
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New prenylated coumarins from the stems of Toddalia asiatica†
Wei Lia,
Jun-Sheng Zhanga,
Jia-Luo Huanga,
Min-Hong Jianga,
You-Kai Xub,
Abrar Ahmeda,
Sheng Yina and
Gui-Hua Tang *a
*a
aSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510006, China. E-mail: tanggh5@mail.sysu.edu.cn; Fax: +86-20-39943043; Tel: +86-20-39943043
bKey Laboratory of Tropical Plant Resource and Sustainable Use, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Menglun, Yunnan 666303, China
First published on 15th June 2017
Abstract
Eight new prenylated coumarins (1a/1b, 2a/2b, and 3–6) including two pairs of enantiomers (1a/1b and 2a/2b), a new phenolic acid derivative, methyl (E)-3,4-bis(4-hydroxyphenyl)-4-oxobut-2-enoate (7), and 33 known compounds (8–40) were isolated from the stems of Toddalia asiatica. Their structures were established from spectroscopic data and by chemical methods. The absolute configurations of two pairs of enantiomers (1a/1b and 2a/2b) were determined by X-ray diffraction analysis together with ECD and specific optical rotation calculations. The inhibitory effects of selected compounds against phosphodiesterase-4 (PDE4) were evaluated, and compounds 12, 19, 21–23, 26, 34, and 35 exhibited PDE4 inhibition activities with IC50 values less than 10 μM.
Introduction
Toddalia is a monotypic genus of the Rutaceae family containing the single species Toddalia asiatica (L.) Lam., which is a woody climber widely distributed in Africa, E, S, and SE Asia, Madagascar, and the Mascarene Islands.1 In China, T. asiatica, well known as “Feilongzhangxue” (Chinese name), is a Traditional Chinese Medicine (TCM), whose roots and barks have been widely used for the treatment of many diseases including traumatic injury, rheumatism, stomach ache, dysmenorrhea, and pyogenic infections.2 Previous phytochemistry investigations on the plants of T. asiatica have revealed the occurrence of the main chemical components including coumarins, alkaloids, triterpenoids, phenolic acids, flavonoids, and lignans.3–5 These chemical components were found to have a wide range of bioactivities such as anti-inflammatory, anti-leukemic, antiplatelet aggregation, antibacterial, antifungal, antimalarial, and cytotoxic activities.4,6–11Recently, our group have found a series of prenylated coumarins including an unusual group of phenylpropenoic acid-coupled prenylated coumarins with potent phosphodiesterase-4 (PDE4) inhibitory activities from the roots of T. asiatica.11 In the course of our ongoing work for natural PDE4 inhibitors from this plant, the EtOH extract of the stems of T. asiatica were subjected to chromatographic procedures to yield eight new prenylated coumarins (1a/1b, 2a/2b, and 3–6) (Fig. 1) including two pairs of enantiomers (1a/1b and 2a/2b), a new phenolic acid derivative (7) (Fig. 1), and 33 known compounds (8–40) (Fig. S1.1, see ESI†) including seven alkaloids (34–40). This paper focuses on the isolation, structural elucidation, and PDE4 inhibitory activities of these isolated compounds.
 | ||
| Fig. 1 Structures of new compounds 1–7 from T. asiatica. | ||
Results and discussion
Compound 1 was obtained as a colorless crystal, which had the molecular formula C16H16O6 with nine degrees of unsaturation as determined by the HRESIMS and 1D NMR data. The characteristic absorption bands at νmax 1727, 1600, 1500, and 1466 cm−1 in the IR spectrum of 1 suggested the presence of ester and aromatic moieties. The 1D NMR spectra of 1 displayed signals for a 5,7-dimethoxy-8-substituted coumarin unit [δH 7.94 (1H, d, J = 9.7 Hz, H-4), 6.29 (1H, s, H-6), 6.11 (1H, d, J = 9.7 Hz, H-3), 3.92 (3H, s, 5-OCH3), and 3.91 (3H, s, 7-OCH3); δC 161.4 (C, C-7), 160.6 (C, C-2), 157.4 (C, C-5), 154.9 (C, C-9), 138.8 (CH, C-4), 111.1 (CH, C-3), 107.0 (C, C-8), 103.9 (C, C-10), 90.4 (CH, C-6), 56.4 (CH3, 7-OCH3), and 56.1 (CH3, 5-OCH3)] as in co-isolated compounds 8–11,12–16 which was further confirmed by the HMBC correlations (Fig. 2). The remaining resonances in the 1D NMR data of 1 (Table 1) were consistent with one singlet methyl group, an oxygenated methylene, two oxygenated methines, and an oxygenated quaternary carbon, indicating the presence of a highly-oxidized isopentenyl derivative moiety. The structure of this highly-oxidized moiety was deduced to be 5-methyl-3,6-dioxabicyclo[3.1.0]hexan-2-yl by analysis of its 2D NMR data including 1H–1H COSY and HMBC correlations (Fig. 2) and the remaining two degrees of unsaturation (seven of nine degrees of unsaturation occupied by the coumarin unit). The HMBC correlations of the proton (δH 5.68, 1H, br. s) of the oxygenated CH-1′ with C-7, C-8, and C-9 confirmed that the moiety 5-methyl-3,6-dioxabicyclo[3.1.0]hexan-2-yl was linked at C-8 of the coumarin unit. | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of 1 and 2. | ||
| Position | (±)-1a | (±)-2b | 3a | 4a | ||||
|---|---|---|---|---|---|---|---|---|
| δH, multi. (J in Hz) | δC, type | δH, multi. (J in Hz) | δC, type | δH, multi. (J in Hz) | δC, type | δH, multi. (J in Hz) | δC, type | |
| a In CDCl3.b In CD3OD. | ||||||||
| 2 | 160.6, C | 162.8, C | 160.5, C | 161.2, C | ||||
| 3 | 6.11, d (9.7) | 111.1, CH | 6.14, d (9.7) | 111.1, CH | 6.30, d (9.7) | 113.7, CH | 6.24, d (9.6) | 112.8, CH |
| 4 | 7.94, d (9.7) | 138.8, CH | 8.09, d (9.7) | 140.8, CH | 7.86, d (9.7) | 138.5, CH | 7.90, d (9.6) | 139.0, CH |
| 5 | 157.4, C | 158.8, C | 155.4, C | 155.6, C | ||||
| 6 | 6.29, s | 90.4, CH | 6.60, s | 92.5, CH | 119.7, C | 116.2, C | ||
| 7 | 161.7, C | 164.2, C | 161.0, C | 161.6, C | ||||
| 8 | 107.0, C | 108.6, C | 6.70, s | 96.7, CH | 6.62, s | 95.6, CH | ||
| 9 | 154.9, C | 155.9, C | 156.3, C | 154.9, C | ||||
| 10 | 103.9, C | 104.9, C | 107.7, C | 107.6, C | ||||
| 1′ | 5.68, br. s | 71.8, CH | 5.47, d (8.5) | 76.8, CH | 6.01, dd (10.4, 7.6) | 64.4, CH | 6.57, br. s | 118.2, CH |
| 2′ | 3.58, br. s | 65.9, CH | 4.45, d (8.5) | 79.8, CH | 6.85, dd (7.6, 1.4) | 153.1, CH | 6.57, br. s | 140.8, CH |
| 3′ | 68.5, C | 78.0, C | 138.9, C | 75.8, C | ||||
| 4′ | 4.18, d (9.3), 3.92, d (9.3) | 71.9, CH2 | 4.20, d (9.2), 3.87, d (9.2) | 79.0, CH2 | 9.44, s | 195.0, CH | 1.38, s | 26.0, CH3 |
| 5′ | 1.75, s | 13.7, CH3 | 1.38, s | 21.9, CH3 | 1.78, d (1.4) | 9.5, CH3 | 1.38, s | 26.0, CH3 |
| 5-OMe | 3.92, s | 56.1, CH3 | 3.99, s | 56.8, CH3 | 3.94, s | 64.6, CH3 | 3.78, s | 62.1, CH3 |
| 7-OMe | 3.91, s | 56.4, CH3 | 3.97, s | 57.0, CH3 | 3.98, s | 56.7, CH3 | 3.89, s | 56.3, CH3 |
| 3′-OMe | 3.24, s | 50.7, CH3 | ||||||
| 1′-OH | 3.29, d (10.4) | |||||||
The small coupling constant of H-1′ (br. s) and H-2′ (br. s) in the 1H NMR spectrum suggested that the dihedral angle between H-1′ and H-2′ was approximately 90°, and arbitrarily assigned H-1′ and H-2′ as β-axial and α-equitorial orientations, respectively.17 The NOE correlation of H-2′/H3-5′ in the NOESY spectrum indicated the same α-orientation of CH3-5′ as H-2′. The single-crystal X-ray crystallographic analysis of 1 (Fig. 3) was performed with a high Flack parameter of 0.42 (19), which showed that 1 were a pair of enantiomers. So, the X-ray result only confirmed the relative configuration of 1 as the same as that deduced by the coupling constant and NOESY experiment.
 | ||
| Fig. 3 Single-crystal X-ray structures of 1 (left: 1a; right: 1b). | ||
Subsequently, compound 1 was subjected to HPLC with a chiral column to obtain the enantiomers 1a and 1b, which had opposite specific rotations ([α]20D = +61.0 for 1a and [α]20D = −61.0 for 1b) and mirror image-like ECD curves (Fig. 4). Unfortunately, no crystals of any of this pair of enantiomers were obtained. To determine the absolute configurations of the enantiomers, the experimental ECD spectra of 1a and 1b were compared with the calculated ECD spectra of (1′S,2′S,3′R)-1 or (1′R,2′R,3′S)-1 by the TDDFT method.
 | ||
| Fig. 4 Experimental ECD spectra of 1a and 1b and calculated ECD spectra (red shifted by 15 nm) of (1′S,2′S,3′R)-1 and (1′R,2′R,3′S)-1. | ||
In Fig. 4, the experimental ECD spectrum of 1a showed an ECD curve with three positive Cotton effects around 316 (+3.01), 261 (+1.40), and 211 (+16.7) nm, which matched the calculated ECD curve for (1′S,2′S,3′R)-1, indicating that 1a possessed the same absolute configuration as (1′S,2′S,3′R)-1. The absolute configuration of 1b was deduced to be the same as that of (1′R,2′R,3′S)-1 on the basis of their matched ECD spectra. Thus, the enantiomers 1a and 1b were determined as shown and named (+)-toddalin E and (−)-toddalin E, respectively.
The molecular formula of 2 was determined as C16H18O7 with 18 mass units more than that of 1 by its HRESIMS and 1D NMR data. Comparison of the 1H and 13C NMR data demonstrated that 2 displayed closely similarity with 1 (Table 1). The obvious differences were the chemical shifts of the C-8 substituent moiety [δC 76.8 (CH, C-1′), 79.8 (CH, C-2′), 78.0 (C, C-3′), 78.9 (CH2, C-4′), and 21.9 (CH3, C-5′) in 2; δC 71.8 (CH, C-1′), 65.9 (CH, C-2′), 68.5 (C, C-3′), 71.9 (CH2, C-4′), and 13.7 (CH3, C-5′) in 1], which may be due to the open loop of the epoxy ring. The molecular formula of 2 with 18 mass units more than that of 1 and one degree of unsaturation less than that of 1 further confirmed the above proposed. Analysis of the 2D NMR data (Fig. 2) determined the gross structure of 2. The NOE cross-peaks of H-2′/H3-5′ and H-4′α indicated that the methyl group, H-2′, and H-4′α were cofacial, and were arbitrarily assigned α-orientation. The large 1H–1H coupling constant (J1′,2′ = 8.5 Hz) suggested the trans-relationship of H-1′ and H-2′.18 Hence, the relative configuration of 2 was assigned to be the same as that of 1. Compound 2 was also a racemic mixture because of its specific rotation approaching zero and no Cotton effect in its ECD spectrum. The enantiomers 2a and 2b, with opposite specific rotations ([α]20D = +116.2 for 2a and [α]20D = −116.2 for 2b) and mirror image-like ECD curves (Fig. 5), were separated by HPLC with a chiral column. The absolute configurations of 2a and 2b were determined as 1′R,2′R,3′S and 1′S,2′S,3′R, respectively, by using the same methods as described above (Fig. 5). Thus, the enantiomers 2a and 2b were given trivial names (+)-toddalin F and (−)-toddalin F, respectively.
 | ||
| Fig. 5 Experimental ECD spectra of 2a and 2b and calculated ECD spectra (bule shifted by 30 nm) of (1′S,2′S,3′R)-2 and (1′R,2′R,3′S)-2. | ||
As one may noticed, 1a and 2a both showed positive specific optical rotation, while their absolute configurations were opposite. In order to confirm the above assignments, calculations of the specific optical rotations for (1′S,2′S,3′R)-1 and (1′S,2′S,3′R)-2 were further carried out. The results showed that (1′S,2′S,3′R)-1 exhibited a theoretical specific optical rotation of +95 (experimental value for 1a: +61) while (1′S,2′S,3′R)-2 exhibited a theoretical specific optical rotation of −117 (experimental value for 2b: −116). The results of specific optical rotation calculations were in accordance with those of ECD calculations.
Compound 3 exhibited a pseudomolecular ion peak at m/z 305.1582 [M + H]+, allowing the molecular formula C16H16O6 to be assigned. Analysis the 1D and 2D NMR data suggested that 3 had a 5,7-dimethoxy-6-substituted coumarin unit as in co-isolated compounds 12–18.11,13,19,20 The remaining signals in the 1D NMR spectra of 3 (Table 1) were consistent with an OH group [δH 3.29 (1H, d, J = 10.4 Hz)], a conjugated aldehyde group [δH 9.44 (1H, s); δC 195.0 (CH)], a trisubstituted double bond [δH 6.85 (1H, dd, J = 7.6 and 1.4 Hz); δC 153.1 (CH) and 138.9 (C)], an oxygenated methine [δH 6.01 (1H, dd, J = 10.4 and 7.6 Hz); δC 64.4 (CH)], and a methyl group [δH 1.78 (1H, d, J = 1.4 Hz); δC 9.5 (CH3)]. According to the 1H–1H COSY correlations of H-1′/H-2′ and 1′-OH and the key HMBC correlations of H-5′ to C-2′, C-3′, and C-4′, H-4′ to C-2′ and C-5′, H-2′ to C-4′ and C-5′, 1′-OH to C-6, and H-1′ to C-5 and C-7, the 6-substituent moiety was determined as shown (Fig. 6). The E-configuration of the double bond between C-2′ and C-3′ was deduced by the NOE correlation of H-4′/H-2′. This compound might be a racemoid because its specific rotation was zero. Thus, compound 3 was given a trivial name (±)-toddalin G.
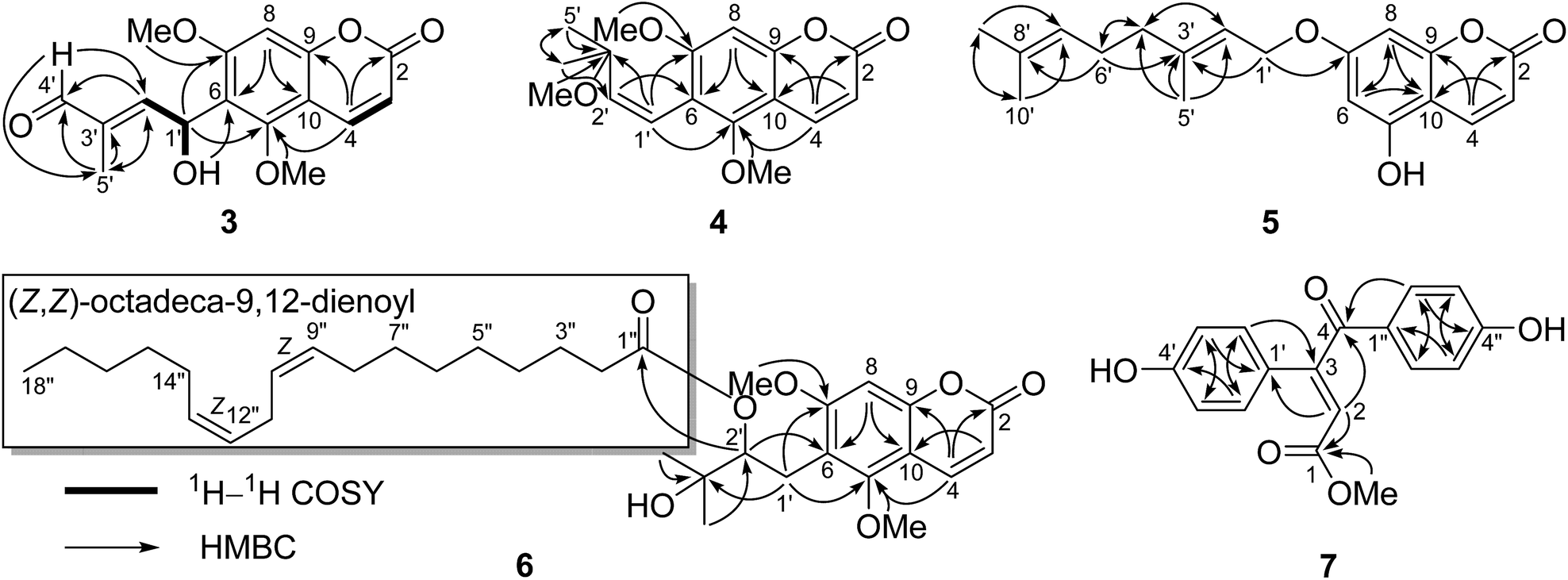 | ||
| Fig. 6 Key 1H–1H COSY and HMBC correlations of compounds 3–7. | ||
Toddalin H (4) was assigned the molecular formula C17H20O5 by its HRESIMS and 1D NMR data. The 1H and 13C NMR spectra of 4 were very similar to those of toddalenol (15),13 except for the presence of an additional methoxy group instead of a hydroxy group at C-3′ and the different configuration of Δ1′. The protons at δH 6.57 (2H, br. s) assigned to carbons C-1′ and C-2′ suggested the double bond between C-1′ and C-2′ was Z-configuration. Analysis of the 2D NMR data of 4 (Fig. 6) further confirmed the structure of 4.
Compound 5 possessed the molecular formula C19H22O4 as determined by its HRESIMS and 1D NMR data. The 1H and 13C NMR data of 5 bore a high resemblance to those of 7-geranyloxy-5-methoxycoumarin14 with the only difference being the absence of the methoxy group. This indicated that 5 was a demethoxy derivative of the known one, which was confirmed by its 2D NMR and MS data. Thus, compound 5 was named as 7-geranyloxy-5-hydroxycoumarin.
The molecular formula of compound 6 was established as C34H50O7 on the basis of its HRESIMS and 1D NMR data. Comparison of the 1H and 13C NMR spectra of 6 with those of ent-toddalolactone (16)11 showed the structural features of a prenylated coumarin identical to 16, except for the presence of an additional fatty acid chain. The signals for the fatty acid chain in the 1D NMR spectra of 6 were almost identical to those of linoleic acid (9Z,12Z-octadecadieneoic acid),21 which was linked to C-2′ by the HMBC correlation of H-2′ to the ester carbonyl (δC 173.2) (Fig. 6). The absolute configuration of 6 was confirmed by analysis of the specific optical rotation of the prenylated coumarin, an alkaline hydrolysis product of 6, which showed a specific optical rotation of −64.0 (−69.0 for ent-toddalolactone). Thus, compound 6 was given a trivial name 2′-O-((Z,Z)-octadeca-9,12-dienoyl)-ent-toddalolactone.
The HRESIMS of 7 exhibited a molecular ion peak at m/z 299.0914 [M + H]+ (calcd 299.0914), corresponding to the molecular formula C17H14O5 with 11 degrees of unsaturation. The 1H NMR spectrum of 7 showed the signals for two p-hydroxyphenyl groups [δH 7.97 (2H, d, J = 8.8 Hz), 7.50 (2H, d, J = 8.8 Hz), 6.87 (2H, d, J = 8.8 Hz), and 6.85 (2H, d, J = 8.8 Hz)], one olefinic proton [δH 7.48 (1H, s)], and a methoxy group [δH 3.91 (3H, s)]. The 13C NMR spectrum, associated with DEPT experiments, resolved 17 carbon resonances attributable to two p-hydroxyphenyls [δC 126.1 (C, C-1′), 130.0 (CH × 2, C-2′ and C-6′), 117.0 (CH × 2, C-3′ and C-5′), 161.8 (C, C-4′), 130.3 (C, C-1′′), 132.3 (CH × 2, C-2′′ and C-6′′), 116.7 (CH × 2, C-3′′ and C-5′′), and 164.9 (C, C-4′′)], a conjugated ketone carbonyl [δC 188.9 (C, C-4)], an ester carbonyl [δC 172.1 (C, C-1)], one trisubstituted double bond [δC 118.9 (CH, C-2) and 148.8 (C, C-3)], and one methoxy [δC 53.0 (CH3, 1-OMe)], which were assigned by the HSQC and HMBC correlations. The HMBC correlations of H-2 to C-1, C-4, and C-1′, H-2′ (6′) to C-3, and H-2′′ (6′′) to C-4 connected the two p-hydroxyphenyls, the conjugated ketone carbonyl, the double bond, and the ester carbonyl to construct the structure of 7 as shown (Fig. 6). The location of the methoxy group at C-1 was deduced by the HMBC correlation of protons at δH 3.91 to the ester carbonyl. The E-configuration of the double bond Δ2 was determined by the NOE correlation of H-2′′ (6′′)/H-2. Thus, compound 7 was elucidated to be methyl (E)-3,4-bis(4-hydroxyphenyl)-4-oxobut-2-enoate.
The known compounds, gleinadiene (8),12 toddalenone (9),13 8-geranyloxy-5,7-dimethoxycoumarin (10),14 5,7,8-trimethoxycoumarin (11),14–16 toddaculin (12),13 6-(3-methyl-1,3-butadienyl)-5,7-dimethoxycoumarin (13),19 toddanol (14),13 toddalenol (15),13 ent-toddalolactone (16),11 (−)-toddalolactone 3′-O-β-D-glucopyranoside (17),11 6-formyllimettin (18),20 8-(3,3-dimethylallyl)-6,7-dimethoxycoumarin (19),13 6-(3-methyl-2-butenyl)-7-hydroxy-5-methoxycoumarin (20),22 6-geranyloxy-7-methoxycoumarin (21),23 norbraylin (22),24 braylin (23),24 toddalins A–C (24–26),11 toddalosin (27),13,25 5-O-(E)-feruloylquinic acid methyl ester (28),26 hycandinic acid ester-1 (29),27 4-O-(E)-feruloylquinic acid methyl ester (30),28,29 trans-p-coumaryl aldehyde (31),30 ferulaldehyde (32),31 trans-sinapaldehyde (33),32 8-acetonyldihydronitidine (34),33 8-acetonyldihydrochelerythrine (35),34 decarine (36),34 4-methoxy-N-methyl-2-quinolone (37),35 γ-fagarine (38),35 haplopine (39),36 and skimmianine (40),36 were identified by comparison of their spectroscopic data with those reported in the literature. The 1H and 13C NMR data of all known compounds are provided in Tables S2.1–S2.10 (see ESI†).
The inhibitory activities of the isolates at an initial concentration of 10 μM against PDE4D2 were screened by using tritium-labelled adenosine 3′,5′-cyclic monophosphate ([3H]-cAMP) as substrate (S3, see ESI†). The IC50 values of the active compounds with inhibition greater than 50% at 10 μM were listed in Table 2. Rolipram, a well-known PDE4 inhibitor, was used as positive control (IC50 = 0.62 μM). As show in Table 2, compounds 12, 19, 21–23, 26, 34, and 35 exhibited moderate activities with IC50 values less than 10 μM toward PDE4D.
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| a Compounds with IC50 > 50 μM were not listed.b Data collected from our previous work.11c Positive control. | |||
| 12 | 9.98 ± 0.63b | 25 | 16.65 ± 1.20b |
| 19 | 6.64 ± 0.02 | 26 | 7.81 ± 0.40b |
| 21 | 3.85 ± 0.34 | 34 | 5.14 ± 0.13 |
| 22 | 2.38 ± 0.14b | 35 | 3.80 ± 0.18 |
| 23 | 0.96 ± 0.10b | Rolipramc | 0.62 ± 0.03 |
Experimental section
General experimental procedures
X-ray data were collected using an Agilent Xcalibur Nova X-ray diffractometer. Melting points were recorded on an X-4 melting instrument and were uncorrected. Optical rotations were detected on a Rudolph Autopol I automatic polarimeter, UV spectra on a Shimadzu UV-2450 spectrophotometer, ECD spectra on an Applied Photophysics Chirascan spectrometer, and IR spectra on PerkinElmer FT-IR C106150 and Bruker Tensor 37 infrared spectrophotometers. 1D and 2D NMR spectra were measured on Bruker AM-400 spectrometers at 25 °C. ESIMS was measured on a Finnigan LCQ Deca instrument, and HRESIMS was performed on a Waters Micromass Q-TOF spectrometer. Silica gel (100–200 and 300–400 mesh, Qingdao Haiyang Chemical Co., Ltd.), Sephadex LH-20 gel (Amersham Biosciences), reversed-phase C18 (RP-C18) (12 nm, S-50 μm, YMC Co., Ltd.), and MCI gel (CHP20P, 75–150 μm, Mitsubishi Chemical Industries Ltd.) were used for column chromatography (CC). A Shimadzu LC-20 AT equipped with a SPD-M20A PDA detector was used for HPLC. An YMC-pack ODS-A column (250 × 10 mm, S-5 μm, 12 nm) and a chiral column (Phenomenex Lux, cellulose-2, 250 × 10 mm, 5 μm) were used for semi-preparative HPLC separation. All solvents used were of analytical grade (Guangzhou Chemical Reagents Company, Ltd.). TLC spots were visualized under UV light and by dipping into 5% H2SO4 in EtOH followed by heating.Plant material
The stems of T. asiatica were collected from Xishuangbanna, Yunnan Province, P. R. China, in October 2012. This material was authenticated by one of the author (Y.-K. Xu) and a voucher specimen (FLZX201211) has been deposited at the School of Pharmaceutical Sciences, Sun Yat-sen University.Extraction and isolation
The air-dried stems of T. asiatica (3.4 kg) were extracted with 95% EtOH (10 L × 3) at rt for three times. After evaporating the solvent, the residue (390 g) was suspended in H2O (2 L) and extracted with EtOAc (2 L × 3) and n-BuOH (2 L × 3), respectively. The EtOAc partition (120 g) was chromatographed over silica gel CC (PE/EtOAc, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 1:1) to give seven sub-fractions (I–VI). Each fraction was subjected to CC over RP-C18 column, silica gel, and Sephadex LH-20 and then further purified by semipreparative HPLC with a YMC-pack ODS-A column or a Phenomenex Lux chiral column to yield pure compounds. Fr. I yielded compound 12 (30 mg). Compounds 1a (2 mg), 1b (2 mg), 5 (9 mg), 10 (24 mg), 19 (315 mg), 20 (5 mg), and 21 (57 mg) were obtained from Fr. II. Fr. III afforded compounds 6 (29 mg), 8 (23 mg), 9 (10 mg), 22 (100 mg), 23 (16 mg), 31 (80 mg), 32 (10 mg), 33 (9 mg), and 36 (27 mg). Fr. IV gave compounds 4 (13 mg), 11 (3 mg), 13 (2 mg), 14 (95 mg), 18 (2 mg), 24 (22 mg), 26 (36 mg), and 37 (8 mg). Compounds 34 (52 mg), 35 (18 mg), 38 (14 mg), 39 (18 mg), and 40 (21 mg) were obtained from Fr. V. Fr. VI afforded compounds 2a (1 mg), 2b (2 mg), 3 (4 mg), 7 (2 mg), 15 (13 mg), 16 (83 mg), 17 (63 mg), 25 (28 mg), 27 (9 mg), 28 (32 mg), 29 (14 mg), and 30 (5 mg). The details on isolation of these compounds are provided in ESI.† Purity of the compounds was tested by using TLC and NMR spectra.
ε) 323 (3.71), 259 (3.52), 251 (3.52), 213 (3.96) nm; ECD (c 3.29 × 10−4 M, MeCN) λmax (Δε) 316 (+3.01), 261 (+1.40), 211 (+16.7) nm; IR (KBr) νmax 1727, 1600, 1500, 1466, 1334, 1253, 1210, 1115 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 327.1 [M + Na]+; HRESIMS m/z 303.0867 [M − H]− (calcd for C16H15O6−, 303.0874).323 (3.60), 260 (3.43), 212 (3.91) nm; ECD (c 3.11 × 10−4 M, MeCN) λmax (Δε) 331 (+0.21), 257 (+6.69), 232 (+3.96), 208 (+7.98) nm; IR (KBr) νmax 3426, 1719, 1604, 1504, 1472, 1434, 1335, 1259, 1222, 1142, 1118 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 323.2 [M + H]+; HRESIMS m/z 345.0961 [M + Na]+ (calcd for C16H18O7Na+, 345.0945).ε) 323 (4.05), 225 (4.24), 206 (4.26) nm; IR (KBr) νmax 3359, 1722, 1654, 1602, 1385, 1353, 1205, 1139 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 305.0 [M + H]+; HRESIMS m/z 303.0851 [M − H]− (calcd for C16H15O6−, 303.0874).ε) 336 (3.54), 313 (3.53), 261 (3.95), 213 (3.78) nm; IR (KBr) νmax 1725, 1597, 1563, 1451, 1419, 1376, 1360, 1203, 1124, 1088, 1070 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 287.1 [M − H2O + H]+, 305.0 [M + H]+; HRESIMS m/z 327.1195 [M + Na]+ (calcd for C17H20O5Na+, 327.1203).ε) 331 (3.52), 210 (3.82) nm; IR (KBr) νmax 3420, 2966, 2927, 1694, 1654, 1611, 1568, 1458, 1428, 1372, 1137 cm−1; 1H NMR (CDCl3, 400 MHz) δH 6.16 (1H, d, J = 9.6 Hz, H-3), 8.01 (1H, d, J = 9.6 Hz, H-4), 6.41 (1H, s, H-6), 6.31 (1H, s, H-8), 4.55 (2H, d, J = 6.6 Hz, H-1′), 5.43 (1H, br. t, J = 6.8 Hz, H-2′), 2.09 (2H, m, H-4′), 1.73 (3H, s, H-5′), 2.11 (2H, m, H-6′), 5.07 (1H, br. t, J = 6.8 Hz, H-7′), 1.66 (3H, s, H-9′), 1.60 (3H, s, H-10′); 13C NMR (CDCl3, 100 MHz) δC 162.2 (C, C-2), 110.7 (CH, C-3), 139.4 (CH, C-4), 156.9 (C, C-5), 94.4 (CH, C-6), 162.9 (C, C-7), 99.0 (CH, C-8), 154.2 (C, C-9), 103.5 (C, C-10), 65.6 (CH2, C-1′), 118.6 (CH, C-2′), 142.5 (C, C-3′), 39.7 (CH2, C-4′), 16.9 (CH3, C-5′), 26.4 (CH2, C-6′), 123.8 (CH, C-7′), 132.1 (C, C-8′), 25.8 (CH3, C-9′), 17.8 (CH3, C-10′); ESIMS m/z 315.2 [M + H]+; HRESIMS m/z 313.1439 [M − H]− (calcd for C19H21O4−, 313.1445).ε) 329 (3.87), 225 (4.03), 210 (4.20) nm; IR (KBr) νmax 3459, 2929, 2855, 1736, 1610, 1565, 1462, 1382, 1205, 1134 cm−1; 1H NMR (CDCl3, 400 MHz) δH 6.23 (1H, d, J = 9.6 Hz, H-1), 7.82 (1H, d, J = 9.6 Hz, H-2), 6.59 (1H, s, H-8), 3.07 (1H, dd, J = 13.6 and 10.3 Hz, H-1′a), 2.90 (1H, dd, J = 13.6 and 3.1 Hz, H-1′b), 5.20 (1H, dd, J = 10.3 and 3.1 Hz, H-2′), 1.29 (3H, s, H-4′), 1.25 (3H, s, H-5′), 3.85 (3H, s, 5-OMe), 3.89 (3H, s, 7-OMe), 2.11 (1H, m, H-1′′′a), 2.03 (1H, m, H-1′′′b), 1.36 (2H, m, H-2′′′), 1.08 (2H, m, H-3′′′), 1.19 (2H, m, H-3′′′), 1.28 (10H, m, H-6′′′, H-7′′′, H-15′′′, H-16′′′, and H-17′′′), 2.02 (4H, m, H-8′′′ and H-14′′′), 5.32 (2H, m, H-9′′′ and H-13′′′), 5.30 (2H, m, H-10′′′ and H-12′′′), 2.76 (2H, t, J = 6.4 Hz, H-11′′′), 0.88 (3H, t, J = 6.0 Hz, H-18′′′); 13C NMR (CDCl3, 100 MHz) δC 161.1 (C, C-2), 112.7 (CH, C-3), 138.8 (CH, C-4), 156.3 (C, C-5), 116.8 (C, C-6), 162.1 (C, C-7), 95.6 (CH, C-8), 155.4 (C, C-9), 107.1 (C, C-10), 23.9 (CH2, C-1′), 77.9 (CH, C-2′), 72.6 (C, C-3′), 25.4 (CH3, C-4′), 26.9 (CH3, C-5′), 63.3 (CH3, 5-OMe), 56.3 (CH3, 7-OMe), 173.2 (C, C-1′′′), 34.3 (CH2, C-2′′′), 25.0 (CH2, C-3′′′), 29.17, 29.18 (CH2, C-4′′′ and 5′′′), 29.3 (CH2, C-6′′′), 29.7 (CH2, C-7′′′), 27.30, 27.34 (CH2, C-8′′′ and 14′′′), 130.4 (CH, C-9′′′), 128.1 (CH, C-10′′′), 25.8 (CH2, C-11′′′), 128.2 (CH, C-12′′′), 130.2 (CH, C-13′′′), 29.5 (CH2, C-15′′′), 31.7 (CH2, C-16′′′), 22.7 (CH, C-17′′′), 14.2 (CH3, C-18′′′); ESIMS m/z 571.4 [M + H]+; HRESIMS m/z 593.3463 [M + Na]+ (calcd for C34H50O7Na+, 593.3449).ε) 349 (4.22), 232 (4.00), 206 (4.07), 194 (3.85) nm; IR (KBr) νmax 3356, 1695, 1638, 1601, 1587, 1563, 1515, 1442, 1371, 1221, 1169 cm−1; 1H NMR (CDCl3, 400 MHz) δH 7.48 (1H, s, H-2), 7.50 (2H, d, J = 8.8 Hz, H-2′ and H-6′), 6.85 (2H, d, J = 8.8 Hz, H-3′ and H-5′), 7.97 (2H, d, J = 8.8 Hz, H-2′′ and H-6′′), 6.87 (2H, d, J = 8.8 Hz, H-3′′ and H-5′′), 3.91 (3H, s, 1-OMe); 13C NMR (CDCl3, 100 MHz) δC 172.1 (C, C-1), 118.9 (CH, C-2), 148.8 (C, C-3), 188.9 (C, C-4), 126.1 (C, C-1′), 130.0 (CH × 2, C-2′ and C-6′), 117.0 (CH × 2, C-3′ and C-5′), 161.8 (C, C-4′), 130.3 (C, C-1′′), 132.3 (CH × 2, C-2′′ and C-6′′), 116.7 (CH × 2, C-3′′ and C-5′′), 164.9 (C, C-4′′), 53.0 (CH3, 1-OMe); ESIMS m/z 299.1 [M + H]+; HRESIMS m/z 299.0914 [M + H]+ (calcd for C17H15O5+, 299.0914).805 reflections measured (4.46° ≤ 2θ ≤ 144.7°), 5326 unique (Rint = 0.0276, Rsigma = 0.0180) which were used in all calculations. The final R1 was 0.0366 (I ≥ 2σ (I)) and wR2 was 0.0979 (all data). Flack parameter = 0.42 (19). Crystallographic data for (1) has been deposited in the Cambridge Crystallographic Data Centre (CCDC number: 1545037†).
:1 → 1:1) to give seven sub-fractions (I–VI). Each fraction was subjected to CC over RP-C18 column, silica gel, and Sephadex LH-20 and then further purified by semipreparative HPLC with a YMC-pack ODS-A column or a Phenomenex Lux chiral column to yield pure compounds. Fr. I yielded compound 12 (30 mg). Compounds 1a (2 mg), 1b (2 mg), 5 (9 mg), 10 (24 mg), 19 (315 mg), 20 (5 mg), and 21 (57 mg) were obtained from Fr. II. Fr. III afforded compounds 6 (29 mg), 8 (23 mg), 9 (10 mg), 22 (100 mg), 23 (16 mg), 31 (80 mg), 32 (10 mg), 33 (9 mg), and 36 (27 mg). Fr. IV gave compounds 4 (13 mg), 11 (3 mg), 13 (2 mg), 14 (95 mg), 18 (2 mg), 24 (22 mg), 26 (36 mg), and 37 (8 mg). Compounds 34 (52 mg), 35 (18 mg), 38 (14 mg), 39 (18 mg), and 40 (21 mg) were obtained from Fr. V. Fr. VI afforded compounds 2a (1 mg), 2b (2 mg), 3 (4 mg), 7 (2 mg), 15 (13 mg), 16 (83 mg), 17 (63 mg), 25 (28 mg), 27 (9 mg), 28 (32 mg), 29 (14 mg), and 30 (5 mg). The details on isolation of these compounds are provided in ESI.† Purity of the compounds was tested by using TLC and NMR spectra.
ε) 323 (3.71), 259 (3.52), 251 (3.52), 213 (3.96) nm; ECD (c 3.29 × 10−4 M, MeCN) λmax (Δε) 316 (+3.01), 261 (+1.40), 211 (+16.7) nm; IR (KBr) νmax 1727, 1600, 1500, 1466, 1334, 1253, 1210, 1115 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 327.1 [M + Na]+; HRESIMS m/z 303.0867 [M − H]− (calcd for C16H15O6−, 303.0874).323 (3.60), 260 (3.43), 212 (3.91) nm; ECD (c 3.11 × 10−4 M, MeCN) λmax (Δε) 331 (+0.21), 257 (+6.69), 232 (+3.96), 208 (+7.98) nm; IR (KBr) νmax 3426, 1719, 1604, 1504, 1472, 1434, 1335, 1259, 1222, 1142, 1118 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 323.2 [M + H]+; HRESIMS m/z 345.0961 [M + Na]+ (calcd for C16H18O7Na+, 345.0945).ε) 323 (4.05), 225 (4.24), 206 (4.26) nm; IR (KBr) νmax 3359, 1722, 1654, 1602, 1385, 1353, 1205, 1139 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 305.0 [M + H]+; HRESIMS m/z 303.0851 [M − H]− (calcd for C16H15O6−, 303.0874).ε) 336 (3.54), 313 (3.53), 261 (3.95), 213 (3.78) nm; IR (KBr) νmax 1725, 1597, 1563, 1451, 1419, 1376, 1360, 1203, 1124, 1088, 1070 cm−1; 1H and 13C NMR data, see Table 1; ESIMS m/z 287.1 [M − H2O + H]+, 305.0 [M + H]+; HRESIMS m/z 327.1195 [M + Na]+ (calcd for C17H20O5Na+, 327.1203).ε) 331 (3.52), 210 (3.82) nm; IR (KBr) νmax 3420, 2966, 2927, 1694, 1654, 1611, 1568, 1458, 1428, 1372, 1137 cm−1; 1H NMR (CDCl3, 400 MHz) δH 6.16 (1H, d, J = 9.6 Hz, H-3), 8.01 (1H, d, J = 9.6 Hz, H-4), 6.41 (1H, s, H-6), 6.31 (1H, s, H-8), 4.55 (2H, d, J = 6.6 Hz, H-1′), 5.43 (1H, br. t, J = 6.8 Hz, H-2′), 2.09 (2H, m, H-4′), 1.73 (3H, s, H-5′), 2.11 (2H, m, H-6′), 5.07 (1H, br. t, J = 6.8 Hz, H-7′), 1.66 (3H, s, H-9′), 1.60 (3H, s, H-10′); 13C NMR (CDCl3, 100 MHz) δC 162.2 (C, C-2), 110.7 (CH, C-3), 139.4 (CH, C-4), 156.9 (C, C-5), 94.4 (CH, C-6), 162.9 (C, C-7), 99.0 (CH, C-8), 154.2 (C, C-9), 103.5 (C, C-10), 65.6 (CH2, C-1′), 118.6 (CH, C-2′), 142.5 (C, C-3′), 39.7 (CH2, C-4′), 16.9 (CH3, C-5′), 26.4 (CH2, C-6′), 123.8 (CH, C-7′), 132.1 (C, C-8′), 25.8 (CH3, C-9′), 17.8 (CH3, C-10′); ESIMS m/z 315.2 [M + H]+; HRESIMS m/z 313.1439 [M − H]− (calcd for C19H21O4−, 313.1445).ε) 329 (3.87), 225 (4.03), 210 (4.20) nm; IR (KBr) νmax 3459, 2929, 2855, 1736, 1610, 1565, 1462, 1382, 1205, 1134 cm−1; 1H NMR (CDCl3, 400 MHz) δH 6.23 (1H, d, J = 9.6 Hz, H-1), 7.82 (1H, d, J = 9.6 Hz, H-2), 6.59 (1H, s, H-8), 3.07 (1H, dd, J = 13.6 and 10.3 Hz, H-1′a), 2.90 (1H, dd, J = 13.6 and 3.1 Hz, H-1′b), 5.20 (1H, dd, J = 10.3 and 3.1 Hz, H-2′), 1.29 (3H, s, H-4′), 1.25 (3H, s, H-5′), 3.85 (3H, s, 5-OMe), 3.89 (3H, s, 7-OMe), 2.11 (1H, m, H-1′′′a), 2.03 (1H, m, H-1′′′b), 1.36 (2H, m, H-2′′′), 1.08 (2H, m, H-3′′′), 1.19 (2H, m, H-3′′′), 1.28 (10H, m, H-6′′′, H-7′′′, H-15′′′, H-16′′′, and H-17′′′), 2.02 (4H, m, H-8′′′ and H-14′′′), 5.32 (2H, m, H-9′′′ and H-13′′′), 5.30 (2H, m, H-10′′′ and H-12′′′), 2.76 (2H, t, J = 6.4 Hz, H-11′′′), 0.88 (3H, t, J = 6.0 Hz, H-18′′′); 13C NMR (CDCl3, 100 MHz) δC 161.1 (C, C-2), 112.7 (CH, C-3), 138.8 (CH, C-4), 156.3 (C, C-5), 116.8 (C, C-6), 162.1 (C, C-7), 95.6 (CH, C-8), 155.4 (C, C-9), 107.1 (C, C-10), 23.9 (CH2, C-1′), 77.9 (CH, C-2′), 72.6 (C, C-3′), 25.4 (CH3, C-4′), 26.9 (CH3, C-5′), 63.3 (CH3, 5-OMe), 56.3 (CH3, 7-OMe), 173.2 (C, C-1′′′), 34.3 (CH2, C-2′′′), 25.0 (CH2, C-3′′′), 29.17, 29.18 (CH2, C-4′′′ and 5′′′), 29.3 (CH2, C-6′′′), 29.7 (CH2, C-7′′′), 27.30, 27.34 (CH2, C-8′′′ and 14′′′), 130.4 (CH, C-9′′′), 128.1 (CH, C-10′′′), 25.8 (CH2, C-11′′′), 128.2 (CH, C-12′′′), 130.2 (CH, C-13′′′), 29.5 (CH2, C-15′′′), 31.7 (CH2, C-16′′′), 22.7 (CH, C-17′′′), 14.2 (CH3, C-18′′′); ESIMS m/z 571.4 [M + H]+; HRESIMS m/z 593.3463 [M + Na]+ (calcd for C34H50O7Na+, 593.3449).ε) 349 (4.22), 232 (4.00), 206 (4.07), 194 (3.85) nm; IR (KBr) νmax 3356, 1695, 1638, 1601, 1587, 1563, 1515, 1442, 1371, 1221, 1169 cm−1; 1H NMR (CDCl3, 400 MHz) δH 7.48 (1H, s, H-2), 7.50 (2H, d, J = 8.8 Hz, H-2′ and H-6′), 6.85 (2H, d, J = 8.8 Hz, H-3′ and H-5′), 7.97 (2H, d, J = 8.8 Hz, H-2′′ and H-6′′), 6.87 (2H, d, J = 8.8 Hz, H-3′′ and H-5′′), 3.91 (3H, s, 1-OMe); 13C NMR (CDCl3, 100 MHz) δC 172.1 (C, C-1), 118.9 (CH, C-2), 148.8 (C, C-3), 188.9 (C, C-4), 126.1 (C, C-1′), 130.0 (CH × 2, C-2′ and C-6′), 117.0 (CH × 2, C-3′ and C-5′), 161.8 (C, C-4′), 130.3 (C, C-1′′), 132.3 (CH × 2, C-2′′ and C-6′′), 116.7 (CH × 2, C-3′′ and C-5′′), 164.9 (C, C-4′′), 53.0 (CH3, 1-OMe); ESIMS m/z 299.1 [M + H]+; HRESIMS m/z 299.0914 [M + H]+ (calcd for C17H15O5+, 299.0914).805 reflections measured (4.46° ≤ 2θ ≤ 144.7°), 5326 unique (Rint = 0.0276, Rsigma = 0.0180) which were used in all calculations. The final R1 was 0.0366 (I ≥ 2σ (I)) and wR2 was 0.0979 (all data). Flack parameter = 0.42 (19). Crystallographic data for (1) has been deposited in the Cambridge Crystallographic Data Centre (CCDC number: 1545037†).Computational section
The details of the ECD and specific optical rotation calculations for compounds 1 and 2 are presented in ESI.†PDE4D inhibitory screening assays
The screening assays for the inhibitory active compounds against PDE4D were performed as we described previously.11,37 For more details about these experimental procedures, please see ESI.†Conclusions
Forty-two compounds including nine new compounds (1a/1b, 2a/2b, and 3–7) were isolated from the stems of T. asiatica. The types of these compounds mainly involved coumarins (1–6 and 8–27), phenolic acid derivatives (7 and 28–33), and alkaloids (34–40). Two racemic mixtures, 1 and 2, were subjected to HPLC with a chiral column to obtain two pairs of enantiomers 1a/1b and 2a/2b, whose absolute configurations were determined by X-ray diffraction analysis and ECD calculation. Interestingly, 1a and 2a both had positive specific optical rotations ([α]20D +61.0 for 1a, +116.2 for 2a), but they had opposite absolute configurations (1′S,2′S,3′R for 1a, 1′R,2′R,3′S for 2a) determined by ECD calculation. The assignments were further confirmed by specific optical rotation calculation ([α]20D +95 for (1′S,2′S,3′R)-1, +117 for (1′R,2′R,3′S)-2). The results of specific optical rotation calculations were in accordance with those of ECD calculations. The PDE4 inhibitory screening assays showed that compounds 12, 19, 21–23, 26, 34, and 35 exhibited moderate activities with IC50 values less than 10 μM, which supported the structure–activity relationship summarized in previous study.11Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 81402813 and 81573302), the Fundamental Research Funds for the Central Universities (No. 14YKPY10), the Science and Technology Planning Project of Guangdong Province, China (No. 2015A020211007), and the National High Technology Research and Development Program of China (863 Project, No. 2015AA020928) for providing financial support to this work.Notes and references
- D. Zhang, G. H. Thomas and J. M. David, Flora of China, 2008, vol. 11, pp. 51–97 Search PubMed.
- Nanjing University of Chinese Medicine, Great Dictionary of Chinese Medicine, Shanghai Scientific & Techincal Publishers, Shanghai, 2nd edn, 2006 Search PubMed.
- S.-M. Fang, G. Fang, R. Liu, Y. Xu and G.-W. Fan, Int. J. Pharm. Res., 2016, 43, 239–248 Search PubMed.
- C. Hirunwong, S. Sukieum, R. Phatchana and C. Yenjai, Phytochem. Lett., 2016, 17, 242–246 CrossRef CAS.
- Z.-H. Tang, Y.-B. Liu, S.-G. Ma, L. Li, Y. Li, J.-D. Jiang, J. Qu and S.-S. Yu, Org. Lett., 2016, 18, 5146–5149 CrossRef CAS PubMed.
- I. L. Tsai, M. F. Wun, C. M. Teng, T. Ishikawa and I. S. Chen, Phytochemistry, 1998, 48, 1377–1382 CrossRef CAS PubMed.
- H. A. Oketch-Rabah, J. W. Mwangi, J. Lisgarten and E. K. Mberu, Fitoterapia, 2000, 71, 636–640 CrossRef CAS PubMed.
- V. Duraipandiyan and S. Ignacimuthu, J. Ethnopharmacol., 2009, 123, 494–498 CrossRef CAS PubMed.
- R. Vazquez, M. E. Riveiro, M. Vermeulen, C. Mondillo, P. H. Coombes, N. R. Crouch, F. Ismail, D. A. Mulholland, A. Baldi, C. Shayo and C. Davio, Phytomedicine, 2012, 19, 737–746 CrossRef CAS PubMed.
- J. Hu, X. Shi, J. Chen, X. Mao, L. Zhu, L. Yu and J. Shi, Food Chem., 2014, 148, 437–444 CrossRef CAS PubMed.
- T.-T. Lin, Y.-Y. Huang, G.-H. Tang, Z.-B. Cheng, X. Liu, H.-B. Luo and S. Yin, J. Nat. Prod., 2014, 77, 955–962 CrossRef CAS PubMed.
- V. Kumar, J. Reisch, D. B. M. Wickremaratne, R. A. Hussain, K. S. Adesina and S. Balasubramaniam, Phytochemistry, 1987, 26, 511–514 CrossRef CAS.
- H. Ishii, J. Kobayashi, M. Ishikawa, J. Haginiwa and T. Ishikawa, Yakugaku Zasshi, 1991, 111, 365–375 CrossRef CAS.
- F. Wang, Y. Xu and J. K. Liu, J. Asian Nat. Prod. Res., 2009, 11, 752–756 CrossRef PubMed.
- M. N. Deshmukh, V. H. Deshpande and A. V. R. Rao, Phytochemistry, 1976, 15, 1419–1420 CrossRef CAS.
- D. Maes, M. E. Riveiro, C. Shayo, C. Davio, S. Debenedetti and N. De Kimpe, Tetrahedron, 2008, 64, 4438–4443 CrossRef CAS.
- M. Ju-Ichi, Y. Takemura, M. Omura, C. Ito and H. Furukawa, Heterocycles, 1999, 51, 851–855 CrossRef.
- J. Suthiwong, U. Sriphana, Y. Thongsri, P. Promsuwan, C. Prariyachatigul and C. Yenjai, Fitoterapia, 2014, 94, 134–141 CrossRef CAS PubMed.
- R. Phatchana and C. Yenjai, Planta Med., 2014, 80, 719–722 CrossRef CAS PubMed.
- H. Ishii, J. Kobayashi and T. Ishikawa, Chem. Pharm. Bull., 1983, 31, 3330–3333 CrossRef CAS.
- R. G. Marwah, M. O. Fatope, M. L. Deadman, Y. M. Al-Maqbali and J. Husband, Tetrahedron, 2007, 63, 8174–8180 CrossRef CAS.
- P. N. Sharma, A. Shoeb, R. S. Kapil and S. P. Popli, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1980, 19, 938–939 Search PubMed.
- M. Nicoletti, F. Delle Monache and G. B. Marini-Bettolo, Planta Med., 1982, 45, 250–251 CrossRef CAS PubMed.
- H. Wei, F. J. Zeng, M. Y. Lu and R. J. Tang, Acta Pharmacol. Sin., 1998, 33, 688–692 CAS.
- H. Ishii, J. Kobayashi, K. Yamaguchi and T. Ishikawa, Chem. Pharm. Bull., 1993, 41, 1655–1656 CrossRef CAS.
- C. M. Smarrito, C. Munari, F. Robert and D. Barron, Org. Biomol. Chem., 2008, 6, 986–987 CAS.
- E. J. Gentry, H. B. Jampani, A. Keshavarz-Shokri, M. D. Morton, D. Vander Velde, H. Telikepalli, L. A. Mitscher, R. Shawar, D. Humble and W. Baker, J. Nat. Prod., 1998, 61, 1187–1193 CrossRef CAS PubMed.
- H. Li, T. Miyahara, Y. Tezuka, T. Namba, N. Nemoto, S. Tonami, H. Seto, T. Tada and S. Kadota, Biol. Pharm. Bull., 1998, 21, 1322–1326 CAS.
- E.-H. Dong, A.-G. Wang, J.-B. Yang, T.-F. Ji and Y.-L. Su, J. Chin. Med. Mater., 2012, 35, 1441–1443 CAS.
- R. R. Stange, J. J. Sims, S. L. Midland and R. E. McDonald, Phytochemistry, 1999, 52, 41–43 CrossRef CAS.
- Y.-X. Zhong, R.-M. Huang, X.-J. Zhou, Y.-H. Zhu, Z.-F. Xu and S.-X. Qiu, Nat. Prod. Res. Dev., 2013, 25, 1515–1518 CAS.
- H. Kim, J. Ralph, F. Lu, S. A. Ralph, A.-M. Boudet, J. J. MacKay, R. R. Sederoff, T. Ito, S. Kawai, H. Ohashi and T. Higuchi, Org. Biomol. Chem., 2003, 1, 268–281 CAS.
- A. P. K. Nissanka, V. Karunaratne, B. M. R. Bandara, V. Kumar, T. Nakanishi, M. Nishi, A. Inada, L. M. V. Tillekeratne, D. S. A. Wijesundara and A. A. L. Gunatilaka, Phytochemistry, 2001, 56, 857–861 CrossRef CAS PubMed.
- B. D. Krane, M. O. Fagbule, M. Shamma and B. Gözler, J. Nat. Prod., 1984, 47, 1–43 CrossRef CAS.
- Y. D. Min, H. C. Kwon, M. C. Yang, K. H. Lee, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2007, 30, 58–63 CrossRef CAS.
- D.-X. Li and Z.-D. Min, Chin. J. Nat. Med., 2004, 2, 285–288 CAS.
- J.-S. Zhang, Y.-H. Zou, Y.-Q. Guo, Z.-Z. Li, G.-H. Tang and S. Yin, RSC Adv., 2016, 6, 53469–53476 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Structures of known compounds, tables of the 1D NMR data assignments of all known compounds, Experimental section, 1D and 2D NMR, MS, HRMS, and IR spectra of new compounds, 1D NMR spectra of known ones. CCDC 1545037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04794k |
| This journal is © The Royal Society of Chemistry 2017 |