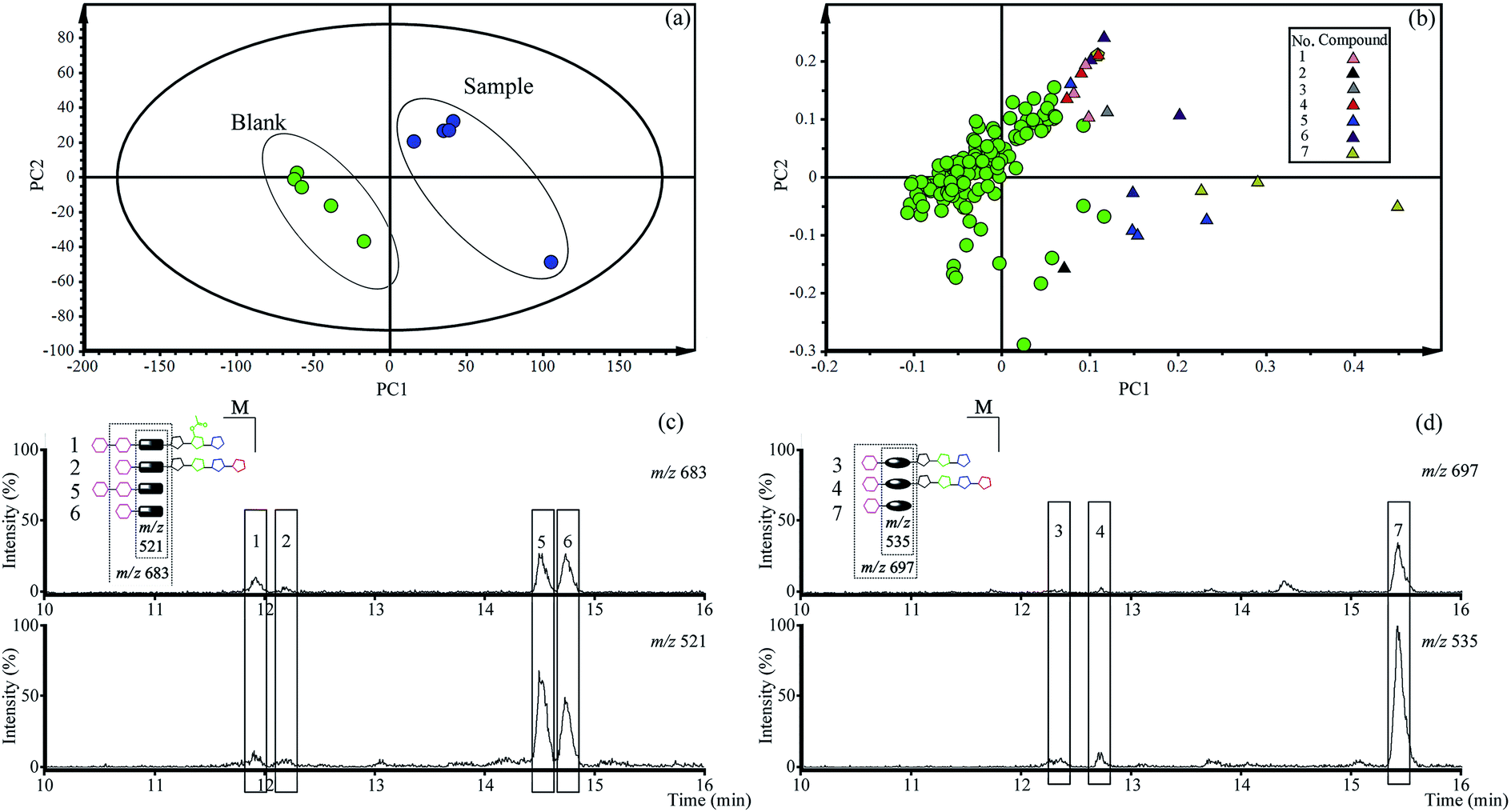 | ||
| Fig. 2 The results of qualitative analysis of total saponins in vivo by PCA. (a) Score plotting; (b) loading plotting; (c) the extracted ion chromatograms of paltcodic acid-type platycosides; (d) the extracted ion chromatograms of platcogenic acid-type platycosides. | ||
According to the retention time and m/z of markers, nineteen markers were identified belong to seven absorbed platycosides (Table 1 and Fig. S1†), which were significantly altered in the experimental group compared to the control group.
| No. | Ret. time (min) | Marker | Formula | Compound |
|---|---|---|---|---|
| 1 | 11.94 | 845, 683, 521 | C60H96O30 | 2′′-O-Deapioacetylplatycodin D2 |
| 2 | 12.26 | 1093 | C57H92O28 | Platycodin D |
| 3 | 12.32 | 517 | C65H104O34 | Platyconic acid C |
| 4 | 12.74 | 1107, 975, 535 | C57H90O29 | Platyconic acid A |
| 5 | 14.55 | 867, 845, 683, 521, 503 | C42H68O17 | Platycoside K |
| 6 | 14.70 | 683, 503 | C36H58O12 | 3-O-β-D-Glucopyranosylplatycodigenin |
| 7 | 15.44 | 719, 697, 535, 517 | C36H56O13 | 3-O-β-D-Glucopyranosylplatyconic acid |
An extracted ion chromatogram (EIC) strategy was established to capture platycosides and determine their relative contents in plasma samples according to the characteristic ion pairs. The ion pair at m/z 521 and 683 was selected as the diagnostic ions for paltcodic acid-type platycosides, and platcogenic acid-type platycosides used m/z 535 and 697 as characteristic ions. The EICs (Fig. 2c and d) were shown that the relative contents of GP and GPA were higher than the primary platycosides in rat plasma after oral administration of total saponins for 30 min. As a result, GP and GPA were determined as target compounds in the subsequent quantitative analysis in vivo.
3.3. Separation and structure identification of GP and GPA
GPA (Fig. 3) was an amorphous powder and the molecular formula was assigned to be C36H56O13 based on the high-resolution spectrum. The spectral features and physic-chemical properties revealed it to be a triterpenoid saponin.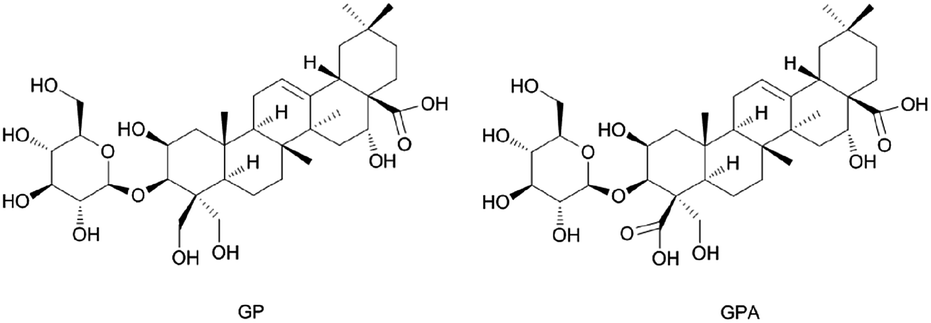 | ||
| Fig. 3 Structures of GP and GPA. | ||
The 1H NMR spectrum (pyridine-d5, 600 MHz) showed five tertiary methyl groups (δ 1.05, 1.09, 1.18, 1.65 and 1.84) and one olefinic proton (δ 5.66, br s). The 13C NMR spectrum (pyridine-d5, 600 MHz) showed five sp3 carbons at δ 15.64, 17.43, 24.72, 27.28 and 33.37, two sp2 olefinic carbons at δ 122.41 and 145.30, two carboxyl carbons at δ 176.31 and 179.99, and four oxygenated methene and methine carbons at δ 69.97, 81.19, 74.72 and 62.67. The information of 1H and 13C NMR spectrum indicated that it possessed a 2,3,16,23-tetrahydroxyolean-12-ene-24,28-dioic acid as aglycon. The chemical shifts of C-3 (δ 81.19) and C-28 (δ 179.99) revealed that it was a monodesmosidic glycoside. The 1H and 13C NMR spectra of it exhibited one sugar anomeric proton at δ 5.40 (1H, d. J = 7.8 Hz), and carbon at δ 106.70. On the basis of the above evidence, 3-O-β-D-glucopyranosyl-platyconic acid was identified to be 3-O-β-D-glucopyranosyl-2,3,16,23-tetrahydroxyolean-12-ene-24,28-dioic acid. Other NMR spectra data were described in Fig. S2–S7 and Table S2.†
GP (Fig. 3) was identified by comparing with the spectra data reported in the literature (1H and 13C NMR are described in Table S2 and Fig. S8 and S9†).27,28
3.4. Method validation for quantitative analysis
The optimized MRM modes were applied for the analysis (Table S3†). No obvious endogenous interference was observed when analytes and IS were detected, and the different retention times and molecule polarity were used to distinguish platycosides that produced the same fragment ions in the plasma samples (Fig. 4).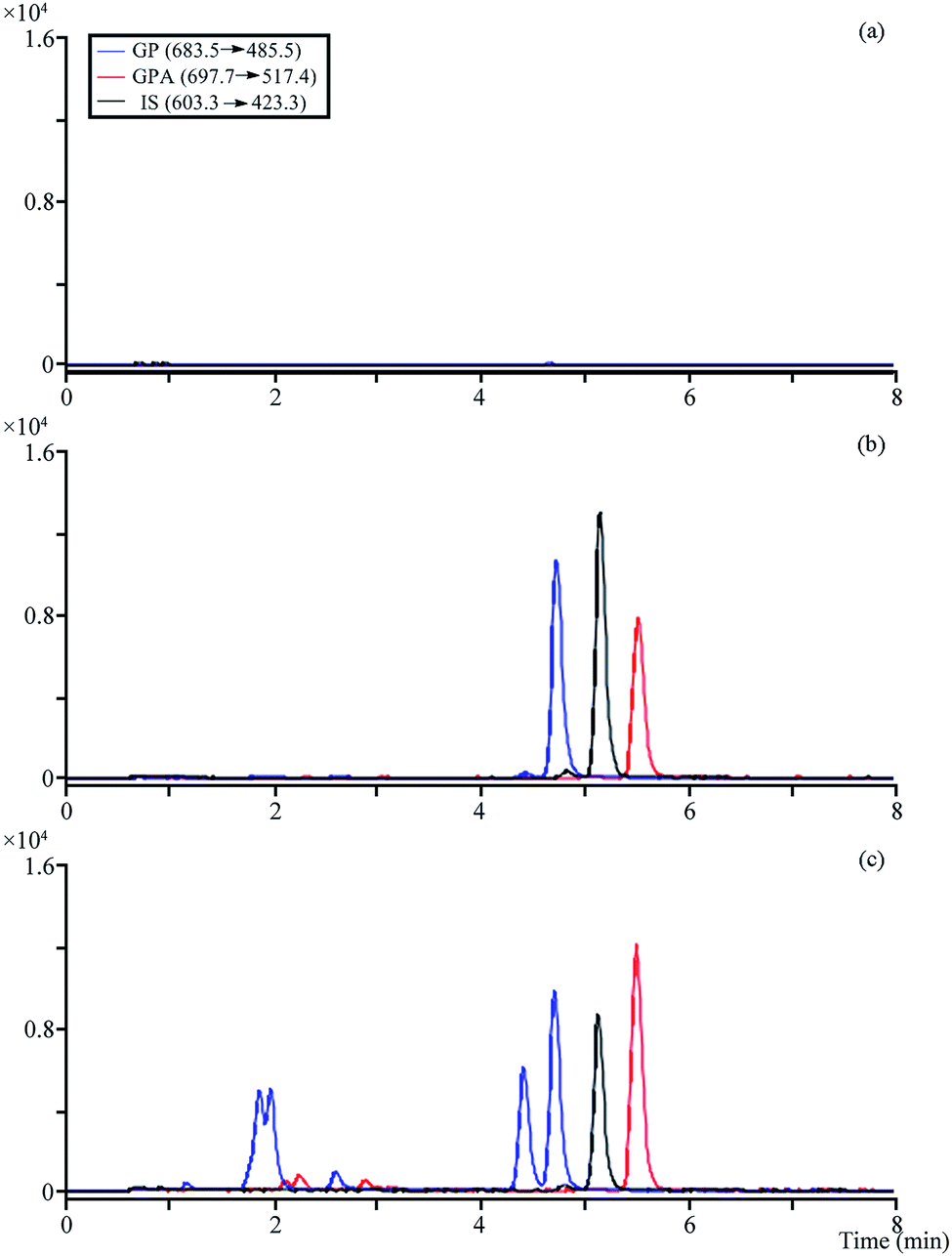 | ||
| Fig. 4 The MRM chromatograms of analytes and IS. (a) Blank plasma; (b) blank plasma spiked with the analytes and IS; (c) 0.5 h after administration of plasma sample. | ||
All calibration curves exhibited good linearity (r2 ≥ 0.9995). Extraction recoveries and matrix effects of analytes and IS were in the range of 91.96–99.94% (RSD ≤ 5.40%) and 93.45–111.65% (RSD ≤ 8.22%), respectively. RSD of intra-and inter-batch precisions were in the range of 1.19–12.28% and 1.09–6.92%, and RE from −1.18 to 9.52%, respectively (Table 2). The detected concentrations for all analytes were in the range of −11.37 to 6.61% of original values, indicating that they were stable in plasma samples after three freeze–thaw cycles, at room temperature and autosampler for 24 h, at −20 °C for 14 days (partial data were described in Tables S4–S6†).
| Analytes | Conc. (μg L−1) | Inter-batch | Intra-batch | ||
|---|---|---|---|---|---|
| Precision (RSD, %) | Accuracy (RE, %) | Precision (RSD, %) | Accuracy (%) | ||
| GP | 2.47 | 3.93 | 5.51 | 4.42 | −0.74 |
| 247.00 | 1.19 | 1.43 | 1.09 | 6.87 | |
| 2470.00 | 2.81 | 4.80 | 2.66 | 9.52 | |
| GPA | 2.50 | 12.28 | 0.44 | 3.37 | −1.18 |
| 249.60 | 2.09 | 0.53 | 2.7 | 1.94 | |
| 2496.00 | 6.35 | 5.07 | 6.92 | 6.03 | |
3.5. Pharmacokinetic study
The validated method described above was successfully applied to the pharmacokinetic study in which plasma concentrations of two analytes were determined for 12 h after the oral administration of total saponins (105 mg kg−1 and 350 mg kg−1, n = 6). The mean plasma concentration time profiles of all analytes (Fig. 5) revealed some similarities in concentration–time curve for them, and the main pharmacokinetic parameters were exhibited in Table 3. All analytes were quickly absorbed within 1 h, and a low peak appeared in the curve due to the enterohepatic circulation at approximately 1.5 h after oral administration. And the five important pharmacokinetic parameters, including T1/2z (h), AUC0–t (μg h L−1), AUC0–∞ (μg h L−1), MRT0–t (h) and MRT0–∞ (h), for GP, were lower than those of GPA at both doses of total saponins. It's worth noting that the Cmax (μg L−1) of GPA with the dosage of 350 mg kg−1 total saponins reached 3673.10 ± 1250.44 μg L−1, which was a high concentration of the drug in the plasma. Pharmacokinetic study results also showed that metabolic behaviors were similar for two secondary saponins, which made sense that GP and GPA had similar metabolic regularity. By comparing the pharmacokinetic parameters and metabolic regularity, GPA was considered to be the main absorbed platycoside in vivo.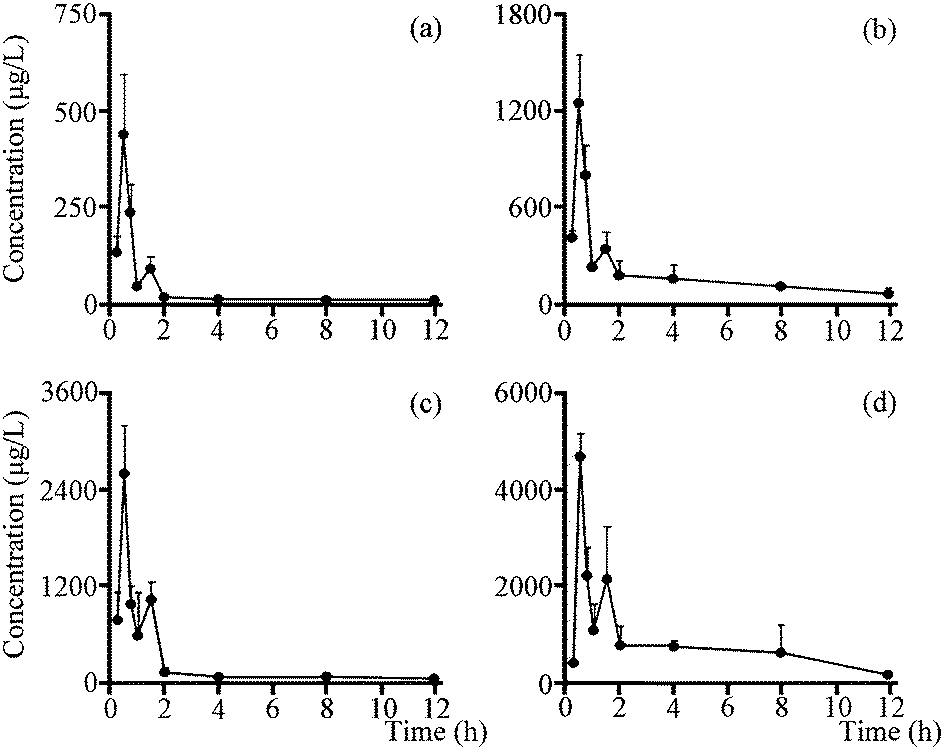 | ||
| Fig. 5 Mean concentration–time profiles of GP and GPA in mice plasma after oral administration of total saponins. (a) GP (105 mg kg−1); (b) GPA (105 mg kg−1); (c) GP (350 mg kg−1); (d) GPA (350 mg kg−1). | ||
| Parameters | 105 mg kg−1 | 350 mg kg−1 | ||
|---|---|---|---|---|
| GP | GPA | GP | GP | |
| AUC0–t (μg h L−1) | 328.57 ± 58.31 | 2119.16 ± 530.84 | 2306.32 ± 438.71 | 8734.07 ± 2362.48 |
| AUC0–∞ (μg h L−1) | 353.77 ± 55.56 | 2391.75 ± 547.42 | 2436.92 ± 421.65 | 9446.18 ± 2426.74 |
| MRT0–t (h) | 2.23 ± 0.42 | 4.10 ± 0.33 | 2.16 ± 0.25 | 3.95 ± 0.82 |
| MRT0–∞ (h) | 3.36 ± 0.81 | 5.76 ± 0.84 | 3.02 ± 0.76 | 4.97 ± 0.79 |
| T1/2z (h) | 3.69 ± 1.20 | 3.90 ± 0.94 | 3.31 ± 1.65 | 3.53 ± 0.38 |
| Tmax (h) | 0.58 ± 0.13 | 0.54 ± 0.10 | 0.46 ± 0.10 | 0.62 ± 0.21 |
| Cmax (μg L−1) | 392.61 ± 127.71 | 1147.75 ± 307.23 | 2434.17 ± 818.94 | 3673.10 ± 1250.44 |
4. Discussion
Jiegeng as the representative medicine of meridian-guiding theory has been shown to promote the absorption and targeted distribution of other drugs in the lungs.29–31 Although the mechanism is not yet clear, the platycosides are considered to be the main active ingredients due to the amphiphilicity.17,32–34 In this study, thirteen platycosides were identified in the total saponins that isolated from the root of platycodi radix extract. Among these saponins, ten saponins belonged to primary saponins and other three were secondary saponins.We found Tmax of platycodin D and platycodin D3 were both less than 1 h after oral administration to rats in the previous pharmacokinetic studies,21–23,25 therefore we selected 30 min as a critical time point in metabolic qualitative research of total saponins. PCA was used qualitatively analyze the metabolites of Jiegeng in vivo, the score plots proved a significant distinction between the experimental and control group. By analyzing the contribution value, m/z and retention time of markers in loading plots, nineteen markers were identified from seven absorbed platycosides.
Although thirteen platycosides were identified in total saponins, only seven among these were confirmed to contribute to the discrimination between experimental and control group. The result demonstrated that most of primary platycosides may be degraded or not absorbed in gastric-intestinal tract, which was in accordance with the previous reports.20,24
Furthermore, some differences between the metabolites in vitro and in vivo were found. The metabolites of platycosides in vivo all contained glucoses at C-3, but in vitro the glucoses could be easily hydrolyzed by enzyme that produced by intestinal bacteria; the secondary platycosides without saccharide chain at C-28 were the main absorbed compounds in vivo, but the saccharide chain at C-28 cloud not be enzymatic hydrolyzed in vitro and the main metabolites of platycosides in vitro were the compounds with the acetylated saccharide at C-28.17,24,35 So the hydrolysis of saccharide chain at C-28 by gastric acid in the stomach was considered to be the main reason of the degradations of primary platycosides and the differences between the metabolites in vitro and in vivo.
By comparing the relative contents obtained from peak areas in EIC, and considering the absorption and degradation of saponins, the two secondary saponins with a glucosyl linked at C-3, including GP and GPA, were used as the representative compounds in pharmacokinetic analysis.
The result of pharmacokinetic study showed that GP and GPA reached Cmax within 1 h after administration in dosage of high and low, which means the rapid absorption and acting in vivo. Although the contents of GP and GPA in the total saponins were 1.03% and 0.45% respectively, the main important pharmacokinetic parameters of GP were lower than GPA. The reason could be that the acidity of GPA is stronger than GP, and the absorption of acidic compounds in the stomach is better due to gastric acid (pH = 1.2).36 On the other hand, this explains why secondary saponins containing carboxyl group at C-28 were more easily absorbed into blood than primary saponins with saccharide chain at C-28.
Based on the qualitative and quantitative results of metabolic analyses of Jiegeng in vivo, the reasons of secondary saponins as the main absorbed components are summarized as follows:
(a) The poor membrane permeability of primary saponins causes difficult to cross a biological barrier by active transport; (b) the acidic secondary saponins are easily absorbed in acidic environment of the stomach; (c) the primary saponins are hydrolysis to produce secondary saponins under the action of gastric acid. Although GPA was proved to be the most important absorbed platycoside in vivo, the further studies about the effect comparison between GP and GPA will be made in the future.
Besides a few studies on pharmacokinetics of platycodin D and D3, there have been no reports about the pharmacokinetics of the absorbed platycosides after the oral administration of Jiegeng. The results of metabolite identification and pharmacokinetics of the absorbed saponins of Jiegeng in vivo will help researchers explore the mechanism of meridian-guiding effect in the subsequent pharmacological studies.
5. Conclusion
This paper developed qualitative and quantitative analyses of bioactive saponins of Jiegeng in vivo. Using the method based on the fragmentation behaviors of platycosides, a total of thirteen platycosides were identified, and seven among these were found in rat plasma after oral administration of total saponins by PCA. A simultaneous quantitative method of GP and GPA, the major absorbed platycosides in mice plasma, was established and validated by LC-QQQ. And the pharmacokinetic parameters of the two platycosides in vivo were revealed. This investigation provided helpful information to clear about bioactive platycosides of Jiegeng in vivo, and provided pharmacokinetic data support for subsequent pharmacological researches.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81374046, 81473403 and 81673616).Notes and references
- L. Zhang, Y. L. Wang, D. W. Yang, C. H. Zhang, N. Zhang, M. H. Li and Y. Z. Liu, J. Ethnopharmacol., 2015, 164, 147–161 CrossRef CAS PubMed.
- T. W. Kim, J. H. Lim, H. K. Lee, M. Giorgi, H. Owen and H. I. Yun, Med. Weter., 2014, 70, 38–41 Search PubMed.
- Y. Xie, H. Pan, H. Sun and D. Li, Vaccine, 2008, 26, 3937–3945 CrossRef CAS PubMed.
- C. H. Jeong, G. N. Choi, J. H. Kim, J. H. Kwak, D. O. Kim, Y. J. Kim and H. J. Heo, Food Chem., 2010, 118, 278–282 CrossRef CAS.
- X. J. Fu, H. B. Liu, P. Wang and H. S. Guan, Am. J. Chin. Med., 2009, 37, 967–975 CrossRef PubMed.
- S. Zheng, W. Li, J. Wang, Y. Chen, W. Hou, W. Gao, Q. Liu and Y. Wang, RSC Adv., 2016, 6, 10606–10614 RSC.
- L. J. Xiu, D. Z. Sun, J. P. Jiao, B. Yan, Z. F. Qin, X. Liu, P. K. Wei and X. Q. Yue, J. Ethnopharmacol., 2015, 172, 155–161 CrossRef PubMed.
- M. Khan, A. Maryam, H. Zhang, T. Mehmood and T. Ma, J. Cell. Mol. Med., 2016, 20, 389–402 CrossRef CAS PubMed.
- N. H. Yim, Y. H. Hwang, C. Liang and J. Y. Ma, J. Ethnopharmacol., 2016, 194, 1060–1068 CrossRef CAS PubMed.
- D. Y. Kwon, Y. S. Kim, S. Y. Ryu, Y. H. Choi, M. R. Cha, H. J. Yang and S. Park, Eur. J. Nutr., 2012, 51, 529–540 CrossRef CAS PubMed.
- H. M. Park, K. T. Park, E. C. Park, S. I. Kim, M. S. Choi, K. H. Liu and C. H. Lee, Nutrients, 2017, 9, e71 CrossRef PubMed.
- Y. P. Hwang, J. H. Choi, H. G. Kim, T. Khanal, G. Y. Song, M. S. Nam, H.-S. Lee, Y. C. Chung, Y. C. Lee and H. G. Jeong, Toxicol. Appl. Pharmacol., 2013, 267, 174–183 CrossRef CAS PubMed.
- J. H. Choi, S. W. Jin, H. G. Kim, T. Khanal, Y. P. Hwang, K. J. Lee, C. Y. Choi, Y. C. Chung, Y. C. Lee and H. G. Jeong, Food Chem. Toxicol., 2013, 56, 231–239 CrossRef CAS PubMed.
- W. Li, Y. Liu, Z. Wang, Y. Han, Y.-H. Tian, G.-S. Zhang, Y.-S. Sun and Y.-P. Wang, Food Funct., 2015, 6, 1418–1427 CAS.
- J. T. Wu, G. W. Yang, W. X. Zhu, W. J. Wen, F. M. Zhang, J. D. Yuan and L. G. An, Biol. Pharm. Bull., 2012, 35, 1216–1221 CAS.
- E. M. Twiner, Z. J. Liu, J. Gimble, Y. Yu and F. Greenway, Adv. Ther., 2011, 28, 857–865 CrossRef CAS PubMed.
- E. Nyakudya, J. H. Jeong, N. K. Lee and Y.-S. Jeong, Prev. Nutr. Food Sci., 2014, 19, 59–68 CrossRef CAS PubMed.
- L. F. Zeng, H. J. Kong, M. Zhu and W. D. Yan, J. Funct. Foods, 2016, 26, 48–56 CrossRef CAS.
- E. K. Jeong, I. J. Ha, Y. S. Kim and Y. C. Na, J. Sep. Sci., 2014, 37, 61–68 CrossRef CAS PubMed.
- J. W. Lee, S. H. Ji, G. S. Kim, K. S. Song, Y. Um, O. T. Kim, Y. Lee, C. P. Hong, D. H. Shin, C. K. Kim, S. E. Lee, Y. S. Ahn and D. Y. Lee, Int. J. Mol. Sci., 2015, 16, 26786–26796 CrossRef CAS PubMed.
- T. H. Kim, B. E. Lee, E. J. Kim, Y. S. Choi, K. S. Lee, H. R. Kim and H. G. Kim, Sci. World J., 2014, 10, DOI:10.1155/2014/231293.
- Q. Zhan, F. Zhang, S.-H. Gao, F. Cai, B. Jiang, L.-N. Sun and W.-S. Chen, Chin. J. Nat. Med., 2014, 12, 154–160 CAS.
- J. Shan, J. Zou, T. Xie, A. Kang, W. Zhou, H. Deng, Y. Mao, L. Di and S. Wang, Pharmacogn. Mag., 2015, 11, 750–755 CrossRef CAS PubMed.
- Y. W. Ha, Y.-C. Na, I. J. Ha, D.-H. Kim and Y. S. Kim, J. Pharm. Biomed. Anal., 2010, 51, 202–209 CrossRef CAS PubMed.
- L. Pei, Y. Bao, L. Ma, Q. Wang, Y. Ye, X. Han, S. Liu and X. Chen, Planta Med., 2012, 78, 244–251 CrossRef CAS PubMed.
- Y. C. Na, Y. W. Ha, Y. S. Kim and K. J. Kim, J. Chromatogr. A, 2008, 1189, 467–475 CrossRef CAS PubMed.
- L. J. Li, Z. H. Liu, Y. Chen and J. K. Tian, China J. Chin. Mater. Med., 2006, 31, 1506–1509 CAS.
- W. W. Fu, D. Q. Dou, W. B. Hou, B. H. Cheng, F. Liu, Y. J. Chen, Y. H. Pei and T. Takeda, China J. Chin. Mater. Med., 2005, 297–301 CAS.
- L. Zhang, L. Zhu, Y. F. Wang, Z. Z. Jiang, X. Chai, Y. Zhu, X. M. Gao and A. D. Qi, J. Pharm. Biomed. Anal., 2012, 62, 203–209 CrossRef CAS PubMed.
- J. S. Yu, C. H. Ho, Y. C. Hsu, J. J. Wang and C. L. Hsieh, J. Integr. Med., 2014, 6, 538–544 CrossRef.
- W. W. Tao, Q. Su, H. Q. Wang, S. Guo, Y. Y. Chen, J. N. Duan and S. M. Wang, Int. Immunopharmacol., 2015, 27, 138–147 CrossRef CAS PubMed.
- X. X. Dai, H. O. Ding, Q. Q. Yin, G. Wan, X. Y. Shi and Y. J. Qiao, J. Mol. Graphics Modell., 2015, 57, 20–26 CrossRef CAS PubMed.
- J. M. Augustin, V. Kuzina, S. B. Andersen and S. Bak, Phytochemistry, 2011, 72, 435–457 CrossRef CAS PubMed.
- J. H. Lorent, J. Quetin-Leclercq and M. P. Mingeot-Leclercq, Org. Biomol. Chem., 2014, 12, 8803–8822 CAS.
- W. Zhang, S. H. Qian, D. W. Qian and S. L. Li, Am. J. Chin. Med., 2016, 44, 817–833 CrossRef CAS PubMed.
- Z. Liu, X. Zheng, Y. Guo, W. Qin, L. Hua and Y. Yang, Fitoterapia, 2016, 113, 27–34 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S6 and Fig. S1–S9. See DOI: 10.1039/c7ra04814a |
| This journal is © The Royal Society of Chemistry 2017 |